
Abstract
In this work, Pt based mono and bimetallic catalysts were tested under conditions of tri-reforming (TR). All the catalysts contained 25% of CeO2 and a metal loading of 2.5 or 5.0% (wt%). The bimetallic catalysts contained 2.5% Pt and 2.5% of Me, where Me = Ni, Co, Mo, Pd, Fe, Re, Y, Cu or Zn. For all the experiments, a synthetic biogas which consisted of 60% CH4 and 40% CO2 (vol.) was mixed with water, S/C = 1.0, and oxygen, O2/CH4 = 0.25, and fed to a fixed bed reactor (FBR) system or a microreactor. The 2.5Pt catalyst was used in order to compare the performance of each reaction system. The tests were performed at reaction temperatures between 700 and 800 °C, and at volume hourly space velocities (VHSV) between 100 LN/(h gcat) and 200 LN/(h gcat) for the FBR system and between 1000 LN/(h gcat) and 2000 LN/(h gcat) for the microreactor, at atmospheric pressure. Then, all catalysts were deposited into microchannel reactors and tested at a constant VHSV of 2000 LN/(h gcat) and reaction temperatures between 700 and 800 °C. Catalysts under investigation were characterized applying the following techniques: inductively coupled plasma optical emission spectroscopy (ICP-OES), N2 Physisorption, Temperature Programmed Reduction (TPR), CO chemisorption, Transmission Electron Microscopy (TEM) and X-ray Photoelectron Spectroscopy (XPS). The microreactor was identified as the most efficient and promising reaction system, and the 2.5(Pt–Pd) catalyst as the bimetallic formulation with the highest activity. Therefore its activity and stability was compared with the reference 5.0Pt catalyst at 700 °C and VHSV of 2000 LN/(h gcat) for more than 100 h. Although slightly lower activity was measured operating with the 2.5(Pt–Pd) catalyst, a significant reduction of the Pt content compared to the reference 5.0Pt catalyst was achieved through the incorporation of Pd.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Biogas is considered a renewable source because it is a carbon dioxide-neutral biofuel and when used as feed in reforming processes two of the most important green-house gases, carbon dioxide and methane, are consumed to produce hydrogen rich gas mixtures [1]. The hydrogen produced from renewable sources like biogas is a clean and sustainable energy vector, and it has a green fuel nature because when it combines with oxygen only water vapor as a reaction product is produced. Therefore, the use of biogas for hydrogen production could provide important environmental benefits, being this one of the main reasons why the scientific community is making huge efforts in terms of looking for alternatives to reduce the cost of the production process and guarantee the supply and distribution, especially for mobile applications.
The bridge that links biogas and hydrogen covers all the possible reforming processes, through which the biogas can easily and effectively be converted into hydrogen rich mixtures. Alves et al. [2] provide an overview of the existing biogas reforming processes based on an extensive literature review. They describe benefits of the tri-reforming (TR) process, also known as autothermal reforming (ATR), like: (i) the improvement of the temperature control in the reactor, (ii) the energetically more economical manufacturing process of H2, (iii) the reduction of the hot-spots formation, avoiding the deactivation of the catalyst, and (iv) the feasibility of the process to generate a desired H2/CO ratio by adapting the CH4/O2/H2O ratio in the feed. In addition, they also mention the most important drawback: the lower H2/CO ratio in comparison with steam reforming (SR). The benefits mentioned above justify the election of the TR process as a very attractive route for hydrogen production from biogas.
The research work developed faces out two important issues related to hydrogen production process by TR. The first one corresponds to study the feasibility of two different reactor systems, the fixed bed reactor (FBR) and microreactors (micro), by comparing their activity in terms of turnover frequency (TOF) and hydrogen productivity (PRO). The main benefits of the microreactors, compared to conventional reaction systems like the FBR, are the lower pressure drop and enhanced heat and mass transfer [3], which are key factors in reactions favored at high temperature and low pressure, like TR. The second objective of this research work focuses on the development of active, stable, selective and economic catalysts for the TR process. For that purpose, all the catalysts prepared contained 25% of CeO2 and a Pt metal loading of 2.5 or 5.0% (wt%). Two reference monometallic catalysts were prepared, denoted as 5.0Pt and 2.5Pt, and the remaining catalysts were bimetallic, denoted as 2.5(Pt–Me), where Me = Ni, Co, Mo, Pd, Fe, Re, Y, Cu or Zn. Indeed, the 2.5Pt catalyst was employed to study the feasibility of both reaction systems.
