
Abstract
The manganese-promoted cobalt supported on gamma alumina was prepared following the incipient wetness impregnation method and employed as a catalyst for carbon dioxide reforming of methane. The addition of manganese has changed the physicochemical properties of the original material. The Co3O4 crystallite size was decreased from 12.14 to 8.66 nm, suggesting a higher metal dispersion. The activation of the catalyst precursor was more effective with less energy required. Notably, the carbon dioxide adsorption over the catalyst surface was greatly enhanced in the presence of 1 wt% manganese. Taken all together, the manganese-promoted catalyst showed an outperformance with higher activity and stability in comparison with the pristine. At 973 K, 1 atm, CH4/CO2 ratio of 1, the addition of 1 wt% Mn into 5Co/Al2O3 led to notable increases in CH4 (15.2%) and CO2 (17.68%) conversions. Also, the stability of Mn-promoted was much higher compared to pristine catalyst with only 0.3 wt% of undesirable coke formed. Meanwhile, it was 4.94 wt% in the case of 5Co/Al2O3 at the same operating condition. Furthermore, the role of carbon dioxide adsorption in carbon dioxide reforming of methane was investigated. At a proper combination of carbon dioxide activation and methane dissociation, the catalytic activity can be significantly enhanced with much lower activation energies for the feedstock gases. Nevertheless, when the adsorption of carbon dioxide was dominant, catalytic activity was much lower and the deactivation occurred rapidly.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The demand for an alternative energy source has drastically grown in recent decades due to the strict environmental regulations, especially the controlling of carbon dioxide emissions generated from fossil fuel combustion. Additionally, oil exploration and agricultural activity release a huge amount of methane—the most harmful greenhouse gas into the atmosphere. The research on potential technologies to reduce carbon footprint by capturing and converting these gases has attracted huge attention from the scientists and government over the world. Among them, the carbon dioxide reforming of methane (CDRM) is the most potential process that can transform two greenhouse gases of CO2 and CH4 at a high temperature into a synthetic gas mixture (syngas) of carbon monoxide (CO) and hydrogen (H2) (cf. Eq. (1)). Compared to other syngas production techniques such as methane steam reforming and partial oxidation, CDRM is advantageous as it can directly employ raw natural gas without requiring a costly CO2 separation [1, 2]. Additionally, the H2/CO ratio in CDRM syngas is close to 1, which is more favorable for Fischer–Tropsch processes to produce synthetic chemicals and fuels [3, 4].
The reforming of the abovementioned greenhouse gases involves the activations of both CH4 and CO2. Methane has a symmetrical structure with four stable C–H bonding (415.5 kJ mol−1) [5, 6]. Meanwhile, due to a linear structure with two bonding of C=O (750 kJ mol−1), CO2 is often considered a relatively inert gas [7]. Based on that, the chemical activations of these feedstock gases require a huge energy supply leading to a highly endothermic CDRM [8]. Therefore, there is a great interest in developing an appropriate catalyst to reduce energy consumption and allow CDRM to be widely commercialized.
Noble metals such as rhodium (Rh) and Ruthenium (Ru) have been employed as catalysts in CDRM because of their excellent performance and high carbon resistance. Nonetheless, the high price and scarcity limit their large-scale applications. In recent years, the catalysts based on cobalt have been considered as a promising substitute due to their good activity and widespread availability compared to noble metals (Fig. 1) [9,10,11]. Nevertheless, as with other catalysts applied in CDRM, this material copes with a serious catalyst deactivation issue relating to the coke formation, sintering, and oxidation of active metal species [12, 13]. Significant attempts have been dedicated to improving stability while retaining the activity of Co-based catalysts. The simplest approach is the doping of third metal to the catalyst acting as a promoter. A proper amount of a promoter in the catalyst could benefit to CDRM towards (i) increasing the number of active sites improving the metal dispersibility [14, 15], (ii) balancing the dissociation of CH4 and CO2 by adjusting basicity/acidity [16,17,18], (iii) effective suppression of the coke formation via gasification reaction [19,20,21].

The number of publications over Co-based catalysts for carbon dioxide reforming of methane from 1998 to 2022 (Source Scopus database accessed on 23 September 2022)
The selection of promoters becomes a crucial link for the catalyst performance in CDRM. Different types of metals, including alkaline and alkaline-earth [13, 17, 22], rare-earth [23,24,25], and transition [19, 26, 27] have been employed and resulted in a great enhancement in catalytic activity. Among them, the metal in the transition group having variable oxidation states is one of the most effective types. Manganese (Mn) has four different oxide forms including MnO, Mn2O3, MnO2, and Mn3O4, posing a high oxygen storage capacity in the crystalline lattice. Hence, it has been used as a promoter for catalytic CDRM [28,29,30,31]. In most of these publications, the addition of Mn showed a significant improvement in reaction performance with better suppression of coke. However, some researchers claimed that the introduction of Mn led to a lower reactivity than pristine catalyst [30, 32].
In this work, the impacts of Mn on the physicochemical properties of Co/γ-Al2O3 were carefully examined using modern techniques. The catalytic evaluation was assessed through CDRM at different operation conditions. The Mn-promoted catalyst showed great enhancements in metal dispersibility, reducibility and basicity. At operation conditions of 973 K, CH4:CO2 = 1:1, atmospheric pressure, the catalyst with Mn dopant gave a much higher activity and better stability. On the other hand, the importance of carbon dioxide activation during CDRM was examined. At a proper combination of carbon dioxide activation and methane dissociation, the catalytic activity considerably increased with much lower activation energies for the feedstock gases. Otherwise, the over-adsorption of CO2 reduced reactant conversions and rapid deactivated of the catalyst.
2 Experimental
2.1 Chemicals
Cobalt (II) nitrate hexahydrate (Co(NO3)2·6H2O) with a purity of 98.0% was supplied by Sigma-Aldrich Chemicals, US. Manganese (II) chloride tetrahydrate (MnCl2·4H2O, 99.0%) was purchased from Xilong Chemical, China. All above chemicals were used as received without any pretreatment. The gamma aluminum oxide (Al2O3) with 99.0% purity was a commercial product of Oakwood Products, Inc.
2.2 Catalyst Preparation and Characterization
2.2.1 The Synthesis of Catalyst Precursors
The catalysts were prepared via the Incipient wetness impregnation method. Precalculated weights of Co(NO3)2·6H2O and MnCl2·4H2O were dissolved in an adequate amount of deionized water. The resulting mixture was impregnated over γ-Al2O3, followed by 1 h sonication (TP-01 ultrasonic bath, Taiwan Total Meter, 40 kHz, 60 W). The slurry was then dried at 353 K, 12 h, and air-calcined at 873 K for 3 h. The calcinated solid was collected separately in the vials and labeled as 5Co/Al2O3 and 1Mn–5Co/Al2O3 catalyst precursors. The cobalt content in the catalysts was fixed at 5 wt% and the manganese contents in the promoted sample were 1 wt%.
2.2.2 Catalyst Characterization
The Brunauer–Emmett–Teller (BET) surface area measurement for all samples was evaluated in a Micromeritics 2020 surface analyzer using N2 adsorption–desorption at 77 K. Before conducting the analysis, the sample was degassed at 523 K for 5 h under vacuum. The pore properties including average diameter and volume were estimated according to Barrett–Joyner–Halenda (BJH) method.
The X-ray powder diffraction (XRD) studies were performed on a Bruker Benchtop X-ray diffraction—D2 Phaser, equipped with a Cu-Kα radiation source (λ = 1.5406 Å). All the samples were analyzed at a high angle between 20° and 80°. The crystalline phase was assigned according to International Centre for Diffraction Data (ICDD).
The reducibility of the catalyst precursor was assessed via temperature-programmed reduction (H2-TPR). The analysis was carried out using an AutoChem II-2920 system. A fixed amount of 0.05 g precursor was placed at the center of an U-tube quartz. Moisture was eliminated from the precursor using a 50 ml min−1 N2, 373 K, 0.5 h. After that, the dried precursor has undergone the reduction in the presence of a 50 ml min−1 of 10% H2/N2 gas mixture. The furnace temperature was programmed to increase to 1173 K with a heating rate of 10 K min−1. At the targeted temperature, the operating conditions were maintained for 30 min to assure the complete reduction. Then, the quartz tube was cooled down to room temperature in an N2 flow of 50 ml min−1. The hydrogen consumption of each sample following temperature was recorded every 1 s.
The carbon dioxide temperature-programmed desorption (CO2-TPD) analysis was employed to estimate the catalyst basicity. The experiment was carried out in the same apparatus as described in the H2-TPR section. In brief, 0.1 g of the precursor was dried at 373 K in the inert gas atmosphere of 50 ml min−1 of He flow, 1 h. The activation of the precursor was conducted at 1023 K using a reducing agent of 50 ml min−1 of 10% H2/N2, 1 h. The quartz tube was then cooled down to 423 K under a 50 ml min−1 Ar flow. The CO2 adsorption was initiated by passing a 20 ml min−1 CO2 (99.99%) through the catalyst bed at 423 K. After 2 h, the unabsorbed CO2 was removed by an Ar flow. Subsequently, furnace temperature was increased from up to 1173 K, 10 K min−1 with the continuous flow of 50 ml min−1 Ar. The desorption of carbon dioxide was recorded using a thermal conductivity detector (TCD).
The morphology and total weight of coke formed on the CDRM used catalyst was estimated using temperature-programmed oxidation (TPO). At the end of the CDRM run, the quartz tube was cooled to 323 K in the Ar atmosphere. The combustion of coke was investigated by increasing of temperature to 1173 K, 10 K min−1 in the 60 ml min−1 of dried air flow. The concentrations of carbon dioxide and carbon monoxide in the outlet gas mixture were quantitatively analyzed time-on-stream using CO2 and CO sensors (Alphasense, UK), respectively.
The morphology of CDRM used catalysts was studied using transmission electron microscopy in a JEOL, JEM-1400 Plus apparatus.
2.3 Catalytic Activity Evaluation
The dry reforming reactions were conducted in a fixed-bed downflow reactor at atmospheric pressure. In a typical run, about 0.1 precursor was loaded in the center of the quartz tube. The reactor temperature was precisely monitored via a proportional–integral–derivative (PID) controller equipped with a K-type thermocouple. Prior to the reaction, in-situ activation of the precursor was carried out at 1023 K using a reducing agent of 40% H2/N2 (60 ml min−1), 2 h. After the in-situ activation, the temperature was adjusted to 973 K under a 60 ml min−1 of N2. The CDRM was then triggered by introducing reactants of CO2 and CH4 at an equal mole ratio and diluted in pure N2. All experiments were carried out in 6 h with a fixed gas hourly space velocity (GHSV) of 36 L gcat−1 h−1. The flows of each reactant and N2 carrier gases were strictly monitored using calibrated mass flow rate controllers (Horiba-MFC). The composition of the output product was analyzed online using Agilent 6890-GC equipped with two thermal conductivity detectors. The operating conditions strictly followed the typical procedure described above, unless otherwise stated.
To ensure accuracy and reproducibility, each experiment was repeated at least two times with an experimental deviation of less than 2.5%. CH4 and CO2 conversions (\({\mathrm{X}}_{{\mathrm{CH}}_{4}}\) \({\mathrm{X}}_{{\mathrm{CH}}_{4}}\) XCH4 and XCO2), product yields (\({\mathrm{Y}}_{\mathrm{CO}}\) \({\mathrm{Y}}_{\mathrm{CO}}\) YCO and YH2), as well as H2/CO ratio were calculated via Eqs. (2)–(6).
and
where \(F_{{}}^{in}\) and \(F_{{}}^{out}\) are the inlet and outlet flowrate (mol s−1), respectively.
The degree of deactivation of CH4 (\(DE_{{{\text{CH}}_{4} }}\)) and CO2 (\(DE_{{{\text{CO}}_{2} }}\)) was computed by Eqs. (7) and (8) as follows:
where \(X_{i}^{in/fi} \,\) is the initial conversion (in) and conversion after 6 h (fi) of CH4 and CO2.
3 Results and Discussion
3.1 Characterization of the Precursor and Fresh Catalyst
3.1.1 Textural Properties
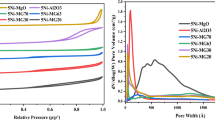
The textural characteristics unpromoted and 1Mn–5Co/Al2O3 precursors were measured by N2 adsorption–desorption isotherm curves as presented in Fig. 2a. The profile of gamma-alumina (γ-Al2O3) that has been investigated in our previous publication [24] was also included as a compared sample with permission. According to IUPAC classification, the physisorption isotherms of all samples were assigned to type IV with H2 hysteresis loops (P/P0 ≥ 0.5) [33]. These features were presentative of the mesoporous materials [34]. The pore size distributions in Fig. 2b confirmed the mesoporous structure of the precursors with pore widths ranging from 2 to 12 nm. The BET surface area (SBET), total pore volume (Vp), and average pore diameter (Dp) are reported in Table 1. For γ-Al2O3, SBET and Vp were 126.9 m2 g−1 and 0.258 cm3 g−1, respectively. Nevertheless, both SBET and Vp were notably decreased to 106.6–109.2 m2 g−1 and 0.202–0.210 cm3 g−1 in the additions of either cobalt or manganese. These declines were indicative of the well dispersed Co and Mn over the pore and surface of the support. This observation is consistent with the findings reported by Bahari et al. [35] and Tran et al. [36]. Remarkably, an increase in surface area of 1Mn–5Co/Al2O3 compared to 5Co/Al2O3 [36] could be explained by the occupation of Mn promoter between Co particles and hence preventing Co agglomeration. Ayodele et al. [37] indicated that the higher dispersion of Co metal on the support resulted in the formation of more fine particles and thus higher BET specific surface area.