The platinum has been widely employed in reforming processes. It is a well-known active metal for reactions involving oxidation processes like water gas-shift (WGS) [4, 5] and also catalyzes in a very effective way feeds containing oxygen, like partial oxidation of methane (POM) [6–8]. In addition, Pt is widely employed in SR and dry reforming (DR) as summarized in [9]. Then, it is generally assumed that Pt catalysts have high activity and stability in the reforming reactions of methane because of: (i) the formation of an interface between metal and support, and (ii) the formation of species that contribute to carbon removal [7]. Furthermore, Pt is a good candidate to avoid the sintering because this effect is associated to the size of Pt particles and the PtOx-oxide support [6]. Therefore, Pt based catalysts combined with an appropriate support seemed to be a promising active metal for TR process. According to this, gamma alumina (γ-Al2O3) was the support selected because of its large surface area and thermal stability. These two properties are essential in order to reach a good Pt dispersion onto the surface of the γ-Al2O3 and preserve the properties of the catalyst when operating at high temperatures. In addition, this support was modified using cerium oxide (CeO2). Ferreira et al. reported an essential contribution to the catalyst activity and stability when adding CeO2 to alumina by wet impregnation method [8]. The benefits of the CeO2, listed elsewhere [10, 11], can be summarized as follows: (i) improvement of metallic dispersion, (ii) enhancement of metal-support interaction (iii) sintering decrease, (iv) improvement of thermal stability, (v) increase of basicity, (vi) enhancement of the adsorption of CO2 which alters the coke formation, and (vii) promotion of the reducibility. In addition, it has been also reported that platinum helps to reduce ceria to Ce3+ and create oxygen vacancies [10].
Once the species of the reference catalysts were determined, the incorporation of several metals was carried out in order to reduce the Pt amount of the reference 5.0Pt catalyst. The intended total metal composition of these catalysts was 5.0 wt% and the catalysts were denoted as 2.5(Pt–Me), where Me = Ni, Co, Mo, Pd, Fe, Re, Y, Cu or Zn. The use of Ni has been widely studied and industrially used because is cheap and very active transition metal, especially for reforming processes as reported in several investigations [11–14]. In addition, Niu et al. affirms that the formation of PtNi alloy has smaller metal crystal size than metal particles in monometallic Pt and Ni catalysts [9]. The rest of metals employed in this work have been also used in reforming processes as promoting metals, but never, to the best of our knowledge, as monometallic catalyst [15–23]. Therefore, their combination with Pt could enhance the properties of the reference 2.5Pt catalyst. Among all the metals under investigation, the Pd has been widely employed in reforming processes [8, 24, 25] showing very high activity. Although Pd is more expensive than Ni, Co, Mo, Fe, Y, Cu or Zn, it is cheaper than Pt [26].
In a previous work of the authors, the activity of microreactor and FBR was also studied using Ni based catalysts [27]. However, the results cannot be compared because the experimental conditions are different; in the previous work, the feed was diluted with nitrogen by simulating an air stream and only experiments at a constant temperature of 800 °C and weight hourly space velocity (WHSV) of around 2000 ggas/(gcat h) were carried out. In the present work, the use of nitrogen was avoided. Even, the dilution with nitrogen could minimized the hot spot formation, this effect is efficiently reduced by operating with microreactors because these systems are able to transfer more efficiently the heat generated [28]. A detailed description of the microreactors employed in this work is available in [29]. On the other hand, the dilution with an inert affects to the activity of the process by reducing the interaction between the reactants and the active sites of the catalyst. Furthermore, there is no need to simulate an air stream because there are adsorption techniques that allows the separation of oxygen from nitrogen in a very cheap and reliable way.
2 Experimental Procedure
2.1 Catalysts Preparation
2.1.1 Reactants Used
The alumina, used as catalysts support, was purchased from Sasol (Puralox, SBa-200). Nickel(II) nitrate hexahydrate [Ni(NO3)2·6H2O, 99%], cobalt(II) nitrate hexahydrate [Co(NO3)2·6H2O, 99.4%), ammonium molybdate tetrahydrate [(NH4)6Mo7O24·4H2O, 99.7%], palladium(II) nitrate solution [Pd(NO3)2, 4.3% Pd], iron(III) nitrate nonahydrate [Fe(NO3)3·9H2O, 99%], copper(II) nitrate trihydrate [Cu(NO3)2·3H2O, >99%], zinc nitrate hexahydrate [Zn(NO3)2·6H2O, 99.5%] and polyvinyl alcohol (PVA) were acquired from Sigma–Aldrich. Alfa Aesar was the provider of chloroplatinic acid hexahydrate (H2PtCl6·6H2O, 40.49%Pt), cerium(III) nitrate hexahydrate [Ce(NO3)3·6H2O, 99.9%], ammonium perrhenate(VII) (NH4ReO4, 99%) and yttrium(III) nitrate hexahydrate [Y(NO3)3·6H2O, 99.9%].
2.1.2 Catalysts Preparation Procedure
All the catalysts were prepared by the incipient wetness impregnation method followed by a wash coating procedure. Briefly, the required amounts of each precursor were completely dissolved in 10 mL of deionized water and, thereafter, co-impregnated into 5.0 g of γ-Al2O3. The obtained mixtures were initially stirred for approximately 3 min and then left for 3 h. Afterwards, the mixtures were calcined at 450 °C for 6 h and finally, the catalysts obtained were manually crushed to a fine powder. For the Mo and Re containing catalysts, the precursors were added in a second impregnation step and calcined thereafter at the same conditions. In the case of the Cu containing catalyst the calcination was carried out at lower temperature, 300 °C, in order to avoid the sintering of Cu particles.
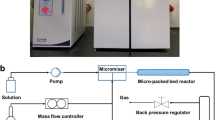
To prepare the catalyst coatings in microreactors, a suspension consisting of H2O, PVA, acetic acid and the catalyst powder was prepared (except for the Cu containing catalyst, for which no acetic acid was used). The whole procedure has been described in more detail previously [30]. Finally, the coated catalysts were calcined again at the conditions specified before. In parallel, additional amounts of these catalysts were prepared in crucibles and treated exactly in the same manner as the micro-channel coatings. These samples were then removed from the crucible and served as samples for physical and chemical characterization. Regarding the FBR, a 316-L stainless-steel reactor (4.57 mm i.d. and 30 cm length) was employed as a bench-scale FBR and 200 mg of calcined catalyst were used in the 0.42–0.50 mm particle size diameter range. The catalyst was diluted with SiC at 1:4.5 w/w in the 0.5–1.0 mm particle size diameter range in order to avoid hot spots in the bed and to easily separate the catalysts after use.
Regarding the composition of the catalysts, all of them contained 25% of CeO2 and a Pt metal loading of 2.5 or 5.0% (wt%). Two reference monometallic catalysts were prepared, denoted as 5.0Pt and 2.5Pt, and the remaining catalysts were bimetallic, denoted as 2.5(Pt–Me), where Me = Ni, Co, Mo, Pd, Fe, Re, Cu, Zn and Y.
2.1.3 Activity Measurements
First, thermodynamic equilibrium calculations were carried out using Aspen Plus software, at a constant CH4/CO2 ratio of 1.5 and several temperatures, from 450 to 800 °C, S/C ratios, from 1.0 to 2.0, and O2/CH4 ratios, from 0.25 to 0.5, in order to determine the most appropriate conditions for hydrogen or synthesis gas production. The carbon in the S/C and O2/CH4 ratios represent the CH4 in the feed, the organic carbon, but not the CO2. As a result of the simulation work, the feed composition was determined to be a synthetic biogas mixed with water, S/C = 1.0, and oxygen, O2/CH4 = 0.25. For the reaction system comparison, the 2.5Pt catalyst was used. All the activity tests were performed at atmospheric pressure, reaction temperatures from 700 to 800 °C, and at VHSV from 100 LN/(h gcat) to 200 LN/(h gcat) for the FBR, and from 1000 LN/(h gcat) to 2000 LN/(h gcat) for the microreactor. Therefore, the only difference regarding the operating conditions between the FBR and microreactors lied in the catalyst amount used. As expected, the operation with microreactor was much satisfactory in terms of activity. Then, all the catalysts synthesized were deposited into microchannel reactors to be tested at a constant VHSV of 2000 LN/(h gcat) and reaction temperatures between 700 and 800 °C. Finally, a long-term experiment was carried out for the two most active formulations. In Table 1, the experimental schedule of the tests described above is shown.
A bench-scale Microactivity plant (PID Eng&Tech) was used for the activity tests. The feed mixture gas flows were adjusted by electronic controllers and a HPLC-Gilson liquid pump was used for the desionizated water injection. Both, fixed bed reactors and microreactors were electrically heated in a furnace to the desired temperature. The outgases were cooled down with a partial condenser and this made possible to collect and weight the condensed water as well as the analysis of the gas phase by a Micro GC equipped with a TCD detector. Three columns, Molsieve 5 Å PLOT, CP–Sil 5 CB and Poraplot Q were used in a series arrangement for the complete separation of hydrogen, carbon monoxide, carbon dioxide, oxygen and methane, and this allowed measuring the following parameters, defined as:
where V i in corresponds to the volumetric flow-rate of reactant i, (mLN/min). V i out corresponds to the volumetric flow-rate of product i, (mLN/min).