N2 adsorption–desorption isotherms for γ-Al2O3 and Co-based catalyst precursors (a) and pore size distribution (b)
3.1.2 XRD Analysis
The crystalline phases of γ-Al2O3, unpromoted, and Mn-promoted precursors were studied via XRD analysis. As depicted in Fig. 3, all XRD diffractograms showed three reflections at 2θ = 37.9°, 45.96°, and 67.02°, assigned to γ-Al2O3 (ICDD 00-004-0858) [39]. In the precursor samples, the diffraction peaks located at 2θ = 31.4°, 37.1°, and 45.2° were ascribed to crystalline Co3O4 (ICDD 00-042-1467) [40] generated due to the thermal decomposition of cobalt salt (Eq. (9)).

XRD profiles of support and Co-based precursor catalysts
Additionally, the existence of CoAl2O4 was verified by the presence of the characteristic peaks at 2θ = 59.6°, 65.4° (ICDD 01-082-2248) [41]. This compound represents the strong interaction between cobalt oxide and the support. In contrast, no signal of manganese oxides was detected in the Mn-promoted precursor. The absence of these peaks might be due to the low dosage and well dispersion of the Mn oxides over γ-Al2O3, agreed with BET analysis. Consequently, the particle size of manganese oxides obtained after air calcination was below the detectable range of XRD. A similar result has been reported in previous publications [26, 42, 43].
The average Co3O4 crystallite size was estimated using the Scherrer equation [38] for the diffraction peak of 2θ = 37.1°. The results are also presented in Table 1. The Co3O4 average size was in the range of 8.66–12.14 nm, confirming the high dispersibility of cobalt over the support. Moreover, the addition of Mn showed a crucial impact on the growth of Co3O4 with a significant decline in Co3O4 average size found on the promoted catalyst precursor. Particularly, the pristine precursor had an average Co3O4 crystallite size of 12.14 nm, while it was only 8.66 nm for 1Mn–5Co/Al2O3. The decrease in this parameter might be explained by the dilution effect, where the promoter act as an inhibiter for Co3O4 sintering during the preparation of the precursor [17, 44, 45]. In other words, the addition of Mn improved the dispersibility of cobalt oxide over gamma-alumina support.
3.1.3 Reducibility Characterization
The redox properties of catalyst precursors were studied via H2-TPR and presented in Fig. 4. The H2-TPR profile of the support showed no peak due to the inert of alumina with H2. In the precursor samples, two major reduction peaks were found, corresponding to the two-step reduction of Co3O4. The first characteristic peak located at a low-temperature range (below 800 K) was assigned to the initial reduction of Co3+ to Co2+ (Eq. (10)) [20, 46]. The second peak between 800 and 1000 K represented to the reaction to convert Co2+ to Co0, as demonstrated in Eq. (11) [47, 48]. Furthermore, the presence of the minor shoulder peak at high temperatures beyond 1000 K was ascribed to the reduction of CoAl2O4 (Eq. (12)) [20], confirming the existence of this crystalline phase as observed from XRD patterns. However, the intensity and area of this CoAl2O4 peak were almost trivial compared to the others, implying the inferior quantity of the CoAl2O4 phase in the precursors. Hence, the potential loss in the activity of the catalyst caused by the incomplete reduction of CoAl2O4 is negligible.

H2-TPR profiles of γ-Al2O3, unpromoted, and 1Mn–5Co/Al2O3
Despite having the same number of reduction peaks, there is a significant difference in the peak center between promoted and pristine precursors. The reduction peak of Co3O4 to CoO considerably shifted to lower temperature, from 724 K (5Co/Al2O3) to 668 K (1Mn-5Co/Al2O3). The decline in the first reduction peak could be attributed to the increased oxygen vacancy mobility owing to the incorporation of the oxide form of dopant, which then facilitated H2 reduction [49]. Meanwhile, the second reduction peak was found to either relocate into a higher temperature range or be broadened in Mn-promoted samples. This phenomenon might be due to the peak overlapping between the second reduction step of cobalt and the reduction of manganese oxides occurring at temperatures above 773 K [50]. Moreover, comparing pristine and Mn-promoted catalysts, 1Mn–5Co/Al2O3 showed a tiny peak at the temperature range above 1000 K, whereas no clear signal was observed in the case of 5Co/Al2O3. It suggested a higher amount of CoAl2O4 in 1Mn–5Co/Al2O3. That might come from the active metal dispersibility effect of Mn to generate a smaller size of cobalt oxide and later induce the formation of CoAl2O4 during the calcination of catalyst precursor [51].
Table 2 presents the quantitative hydrogen uptake of precursors. The unpromoted precursor had a low H2 uptake of 0.127 mmol g−1. An addition of 1 wt% Mn showed a positive effect on the reducibility of precursor with a notable increase up to 0.189 mmol g−1. It suggested that the doping of Mn was beneficial to the activation of the catalyst precursor.
3.1.4 Basicity Measurement
The acidity/basicity of unpromoted and Mn-promoted Co/Al2O3 catalysts was evaluated using CO2-TPD analysis. The results are depicted in Fig. 5. In general, the basic site can be cataloged into three distinct groups based on the CO2 desorption temperature. They are weak (300–400 K), moderate (400–500 K), and strong (> 500 K) [52]. As illustrated in Fig. 5, all synthesized catalysts showed three distinct desorption peaks in the range of 300–600 K, with the majority located in the moderate basic site. Compared to the unpromoted catalyst, the peak found on 1Mn–5Co/Al2O3 showed big increases in intensity and area, implying the enhancement of CO2 chemisorption. Moreover, the quantitative analysis of the catalyst was estimated and summarized in Table 2. The surface basicity of 5Co/Al2O3 was 317.7 μmol CO2 g−1, meanwhile it was 600.9 μmol CO2 g−1 in the case of Mn-promoted catalyst. The big difference between these two cases is due to the basic nature of manganese oxide, which has a good affinity with CO2.

CO2-TPD profiles for precursor catalysts
3.2 Carbon Dioxide Reforming of Methane Performance
3.2.1 Promotional Effect of Manganese
CDRM experiments were performed at 973 K, CH4:CO2 = 1 (mole/mole) and GHSV = 36 L gcat−1 h−1 for 6 h. Figure 6a and b present CH4 and CO2 conversions versus time-on-stream for the unpromoted and 1Mn–5Co/Al2O3 catalysts. As depicted in Fig. 6, the catalyst activity of the Mn-promoted material was outperformed in comparison to the original. Particularly, the initial CH4 conversion was notably changed from 50.6 to 59.7% when 5Co/Al2O3 was switched to 1Mn–5Co/Al2O3. Since the dissociation of methane widely accepted by scientists, is occurred over the active metal [12], the increase in CH4 conversion is correlated with the higher number of catalytic sites. That agreed with the XRD results where the average size of Co3O4 was substantially decreased in the addition of 1 wt% of Mn. In terms of CO2 conversion, a remarkable enhancement from 62.4% (5Co/Al2O3) to 75.8% (1Mn–5Co/Al2O3) was observed. The CO2 conversion of promoted catalyst even exceeded 66.3% expected for the system at thermodynamic equilibrium (CH4:CO2 = 1, 973 K and 1 atm) [53]. This might be due to the occurrence of the reverse water gas shift reaction (RWGS) (Eq. (13)), as reported in other studies [32, 42].