The turnover frequency, TOF (moles of converted methane or carbon dioxide per mole of active metal and second), and catalyst productivity, PRO, (moles of hydrogen produced per mole of active metal and second) are defined as the velocity of the reaction measured at the catalytic surface [32–34] which reflects the activity level of available catalytically active sites on the surface. The TOF and PRO parameters will be used in the reaction systems comparison employing the 2.5Pt monometallic catalyst d. In the case of the catalysts screening study, the bimetallic character of most of the tested catalysts makes impossible to attribute the metallic dispersion to a specific metal of the catalyst and therefore, the activity of this study was measured in terms of CH4 and CO2 conversion and hydrogen yield. The TOF and PRO parameters were defined as follows:
where N i corresponds to the molar flow of i (CH4, CO2 or H2), (mol/s). W cat is the catalyst weight in each reactor system, (g). Wt Me is the elemental weight composition given by ICP-OES, (gPt/gcat). D Me is the metal dispersion for the catalyst, (%). Pm Me is the metal molecular weight, (g/mol).
2.2 Catalysts Characterization
Fresh and calcined catalysts were characterized by different techniques. An inductively coupled plasma optical emission spectroscopy (ICP-OES) instrument, model 2000-DV (Perkin Elmer), was used for the determination of the metallic compounds in the catalysts. The catalysts were properly dissolved by a solution that consisted of H2O2 and HNO3 [31] and using a digester. Thereafter the samples were analysed by the ICP-OES instrument.
The textural properties, BET surface area, pore volume (Pv) and pore radius (Pr) of the calcined and outgassed (at 100 °C for 5 h) catalysts were evaluated by means of N2 adsorption obtained at 77 K using a Sorptomatic equipment. The specific surface area was calculated using the BET method while the Pr and Pv were derived using the Barrett–Joyner–Halenda (BJH) method.
Temperature programmed reduction (TPR) measurements were performed in order to determine the reduction temperature of the reducible species formed during the calcination of the catalysts. These analyses were carried out using an AutoChem II 2920 apparatus that was equipped with a thermal conductivity detector (TCD). A continuous flow of 5.0% H2/Ar (40.0 mLN/min) was fed over the calcined catalyst powder, and the temperature was increased from room temperature to 450 °C at a rate of 5.0 C/min. All the samples were previously outgassed at 40 °C during 30 min.
The same equipment was used for the CO chemisorption experiments in order to determine the metal dispersion (M.D.), metallic surface area (M.S.A.) and cubic crystallite size (C.C.S.). The catalysts were previously reduced under 5.0 vol% hydrogen in argon flow of 50.0 mLN/min for 4 h at 800 °C and then, the CO-pulse chemisorption was carried out at 100 °C. Metal dispersion was determined by assuming a stoichiometric ratio of (CO/metal = 1.0).
X-ray photoelectron spectroscopy (XPS) technique was used to evaluate the surface characteristics of the fresh catalysts. The measurements were carried out with a SPECS system (Berlin, Germany) equipped with a Phoibos 150 1D-DLD analyser and an Al Kα (hυ = 1486.6 eV) X-ray source. The analyses of the catalysts were carried out at the following experimental conditions: step energy of 1.0 eV, dwell time of 0.1 s and pass energy of 40 eV. Thereafter, a detailed scan of the detected compound was performed under the following conditions: step energy of 0.1 eV, dwell time of 0.1 s, pass energy of 20 eV and with an outlet electron angle of 90°. The obtained spectra were adjusted using CasaXPS 2.3.16 software which modelled the Gauss-Lorentzian contributions, after a background subtraction.
Finally, transmission electron micrographs were acquired on a Philips CM 200 transmission electron microscope (TEM) at an acceleration voltage of 200 kV with a LaB6 filament. Typically, a small amount of sample was suspended in pure ethanol, sonicated and dispersed over a Cu grid with a carbon coated cellulose acetate-butyrate holey film. TEM images were recorded using a 4k × 4k TVIPS CCD camera at different magnifications. Due to the limitations of this technique for the metals identification, another transmission electron analysis was carried out on a TECNAI G2 20 TWIN transmission electron microscope (TEM) at an acceleration voltage of 200 kV with LaB6 filament and equipped with EDAX EDS microanalysis system and high angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM). For this equipment, the same sample preparation procedure was applied.