Effects of Mn promoter on (a) CH4 conversion and (b) CO2 conversion of 5Co/Al2O3 at 973 K
The stability of the catalyst was justified based on the degree of deactivation (DE) in activity shown in Table 3. The incorporation of Mn improved the stability of the catalyst. Specifically, 5Co/Al2O3 suffered high deactivation even in a short period of 6 h. The \(DE_{{{\text{CH}}_{4} }}\) and \(DE_{{{\text{CO}}_{2} }}\) were 83.4% and 100%, respectively. The declines in activity were mainly caused by the coke deposition, as reported in the literature [26, 54]. The coverage of coke over catalyst inhibited the adsorption as well as dissociation of carbon dioxide, leading to the complete suppression of CO2 conversion. Meanwhile, the thermal cracking of C–H bonding requires less energy and possibly continues to occur but with a slow rate. When 1Mn–5Co/Al2O3 was employed, these parameters were found to be much lower with 19.1% and 8.1% corresponding to \(DE_{{{\text{CH}}_{4} }}\) and \(DE_{{{\text{CO}}_{2} }}\). The notable enhancement in the stability of 1Mn–5Co/Al2O3 could be due to the basic nature of manganese oxide that facilitates CO2 adsorption over the catalyst (see Table 2). Consequently, the reverse-Boudouard reaction (Eq. (14)) was boosted to eliminate the carbon deposited on the catalytic active center. It is in line with the higher CO2 conversion observed in 1Mn–5Co/Al2O3 compared to 5Co/Al2O3 (cf. Fig. 6b). In addition, the higher resistance to coke of Mn-promoted catalyst in comparison with the pristine also came from the better dispersion of cobalt metal over γ-Al2O3 (see Table 1). According to Christensen, a smaller active metal crystallite size possibly resulted in a greater saturation concentration of carbon and hence a lower driving force for diffusion of carbon through active metal particles [55].
The yields of products and the ratio between H2 and CO in the outlet gas are presented in Fig. 7 and Table 3. The initial yields of H2 and CO over 5Co/Al2O3 were 35.8% and 58.9%, respectively. However, following the rapid deactivation of the catalyst, no sign of these gases was detected after 6 h of operation. In contrast, over 1Mn–5Co/Al2O3, the productions of hydrogen and carbon monoxide were higher with 58.9% H2 and 68.5% CO at the beginning of the process. Additionally, much lower deactivation degree was observed as presented in Table 3. Regarding H2/CO ratio, it was lower than the theoretical values of 1 due to the parallel occurrence of the side reactions, typically RWGS [56, 57]. The addition of Mn has a huge impact on the H2/CO yield. The initial H2/CO was increased from 0.60 to 0.64 and after 6 h of reaction time, it was 0.56 compared to 0.11 in 5Co/Al2O3. In other words, the addition of 1 wt% of Mn was beneficial for not only yield but also the quality of the product with a higher H2/CO ratio.

Effects of Mn promoter on (a) H2 yield and (b) CO yield of 5Co/Al2O3 at 973 K
Table 4 demonstrates the DRM performance of the Co-based catalysts in our work and other studies. Generally, the catalyst activity of 1Mn–5Co/Al2O3 was much higher than 5Co/Al2O3. The performance of 1Mn–5Co/Al2O3 was comparable and even greater than that of other rare-earth meta-containing catalysts, recently reported in the literature. With low-cost starting material and efficient performance, 1Mn–5Co/Al2O3 could be a potential DRM catalyst for practical application.
3.2.2 Effect of Operating Temperature on CDRM Catalytic Performance
The catalytic activities of unpromoted and Mn-promoted catalysts in terms of CH4 (a) and CO2 conversion (b) were investigated at three temperatures of 923 K, 973 K and 1023 K. In this section, all reforming runs were performed at constant atmospheric pressure, CH4:CO2 = 1:1 and GHSV = 36 L gcat−1 h−1. Results are presented in Fig. 8. The operating temperature plays a key role in the conversion of feedstock. For all catalysts, the feedstock conversions were always proportional to the reaction severity, confirming the endothermic characteristic of CDRM. Regardless of catalyst and temperatures, the CO2 conversions were higher than those of CH4 because of the presence of RWGS [59, 60]. Between the two catalysts, the experiments using 1Mn–5Co/Al2O3 always had higher feedstock conversions in comparison with 5Co/Al2O3 at similar operating conditions.

Effect of operating temperature on CH4 (a) and CO2 (b) conversion of 5Co/Al2O3 and 1Mn–5Co/Al2O3
The apparent activation energies (Ea) were predicted from the slopes of Arrhenius plots, as presented in Fig. S1 of the Supporting Information. Ea of the feedstocks showed a large dependence on the catalyst. For 5Co/Al2O3, the Ea of CH4 and CO2 consumptions were 80.86 and 63.56 kJ mol−1, whilst 1Mn–5Co/Al2O3 were 49.13 and 30.17 kJ mol−1, respectively. The Ea values for 1Mn–5Co/Al2O3 were lower than those for 5Co/Al2O3, indicating the efficient catalytic of the Mn-promoted catalyst.
3.2.3 Effect of Feedstock Composition
The impacts of carbon dioxide partial pressure on CDRM were studied by changing the composition of the feedstock. In particular, the ratios between CH4 and CO2 in the feedstock were adjusted to 1:1, 1:1.5, and 1:2 (kPa kPa−1). All experiments in this section were carried out at 973 K, 1 atm, and GHSV of 36 L gcat−1 h−1, 6 h. The effect of feedstock composition on initial methane conversion (\(X_{{{\text{CH}}_{4} }}^{in}\)) is shown in Fig. 9.

The effect of feedstock composition on initial CH4 conversion
Different trends were observed in Mn-promoted and unpromoted catalysts. In the case of 5Co/Al2O3, the \(X_{{{\text{CH}}_{4} }}^{in}\) increased significantly from 50.6 to 73.5% when the CH4:CO2 ratio was increased from 1:1 to 1:1.5. Nevertheless, it dropped to 59.7% at the time of CH4:CO2 = 1:2. In contrast, a downtrend was found in the case of 1Mn–5Co/Al2O3. The \(X_{{{\text{CH}}_{4} }}^{in}\) continued to decrease from 59.72% (CH4:CO2 = 1:1) to 51.75% (CH4:CO2 = 1:1.5) and 19.54% (CH4:CO2 = 1:2). The different behaviors of CDRM performance were possibly due to the basicity of the two materials. In the case of pristine catalyst, it had relatively low basicity as estimated using the CO2-TPD technique. The increase in CO2 partial pressure enhanced the adsorption of this gas over the cobalt active metal. In CDRM, the activations of methane and carbon dioxide are highly endothermic and competing processes [12]. Hence, the enhancement of CO2 adsorption would impede the CH4 dissociation, which was a thermally preferable reaction because of having lower C–H bonding energy (415.5 kJ mol−1) than C=O (750 kJ mol−1). When CO2 activation and CH4 dissociation occurred at proper rates, the catalytic activity can be significantly enhanced [61]. The continued increase in CO2 partial pressure led to over-adsorption of CO2, reduced the accessibility of CH4 to Co0 active metal, and finally decreased the X0CH4 [17, 20]. Similar trends were shown in the previous publications [62, 63]. In the case of 1Mn–5Co/Al2O3, this material had high basicity of 600.9 μmol CO2 g−1 (see Table 2). The adsorption of CO2 was already boosted at the run having the lowest CH4:CO2 ratio of 1. The continued increase of CO2 partial pressure surely led to the over-adsorption of this gas and resulted in a dramatic decrease \(X_{{{\text{CH}}_{4} }}^{in}\). This result agreed with the literature where a high basicity catalyst was employed and witnessed a clear decrease in \(X_{{{\text{CH}}_{4} }}^{in}\) along with the increase in CO2:CH4 ratio [37].
The effect of feedstock ratio on the deactivation degree (calculated based on the decrease in CH4 conversion) is shown in Fig. 10. The pristine catalyst exhibited a very high DECH4 of 83.44% at CH4:CO2 = 1:1. The increase of CO2 partial pressure to CH4:CO2 = 1:1.5 significantly mitigated the catalyst deactivation to only 11.82% of DECH4. However, it was raised to 47.02% at a further increase of CH4:CO2 = 1:2. On the other hand, the DECH4 found in experiments using 1Mn–5Co/Al2O3 catalyst, exhibited a continued uptrend. Especially, this catalyst was completely deactivated at CH4:CO2 = 1:2. This phenomenon was attributed to the adsorption of CO2 over the catalyst surface. When it was below the adsorption limit, either enhancement of basicity or increase of CO2 partial pressure led to an improvement in not only activity but also stability of the catalyst. Over that limitation, it resulted in reducing the accessibility of methane or catalyst deactivation by the oxidation of active meal [64, 65]. Hence, the addition of a promoter can cause different effects on catalyst performance depending on its loading or operating conditions. That might explain the contradictory results that have been reported in the promoter studies.

The effect of feedstock ratio on the deactivation degree
3.3 Spent Catalysts Characterization
It is well known that coke is the main cause of catalyst decay. In this study, the coke formed on the CDRM spent catalyst (CH4:CO2 = 1:1, 973 K and 6 h) was characterized using TPO with the result shown in Fig. 11 and Table 5. Along with the quantity of coke, its morphology also has a critical impact on catalyst lifetime. High amorphous carbon is relatively active and can be easily removed through the gasification reaction (Eq. (14)) [66]. However, this type of coke has a high specific area and might cover the Co0 particle leading to the decay of the catalyst [67, 68]. In contrast, the carbon with a low amorphous degree has a lower specific area and is lesser harmful to the catalyst. The disadvantage of this carbon is the difficulty to remove and requires a high operating temperature during catalyst generation. From TPO data, the combustion temperature is indicative of distinguishing between these two types of coke.

TPO profiles of spent 5Co/Al2O3 and 1Mn–5Co/Al2O3. Operating conditions: 973 K, CH4:CO2 = 1:1, GHSV = 36 L gcat−1 h−1
According to the TPO profiles shown in Fig. 11, regardless of catalyst, the combustion temperature of coke lies in the range of 425–900 K, corresponding to the high and low amorphous carbon. For 5Co/Al2O3, two separated peaks with centers located at around 490 K and 800 K were discovered. Meanwhile, the addition of 1 wt% Mn witnessed the suppression of the high amorphous carbon peak. Based on the peak intensities, the yield of coke formed on spent 5Co/Al2O3 and 1Mn–5Co/Al2O3 was estimated, and the results were presented in Table 5. A high coke of 4.94 wt% with a rapid formation rate of 1.37 × 10–4 (gcarbon gcat−1 min−1) was found when an unpromoted catalyst was used. A notable decrease in carbon deposition was achieved in the case of 1Mn–5Co/Al2O3 with only 0.30 wt%. The great deduction of coke yield and slower coke formation rate observed in Mn-promoted catalysts were possibly due to the high basicity leading to facilitating carbon gasification [69,70,71]. Since the high amorphous carbon is much more active than the other, it was rapidly wiped out of the catalyst surface and resulted in the almost disappearance of the low-temperature peak on the TPO profile of spent 1Mn–5Co/Al2O3.
Morphology of carbon deposited on spent catalysts was studied using TEM and the result was displayed in Fig. 12. As reported in TPO analysis, TEM images also showed the presence of amorphous and graphitic carbon in both spent catalysts. This observation was in line with other works [36, 72]. The amorphous carbon was easily seen like a mist covering the catalyst in the used 5Co/Al2O3. In contrast, the TEM of 1Mn–5Co/Al2O3 was much clearer with major coke in the form of graphitic carbon. Hence, in a manner of speaking, Mn serves as a coke inhibitor that allows the catalyst to have a higher in both activity and stability.

TEM micrographs for (a) 5Co/Al2O3 and (b) 1Mn–5Co/Al2O3
Based on the results, the role of Mn in the CDRM catalytic performance of cobalt-based catalyst was proposed in Scheme 1. The Mn-promoted catalyst had a better active metal dispersibility and then gave a higher initial feedstock conversion. In addition, owing to the basic nature of the promoter, manganese oxide became additional active sites for CO2 adsorption and activation. Consequently, it facilitated the carbon gasification to suppress coke formation and then improve catalyst stability.

The role of Mn in the performance of Co-based catalysts
4 Conclusions
In this study, the role of carbon dioxide adsorption and activation was studied. The doping of Mn over the Co/Al2O3 notably improved the dispersibility and reducibility of cobalt oxides over the gamma alumina. Especially, with a low loading of 1 wt% Mn, the basicity of catalyst was found twice compared to the original. That led to much better adsorption of CO2 and resulted in a great CDRM performance of 1Mn–5Co/Al2O3 at 973 K, CO2:CH4 = 1:1. At this condition, the coke resistance of Mn-promoted catalyst was also enhanced. The good adsorption of CO2 onto the catalyst surface also led to the suppression of coke, typically the amorphous type. Both carbon deposition and formation rate were decreased from 4.94 wt% and 1.37 × 10–4 gcarbon gcat−1 min−1 (5Co/Al2O3) to only 0.30 wt% and 8.35 × 10–6 gcarbon gcat−1 min−1 (1Mn–5Co/Al2O3), respectively. At a proper combination of CO2 activation and CH4 dissociation, the catalytic activity can be significantly enhanced with much lower activation energies for the feedstock gases. However, when the adsorption of CO2 was dominant, catalytic activity was much lower and the deactivation occurred rapidly.