3 Results and Discussion
3.1 Catalysts Characterization Results
3.1.1 Chemical Compositions, Textural Properties and CO-Chemisorption Results
Chemical compositions and textural properties were determined for the fresh calcined catalysts. In the case of CO-chemisorption experiments, the catalysts were pre-reduced at 800 °C in order to simulate the surface characteristics at the highest temperature reached during the activity tests. The atomic ratios were calculated based on the ICP-OES and XPS results:
The metallic components of the catalysts were determined applying the procedure described in [31], especially for the Ce quantification, in order to avoid the formation of CeF2 precipitates. The results obtained showed significantly lower Pt content, compared to the intended values, which could be originated because of the hygroscopic character of the precursor used; probably certain amount of water was adsorbed to the Pt precursor during the preparation procedure, and consequently decreased the Pt content of the catalysts. Regarding the remaining components, real and nominal amounts are close.
The highest surface area was measured for the monometallic catalysts followed closely by the 2.5(Pt–Re) and 2.5(Pt–Cu) catalysts. The pore radius and volume do not show significant differences. All catalysts are ranging between 2.40 and 3.19 for the Pr and between 0.26 and 0.34 for the Pv respectively, which is not regarded as a significant difference. However, much higher differences can be observed concerning the M.D., M.S.A. and C.C.S. measured by CO chemisorption. Nevertheless, the comparison of the measured parameters for the bimetallic catalysts it is merely descriptive because it is impossible to attribute each result to the interaction of each metal. Despite, for the 5.0Pt monometallic catalyst the highest M.D. and M.S.A. were measured because of the good dispersion achieved by this metal on supports with large surface area available such as γ-Al2O3. If a descriptive comparison between bimetallic catalysts is possible, the 2.5(Pt–Re) and 2.5(Pt–Pd) catalysts stands out from the rest of the bimetallic catalysts because they showed the highest M.D. and M.S.A. as well as the smallest C.C.S. measured.
Table 2 also shows the relationship between the surface atomic ratios measured by XPS and atomic ratios measured by ICP-OES. High Pt dispersion was measured for all catalysts prepared apart from the Y based catalyst as indicated by the higher atomic ratios measured by ICP-OES than for XPS. However, this was expected because noble metals can be easily dispersed on materials with large surface area, such as Al2O3. The ICP-OES atomic ratios measured for the Ni and Co containing catalysts were higher than those obtained by XPS indicating a poor dispersion for these metals, while no Zn was detected by XPS. However, Pd and Y containing catalysts obtained a higher XPS surface atomic ratio and this is in good agreement with the dispersion and M.S.A. measured by CO chemisorption.
3.1.2 Temperature Programmed Reduction
Figure 1 shows the TPR profiles obtained for the calcined catalysts. The temperature of the experiments was increased from 50 to 800 °C because this was the highest temperature applied in the experimental tests. The profiles observed are very diverse because of the different metal-support interactions. In addition, only the profiles of the most promising catalysts are depicted in this figure. On the one hand, it has been reported that the Al2O3 and the Al2O3–CeO2 do not consume significant amounts of hydrogen at temperatures below 450 °C [32]. It has also been reported that CeO2 content higher than 27% decreases the temperature of the Al2O3–CeO2 reduction peak [33]. However, all the catalysts prepared contain less than 25 wt% of CeO2. For the monometallic catalysts, the profile obtained for the 5.0Pt is very similar those obtained by other researchers [34], with the main reduction peaks around 150 and 300 °C. In [35] it is stated that the reduction peaks located at the temperature interval from 150 to 280 °C could be attributed to the reduction of surface Pt oxides, and those comprehended between 250 and 350 °C to the reduction of CeO2 closely contacting Pt oxides. Therefore, it can be concluded that Pt contained in the 5.0Pt catalyst interacts with part of the CeO2 (the reduction peak located at around 304 °C). In the case of the 2.5Pt catalyst, its lower Pt amount makes the peaks appearing together [36]. Therefore, it can also be assumed that the metals present in the bimetallic catalysts interact with the Pt at temperature lower than 280 °C because reduction peaks were recorded at 320 and 300 °C respectively.

Left TPR profiles of the most promising calcined catalysts. Right deconvoluted profiles of the 2.1Pt (top) and 3.6Pt (down) catalysts
The total hydrogen consumption was determined for the catalysts shown in Fig. 1. In addition, the amount of hydrogen needed for the reduction of Pt4+ to Pt0 and the one needed for the reduction of Ce4+ to Ce3+ was calculated for the monometallic catalysts by the corresponding peaks deconvolution, as shown in Fig. 1. The hydrogen consumption attributed to the reduction of surface Pt oxides contained in 5.0Pt and 2.5Pt catalysts corresponded to 640.7 and 325.2 µmol H2/gcat consecutively. On the other hand, the amount of hydrogen attributed to the reduction of CeO2 closely contacting with the Pt oxides to Ce2O3 that contained the 5.0Pt and 2.5Pt catalysts corresponded to 105.5 and 55.7 µmol H2/gcat respectively. Therefore, while the entire Pt was reduced at low temperatures, only part of the CeO2 present in the sample is reduced to Ce2O3 (the 40.7 and 13.0% of the total CeO2 of the 5.0Pt and 2.5Pt catalysts respectively) being the rest of CeO2 reduced at higher temperature. Regarding the 2.5(Pt–Y) and 2.5(Pt–Pd) catalysts, the total hydrogen consumption was 379.6 cat and 260.3 µmol H2/g cat respectively.