Abbreviations
- CDRM:
-
Carbon dioxide reforming of methane
- GHSV:
-
Gas hourly space velocity
- XRD:
-
X-ray diffraction
- BET:
-
Brunauer–Emmett–Teller
- BJH:
-
Barrett–Joyner–Halenda
- H2-TPR:
-
Hydrogen temperature-programmed reduction
- CO2-TPD:
-
Carbon dioxide temperature-programmed desorption
- TPO:
-
Temperature-programmed oxidation
- TEM:
-
Transmission electron microscopy
- TOS:
-
Time-on-stream
- RWGS:
-
Reverse water gas shift
References
Sun Y, Zhang G, Xu Y et al (2019) Comparative study on dry reforming of methane over Co-M (M = Ce, Fe, Zr) catalysts supported on N-doped activated carbon. Fuel Process Technol 192:1–12. https://doi.org/10.1016/j.fuproc.2019.04.017
Kawi S, Kathiraser Y, Ni J et al (2015) Progress in synthesis of highly active and stable nickel-based catalysts for carbon dioxide reforming of methane. ChemSusChem 8:3556–3575. https://doi.org/10.1002/cssc.201500390
Serrano-Lotina A, Daza L (2014) Influence of the operating parameters over dry reforming of methane to syngas. Int J Hydrogen Energy 39:4089–4094. https://doi.org/10.1016/j.ijhydene.2013.05.135
Rostrup-Nielsen JR (2000) New aspects of syngas production and use. Catal Today 63:159–164. https://doi.org/10.1016/S0920-5861(00)00455-7
Tang P, Zhu Q, Wu Z, Ma D (2014) Methane activation: the past and future. Energy Environ Sci 7:2580–2591. https://doi.org/10.1039/c4ee00604f
Hammond C, Conrad S, Hermans I (2012) Oxidative methane upgrading. ChemSusChem 5:1668–1686. https://doi.org/10.1002/cssc.201200299
North M (2015) What is CO2? Thermodynamics, basic reactions and physical chemistry. Carbon Dioxide Util Closing Carbon Cycle First Ed. https://doi.org/10.1016/B978-0-444-62746-9.00001-3
Pakhare D, Spivey J (2014) A review of dry (CO2) reforming of methane over noble metal catalysts. Chem Soc Rev 43:7813–7837. https://doi.org/10.1039/c3cs60395d
Taherian Z, Yousefpour M, Tajally M, Khoshandam B (2017) Promotional effect of samarium on the activity and stability of Ni-SBA-15 catalysts in dry reforming of methane. Microporous Mesoporous Mater 251:9–18. https://doi.org/10.1016/j.micromeso.2017.05.027
Chen S, Zaffran J, Yang B (2020) Dry reforming of methane over the cobalt catalyst: theoretical insights into the reaction kinetics and mechanism for catalyst deactivation. Appl Catal B Environ 270:118859. https://doi.org/10.1016/j.apcatb.2020.118859
Zhang Y, Liang Z, Zhang G et al (2022) Highly active and stable cobalt catalysts with a tungsten carbide-activated carbon support for dry reforming of methane: effect of the different promoters. Catal Sci Technol 12:4871–4883. https://doi.org/10.1039/d2cy00833e
Budiman AW, Song SH, Chang TS et al (2012) Dry reforming of methane over cobalt catalysts: a literature review of catalyst development. Catal Surv Asia 16:183–197. https://doi.org/10.1007/s10563-012-9143-2
Fakeeha AH, Naeem MA, Khan WU, Al-Fatesh AS (2014) Syngas production via CO2 reforming of methane using Co-Sr-Al catalyst. J Ind Eng Chem 20:549–557. https://doi.org/10.1016/j.jiec.2013.05.013
Das S, Sengupta M, Bag A et al (2021) Gd-Ru nanoparticles supported on Zr0.5Ce0.5O2 nanorods for dry methane reforming. ACS Appl Nano Mater 4:2547–2557. https://doi.org/10.1021/acsanm.0c03140
Fakeeha AH, Al Fatesh AS, Ibrahim AA et al (2021) Yttria modified ZrO2 supported Ni catalysts for CO2 reforming of methane: the role of Ce promoter. ACS Omega 6:1280–1288. https://doi.org/10.1021/acsomega.0c04731
Hambali HU, Jalil AA, Abdulrasheed AA et al (2020) Effect of Ni-Ta ratio on the catalytic selectivity of fibrous Ni-Ta/ZSM-5 for dry reforming of methane. Chem Eng Sci 227:115952. https://doi.org/10.1016/j.ces.2020.115952
Vo CM, Cao ANT, Saleh Qazaq A et al (2022) Toward syngas production from simulated biogas dry reforming: promotional effect of calcium on cobalt-based catalysts performance. Fuel 326:125106. https://doi.org/10.1016/j.fuel.2022.125106
Dama S, Ghodke SR, Bobade R et al (2018) Active and durable alkaline earth metal substituted perovskite catalysts for dry reforming of methane. Appl Catal B Environ 224:146–158. https://doi.org/10.1016/j.apcatb.2017.10.048
Lu Y, Kang L, Guo D et al (2021) Double-site doping of a v promoter on Nix-V-MgAl catalysts for the DRM reaction: simultaneous effect on CH4 and CO2 activation. ACS Catal 11:8749–8765. https://doi.org/10.1021/acscatal.1c01299
Cao ANT, Pham CQ, Pham LKH et al (2021) Boosted methane dry reforming for hydrogen generation on cobalt catalyst with small cerium dosage. Int J Hydrogen Energy. https://doi.org/10.1016/j.ijhydene.2021.11.077
Luisetto I, Tuti S, Romano C et al (2019) Dry reforming of methane over Ni supported on doped CeO2: new insight on the role of dopants for CO2 activation. J CO2 Util 30:63–78. https://doi.org/10.1016/j.jcou.2019.01.006
Wysocka I, Mielewczyk-Gryń A, Łapiński M et al (2021) Effect of small quantities of potassium promoter and steam on the catalytic properties of nickel catalysts in dry/combined methane reforming. Int J Hydrogen Energy 46:3847–3864. https://doi.org/10.1016/j.ijhydene.2020.10.189
Rodriguez-Gomez A, Lopez-Martin A, Ramirez A et al (2021) Elucidating the promotional effect of cerium in the dry reforming of methane. ChemCatChem 13:553–563. https://doi.org/10.1002/cctc.202001527
Cao ANT, Pham CQ, Nguyen TM et al (2022) Dysprosium promotion on Co/Al2O3 catalysts towards enhanced hydrogen generation from methane dry reforming. Fuel 324:124818. https://doi.org/10.1016/j.fuel.2022.124818
Zhang G, Wang Y, Li X et al (2018) Effect of Gd promoter on the structure and catalytic performance of mesoporous Ni/Al2 O3-CeO2 in dry reforming of methane. Ind Eng Chem Res 57:17076–17085. https://doi.org/10.1021/acs.iecr.8b03612
Al-Fatesh AS, Naeem MA, Fakeeha AH, Abasaeed AE (2015) The effect of Sc promoter on the performance of Co/TiO2-P25 catalyst in dry reforming of methane. Bull Korean Chem Soc 36:2081–2088. https://doi.org/10.1002/bkcs.10408
Sajjadi SM, Haghighi M, Eslami AA, Rahmani F (2013) Hydrogen production via CO2-reforming of methane over Cu and Co doped Ni/Al2O3 nanocatalyst: impregnation versus sol-gel method and effect of process conditions and promoter. J Sol-Gel Sci Technol 67:601–617. https://doi.org/10.1007/s10971-013-3120-8
Wang Y, Li L, Cui C et al (2022) The effect of adsorbed oxygen species on carbon-resistance of Ni-Zr catalyst modified by Al and Mn for dry reforming of methane. Catal Today 384–386:257–264. https://doi.org/10.1016/j.cattod.2021.03.004
Shahnazi A, Firoozi S (2021) Improving the catalytic performance of LaNiO3perovskite by manganese substitution via ultrasonic spray pyrolysis for dry reforming of methane. J CO2 Util 45:101455. https://doi.org/10.1016/j.jcou.2021.101455
Liu H, Hadjltaief HB, Benzina M et al (2019) Natural clay based nickel catalysts for dry reforming of methane: on the effect of support promotion (La, Al, Mn). Int J Hydrogen Energy 44:246–255. https://doi.org/10.1016/j.ijhydene.2018.03.004
Özkara-Aydinoǧlu Ş, Aksoylu AE (2010) Carbon dioxide reforming of methane over Co-X/ZrO2 catalysts (X = La, Ce, Mn, Mg, K). Catal Commun 11:1165–1170. https://doi.org/10.1016/j.catcom.2010.07.001
Shi C, Zhang P (2012) Effect of a second metal (Y, K, Ca, Mn or Cu) addition on the carbon dioxide reforming of methane over nanostructured palladium catalysts. Appl Catal B Environ 115–116:190–200. https://doi.org/10.1016/j.apcatb.2011.12.002
Sing KSW, Everett DH, Haul RAW et al (1985) Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl Chem 57:603–619. https://doi.org/10.1351/pac198557040603
Leofanti G, Padovan M, Tozzola G, Venturelli B (1998) Surface area and pore texture of catalysts. Catal Today 41:207–219. https://doi.org/10.1016/S0920-5861(98)00050-9
Bahari MB, Phuc NHH, Alenazey F et al (2017) Catalytic performance of La-Ni/Al2O3 catalyst for CO2 reforming of ethanol. Catal Today 291:67–75. https://doi.org/10.1016/j.cattod.2017.02.019
Tran NT, Van Le Q, Van Cuong N et al (2020) La-doped cobalt supported on mesoporous alumina catalysts for improved methane dry reforming and coke mitigation. J Energy Inst 93:1571–1580. https://doi.org/10.1016/j.joei.2020.01.019
Ayodele BV, Hossain SS, Lam SS et al (2016) Syngas production from CO2 reforming of methane over neodymium sesquioxide supported cobalt catalyst. J Nat Gas Sci Eng 34:873–885. https://doi.org/10.1016/j.jngse.2016.07.059
Patterson AL (1939) The scherrer formula for X-ray particle size determination. Phys Rev 56:978–982. https://doi.org/10.1103/PhysRev.56.978
Feng Y, Zhang H, Fang L et al (2016) Novel three-dimensional flower-like porous Al2O3 nanosheets anchoring hollow NiO nanoparticles for high-efficiency lithium ion batteries. J Mater Chem A 4:11507–11515. https://doi.org/10.1039/c6ta04323b
Mohammadi SZ, Beitollahi H, Allahabadi H, Rohani T (2019) Disposable electrochemical sensor based on modified screen printed electrode for sensitive cabergoline quantification. J Electroanal Chem 847:113223. https://doi.org/10.1016/j.jelechem.2019.113223
Fayaz F, Bach LG, Bahari MB et al (2019) Stability evaluation of ethanol dry reforming on Lanthania-doped cobalt-based catalysts for hydrogen-rich syngas generation. Int J Energy Res 43:405–416. https://doi.org/10.1002/er.4274
Amin MH, Mantri K, Newnham J et al (2012) Highly stable ytterbium promoted Ni/γ-Al2O3 catalysts for carbon dioxide reforming of methane. Appl Catal B Environ 119–120:217–226. https://doi.org/10.1016/j.apcatb.2012.02.039
Park JH, Yeo S, Chang TS (2018) Effect of supports on the performance of Co-based catalysts in methane dry reforming. J CO2 Util 26:465–475. https://doi.org/10.1016/j.jcou.2018.06.002
Movasati A, Alavi SM, Mazloom G (2017) CO2 reforming of methane over Ni/ZnAl2O4 catalysts: influence of Ce addition on activity and stability. Int J Hydrogen Energy 42:16436–16448. https://doi.org/10.1016/j.ijhydene.2017.05.199
Zhang S, Muratsugu S, Ishiguro N, Tada M (2013) Ceria-doped Ni/SBA-16 catalysts for dry reforming of methane. ACS Catal 3:1855–1864. https://doi.org/10.1021/cs400159w
Abasaeed AE, Al-Fatesh AS, Naeem MA et al (2015) Catalytic performance of CeO2 and ZrO2 supported Co catalysts for hydrogen production via dry reforming of methane. Int J Hydrogen Energy 40:6818–6826. https://doi.org/10.1016/j.ijhydene.2015.03.152
Al Abdulghani AJ, Park JH, Kozlov SM et al (2020) Methane dry reforming on supported cobalt nanoparticles promoted by boron. J Catal 392:126–134. https://doi.org/10.1016/j.jcat.2020.09.015
Rahmati M, Huang B, Mortensen MK et al (2018) Effect of different alumina supports on performance of cobalt Fischer–Tropsch catalysts. J Catal 359:92–100. https://doi.org/10.1016/j.jcat.2017.12.022
Yao L, Galvez ME, Hu C, Da Costa P (2018) Synthesis gas production via dry reforming of methane over manganese promoted nickel/cerium-zirconium oxide catalyst. Ind Eng Chem Res 57:16645–16656. https://doi.org/10.1021/acs.iecr.8b04183
Shen B, Zhu S, Zhang X et al (2018) Simultaneous removal of NO and Hg0 using Fe and Co co-doped Mn-Ce/TiO2 catalysts. Fuel 224:241–249. https://doi.org/10.1016/j.fuel.2018.03.080