3.1.3 Transmission Electron Microscopy
In Fig. 2 TEM and STEM micrographs of the most promising catalysts are shown. Initially TEM micrographs of benchmark catalysts were only acquired in order to observe the dispersion of the Pt particles. For these two catalysts, Pt particles of several sizes are observed; from 2.0 to 7.0 nm for the 5.0Pt catalyst and from 1.4 to 5.0 nm for the 2.5Pt catalyst. Particles with lower size and better dispersion were expected for the Pt, because it is a noble metal. However, Pt particles could have suffered from sintering, especially the catalyst containing 5.0Pt owing to its high Pt concentration. When the bimetallic catalysts were analysed by this technique, it was not possible to distinguish the metals using bright field images. Therefore, EDAX EDS microanalysis was carried out, for which a high angle annular dark-field-scanning transmission electron detector (HAADF-STEM) was used. With the micrographs taken, it was possible to observe and analyze the metals present on each catalyst, as well as their dispersion. All the micrographs acquired by this technique are collected in the Fig. 2:


TEM and STEM micrographs of the most promising catalysts
All the analyses are represented by the corresponding square and scale in each micrograph. Focusing on the TEM and STEM micrographs acquired for the 2.5Pt monometallic catalyst, even if they correspond to different areas, similar Pt particle size and dispersion were observed. The STEM images of the 2.5(Pt–Ni) catalyst show better CeO2 dispersion on alumina than the observed for 2.5 and 5.0 Pt. For the 2.5(Pt–Ni) catalyst, good dispersion of both active metals was observed while less Pt particles were observed and an effective dispersion of all the ingredients was achieved. However, for the 2.5(Pt–Co) catalyst larger Pt and Co particles were detected. The 2.5(Pt–Y) catalyst shows the largest Pt particles measured among all the catalysts characterized, with a size of about 10.0 nm, which are much larger than the Y particles present in the same catalyst. For this catalyst only Y was properly dispersed onto the alumina support. Finally, the micrographs acquired for the 2.5(Pt–Pd) catalyst are presented, in which the Pt and Pd particles formed were also small. However, the Pd particles are bigger than the Pt ones. Overall, among all the bimetallic catalysts characterized, the 2.5(Pt–Ni) catalyst showed the smallest particle sizes of the active metals present in the catalysts, and showed, together with CeO2, good dispersion over the alumina support. In addition, there was no evidences of alloy formation with the characterization techniques used.
3.1.4 Catalyst Activity Tests
Prior to start with the experiments, a sensitivity analysis was carried out using Aspen Plus software in order to determine the most appropriate O2/CH4 and S/C feed ratios (vol%) to obtain the highest hydrogen yield, at a constant CH4/CO2 ratio of 1.5 (vol%) [4]. Based on the results of this study, Fig. 3, it was concluded that the ratios of O2/CH4 = 0.25 and S/C = 1.0 are the most suitable ones, which is in good agreement with previous investigation of the authors [27]. On the one hand, at increasing O2/CH4 ratio, the higher partial pressure of oxygen would favor the methane partial or total oxidation reaction, thus, reducing the interaction of methane for hydrogen production reactions, like SR or DR. Consequently, most of the CO2 remains in the feed without being converted through DR reaction. On the other hand, at increasing S/C ratio, the higher water content results in a lower hydrogen yield, as defined above in Sect. 2.2. Nevertheless, if hydrogen yield is considered as the amount of hydrogen produced by the amount of hydrogen produced if all the methane fed reacted to produce carbon dioxide and hydrogen, the obtained yield results are different. As an example, the results at 800 °C were 56.4% at O2/CH4 = 0.25 and S/C = 1.0, 63.3% at O2/CH4 = 0.25 and S/C = 2.0, 48.6% at O2/CH4 = 0.50 and S/C = 1.0 and 53.5% at O2/CH4 = 0.50 and S/C = 2.0. Therefore, the highest hydrogen yield would be achieved at the highest S/C ratio and lower O2/CH4 ratio. Regardless to the hydrogen yield definition, at the lowest O2/CH4 ratio, the reactions that produce hydrogen are promoted; DR and SR. However, the use of low O2/CH4 and S/C ratios plays an adverse effect regarding the stability of the catalysts: the appearance of carbon deposits onto the catalytic surface is more probable. Hence, the operating conditions selected would be also appropriate to observe in advance the possible deactivation phenomenon, thus, allowing a better catalysts activity and stability comparison.