Horlyck J, Sara M, Lovell EC et al (2019) Effect of metal-support interactions in mixed Co/Al catalysts for dry reforming of methane. ChemCatChem 11:3432–3440. https://doi.org/10.1002/cctc.201900638
Wang F, Ta N, Shen W (2014) MgO nanosheets, nanodisks, and nanofibers for the Meerwein–Ponndorf–Verley reaction. Appl Catal A Gen 475:76–81. https://doi.org/10.1016/j.apcata.2014.01.026
Nikoo MK, Amin NAS (2011) Thermodynamic analysis of carbon dioxide reforming of methane in view of solid carbon formation. Fuel Process Technol 92:678–691. https://doi.org/10.1016/j.fuproc.2010.11.027
Bahari MB, Setiabudi HD, Duy Nguyen T et al (2020) Insight into the influence of rare-earth promoter (CeO2, La2O3, Y2O3, and Sm2O3) addition toward methane dry reforming over Co/mesoporous alumina catalysts. Chem Eng Sci 228:115967. https://doi.org/10.1016/j.ces.2020.115967
Christensen KO, Chen D, Lødeng R, Holmen A (2006) Effect of supports and Ni crystal size on carbon formation and sintering during steam methane reforming. Appl Catal A Gen 314:9–22. https://doi.org/10.1016/j.apcata.2006.07.028
Damyanova S, Shtereva I, Pawelec B et al (2020) Characterization of none and yttrium-modified Ni-based catalysts for dry reforming of methane. Appl Catal B Environ 278:119335. https://doi.org/10.1016/j.apcatb.2020.119335
Gould TD, Montemore MM, Lubers AM et al (2015) Enhanced dry reforming of methane on Ni and Ni-Pt catalysts synthesized by atomic layer deposition. Appl Catal A Gen 492:107–116. https://doi.org/10.1016/J.APCATA.2014.11.037
Shah M, Das T, Mondal P (2022) Development of nanocrystalline mesoporous Pt promoted Co-based catalysts for carbon dioxide reforming of methane. Fuel 313:122683. https://doi.org/10.1016/j.fuel.2021.122683
Vafaeian Y, Haghighi M, Aghamohammadi S (2013) Ultrasound assisted dispersion of different amount of Ni over ZSM-5 used as nanostructured catalyst for hydrogen production via CO2 reforming of methane. Energy Convers Manag 76:1093–1103. https://doi.org/10.1016/j.enconman.2013.08.010
Fakeeha AH, Khan WU, Al-Fatesh AS, Abasaeed AE (2013) Stabilities of zeolite-supported Ni catalysts for dry reforming of methane. Chin J Catal 34:764–768. https://doi.org/10.1016/s1872-2067(12)60554-3
Huang H, Yu Y, Zhang M (2020) Mechanistic insight into methane dry reforming over cobalt: a density functional theory study. Phys Chem Chem Phys 22:27320–27331. https://doi.org/10.1039/c9cp07003f
Omoregbe O, Danh HT, Nguyen-Huy C et al (2017) Syngas production from methane dry reforming over Ni/SBA-15 catalyst: effect of operating parameters. Int J Hydrogen Energy 42:11283–11294. https://doi.org/10.1016/j.ijhydene.2017.03.146
Ayodele BV, Khan MR, Cheng CK (2015) Syngas production from CO2 reforming of methane over ceria supported cobalt catalyst: effects of reactants partial pressure. J Nat Gas Sci Eng 27:1016–1023. https://doi.org/10.1016/j.jngse.2015.09.049
Ruckenstein E, Wang HY (2002) Carbon deposition and catalytic deactivation during CO2 reforming of CH4 over Co/γ-Al2O3 catalysts. J Catal 205:289–293. https://doi.org/10.1006/jcat.2001.3458
Nagaoka K, Takanabe K, Aika KI (2004) Modification of Co/TiO2 for dry reforming of methane at 2 MPa by Pt, Ru or Ni. Appl Catal A Gen 268:151–158. https://doi.org/10.1016/j.apcata.2004.03.029
Son IH, Lee SJ, Song IY et al (2014) Study on coke formation over Ni/γ-Al2O3, Co-Ni/γ-Al2O3, and Mg-Co-Ni/γ-Al2O3 catalysts for carbon dioxide reforming of methane. Fuel 136:194–200. https://doi.org/10.1016/j.fuel.2014.07.041
Zhu X, Huo P, Zhang YP et al (2008) Structure and reactivity of plasma treated Ni/Al2O3 catalyst for CO2 reforming of methane. Appl Catal B Environ 81:132–140. https://doi.org/10.1016/j.apcatb.2007.11.042
Damyanova S, Pawelec B, Arishtirova K et al (2009) MCM-41 supported PdNi catalysts for dry reforming of methane. Appl Catal B Environ 92:250–261. https://doi.org/10.1016/j.apcatb.2009.07.032
Arora S, Prasad R (2016) An overview on dry reforming of methane: strategies to reduce carbonaceous deactivation of catalysts. RSC Adv 6:108668–108688. https://doi.org/10.1039/c6ra20450c
Sun H, Zhang Q, Wen J et al (2020) Insight into the role of CaO in coke-resistant over Ni-HMS catalysts for CO2 reforming of methane. Appl Surf Sci 521:146395. https://doi.org/10.1016/j.apsusc.2020.146395
Azancot L, Bobadilla LF, Centeno MA, Odriozola JA (2021) IR spectroscopic insights into the coking-resistance effect of potassium on nickel-based catalyst during dry reforming of methane. Appl Catal B Environ 285:119822. https://doi.org/10.1016/j.apcatb.2020.119822
Amin MH, Putla S, Hamid SBA, Bhargava SK (2015) Understanding the role of lanthanide promoters on the structure-activity of nanosized Ni/γ-Al2O3 catalysts in carbon dioxide reforming of methane. Appl Catal A Gen 492:160–168. https://doi.org/10.1016/j.apcata.2014.12.038
Acknowledgements
The study was supported by The Youth Incubator for Science and Technology Programme, managed by Youth Development Science and Technology Center—Ho Chi Minh Communist Youth Union and Department of Science and Technology of Ho Chi Minh City, the contract number is “08/2021/HĐ-KHCNT-VƯ” signed on 8th, December, 2021.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cao, A.N.T., Le Phuong, D.H., Phuong, P.T.T. et al. Carbon Dioxide Reforming of Methane Over Co/Al2O3 Catalysts Doped with Manganese. Top Catal 66, 247–261 (2023). https://doi.org/10.1007/s11244-022-01709-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-022-01709-z