The negative carbon dioxide conversion measured is related to the WGS reaction promotion. Thus, the outgas CO2 concentration increased with the O2/CH4 and/or S/C ratios, due to net carbon dioxide production.

Thermodynamic equilibrium calculations for CH4 conversion, CO2 conversion and H2 yield at several temperatures
Then, the performance of the catalysts at several temperatures, from 700 to 800 °C, and VHSV, from 500 to 2000 LN/gcat·h, was studied with the 2.5Pt catalyst in order to determine the best suited experimental conditions that allow a better comparison between reaction systems. All these experiments were carried out at O2/CH4 = 0.25 and S/C = 1.0. In Fig. 4, the activity results measured for each reaction system, operating at a constant VHSV and several temperatures, can be observed. If conversion and yield are compared, left of the figure, similar values were achieved, being slightly higher the ones measured for the FBR. In addition, the activity of the 2.5Pt catalyst increased with the temperature, from values far from the equilibrium conversion, until values almost at equilibrium conversion, at 800 °C. At the right hand of Fig. 4, CH4 TOF, CO2 TOF and H2 PRO are shown in order to elucidate the real activity of each reaction system. This figure clearly demonstrated the higher activity achieved operating with microreactors.

Left CH4 conversion (black), CO2 conversion (green) and H2 yield (red) measured at several temperatures, O2/CH4 = 0.25 and S/C = 1.0. Right CH4 TOF, CO2 TOF and H2 PROD measured at several temperatures. The experiments were carried out using both FBR [(triangle), VHSV = 200 LN/(h gcat)] and microreactors [circle, 2000 LN/(h gcat)] in the temperature range between 700 < T [ºC] < 800. The (squares) represent the measured parameters at thermodynamic equilibrium
Figure 5 shows the activity results measured for the 2.5Pt catalyst operating at O2/CH4 = 0.25 and S/C = 1.0, constant temperature and several VHSV. The temperature was fixed at 700 °C because the activity measured at this temperature ensures the operation far from the equilibrium calculation, as shown in Fig. 4, and therefore allows a better comparison between reaction systems. It was not possible to operate at the lowest VHSV using the FBR because of the flow limitation of the mass flow controllers, as shown in the left of Fig. 5. In this case, the highest conversion values, close to the ones calculated for the equilibrium conversion, were measured operating at the lowest VHSV. Then, at increasing VHSV, the conversion of reactants decreased because of their lower residence time inside the reaction systems. Once again, the real activity calculated by CH4 TOF, CO2 TOF and H2 PRO shows higher activity for the microreactor system.

Left CH4 conversion (black), CO2 conversion (green) and H2 yield (red) measured at 700 °C, O2/CH4 = 0.25, S/C = 1.0 and several VHSV. Right CH4 TOF, CO2 TOF and H2 PROD measured at 700 °C and several VHSV. The experiments were carried out using both FBR (triangle), 100 < VHSV [LN/(h gcat)] < 200 and microreactors (circle), 1000 < VHSV [LN/(h gcat)] < 2000. The (squares) represent the measured parameters at thermodynamic equilibrium
As defined above in Sect. 2.2, the TOF and PRO parameters truly reflects the activity level of available catalytically active sites on the surface. The activity measured for the catalyst coated onto microreactor is about one order of magnitude higher than the one measured for the catalyst used in the fixed bed reactor, when the same catalyst is tested in both reaction systems, 2.5Pt. The higher performance of the microreactors is assigned to the higher heat and mass transfer capacities, which is related to the size of the microchannels that allows a close contact between reacting molecules and catalyst surface. As a result, catalyst containing smaller amount of noble metal can be used in microreactors, allowing a better performance, including the stability during the time on stream. Because of the previously stated properties, these systems allow a better utilization of the catalyst, and this is one the main reasons of using Pt as a common active metal for all the catalysts prepared [37, 38].
The results of Figs. 4 and 5 showed activity values far from those predicted by the thermodynamic calculations when operating at the lowest temperature, 700 °C, and the highest VSHV, 2000 LN/gcat·h. In addition, the better performance in terms of catalytic activity was clearly demonstrated operating with microreactors. Therefore, all the monometallic and bimetallic catalysts synthetized were prepared and coated onto the microchannels of the microreactors and were tested at several temperatures and the highest VHSV, 2000 LN/gcat·h, according to the schedule of Table 1a.
In Fig. 6, the results measured for the catalysts that showed superior performance are depicted. All the catalysts tested showed a similar tendency: their activity increased with the temperature because of the endothermic nature of the main reactions involved in the TR process. If the catalysts’ activity results are compared with the ones calculated for the equilibrium, at 700 °C the activity measured was low, and increased with the temperature. At 800 °C, all catalysts almost reached equilibrium conversion values. The Y containing catalyst showed lower methane conversion capacity and the 2.5Pt catalyst lower carbon dioxide conversion capacity. Therefore, these two catalysts were not able to reach the values measured for the equilibrium in terms of hydrogen yield. Concerning this parameter, the small errors corresponding to the measurements with the analytical instruments could be the reason why there are activity values higher than the ones calculated for the equilibrium. Nevertheless, the absolute error calculated for the carbon balance was always lower than 5.0 mol.%.

CH4 conversion, CO2 conversion and H2 yield as determined for a VHSV of 2000 LN/(h gcat), at O2/CH4 = 0.25, S/C = 1.0 and different temperatures for the most promising catalysts tested. The (squares) represent methane conversion, XCH4, (triangles) carbon dioxide conversion, XCO2 and (circles) hydrogen yield, H2 yield
In Fig. 7, the results measured for the rest of the catalysts under investigation can be observed. For all of them, the combination of Pt with the corresponding metals results in a catalyst with a lower performance, compared to the 2.5Pt monometalic one. Therefore, the systhesis of these catalysts was unsatisfcatory in terms of catalytic activity. In addition, the results measured for the Re and Cu containing catalysts are not shown because they reached the worst activity results. Among all the catalysts prepared, the results measured for the Ni containing catalysts were unexpected because Ni is considered, and also used, as a very active metal in reforming processes. However, the low Ni content in the catalyst was probably the reason of the low activity measured. This could be related to the nature of the reaction system used, because when microreactors are used very active metals or very high content of less active metals are required. Therefore, the concentration of Ni, and also Co, Zn, Fe and Mo, could be low for the experimental conditions of the tests carried out.

CH4 conversion, CO2 conversion and H2 yield as determined for a VHSV of 2000 LN/(h gcat), at O2/CH4 = 0.25, S/C = 1.0 and different temperatures for the remaining of catalysts tested. The (squares) represent methane conversion, XCH4, (triangles) carbon dioxide conversion, XCO2 and (circles) hydrogen yield, H2 yield
Once the influence of the temperature was studied, a medium term experiments were conducted under the most severe experimental conditions: the lowest temperature, 700 °C, and the highest space velocity, 2000 LN/(h gcat). The results, represented in Fig. 8, showed the average of the last two measurements taken for hydrogen yield after 26 h on stream. If these results are compared to the ones shown in Figs. 6 and 7, which were obtained operating under the same conditions, it is evident that all catalysts suffered from deactivation. The hydrogen yield measured for the 5.0Pt and 2.5(Pt–Pd) catalysts at 2 h was 58.6 and 52.4%, and after 26 h was 51.0 and 47.5% consecutively. For the other catalysts, the deactivation was even more evident. The most probable reason that could justify the deactivation is the morphological change of the surface of the catalysts due to the high temperature. On the one hand, it could affect to the structural properties of the catalysts’ support surface; the gamma alumina, when exposed to high temperatures, suffers continuous transitions from gamma to alpha, being the reduction of its surface area one of the most important consequences. This phenomenon could occlude structural pores in which active metals were initially deposited, thus reducing the number of active sites along the surface of the catalyst support and therefore, their activity. On the other hand, the high temperatures also promoted the sintering effect of the active metals, resulting in metal active sites reduction. Therefore, the high temperatures could justify the deactivation measured for all the catalysts tested. However, the deactivation by carbon deposition is also probable because of the low oxygen and water concentration in the feed. The reaction of methane oxidation, which is the fastest one among all the reactions present in this system, could consume all the oxygen contained in the feed, and therefore, it might not be oxygen available to oxidize the possible carbon deposited onto the surface of the catalysts. The mentioned two phenomenon, temperature and carbon deposition, could result in the observed catalyst deactivation.

H2 yield determined for all catalysts tested after 26 h test duration at O2/CH4 = 0.25, S/C = 1.0, VHSV of 2000 LN/(h gcat) and 700 °C
The influence of the temperature was studied for the 5.0Pt catalyst by CO-chemisorption experiments carried out at three simulated reaction temperatures. The results are collected in the following Table 3:
The results shown in Table 3 clearly indicates that the sintering process is more evident as the reaction temperature was increased. The C.Q reflects the CO mmol that are able to interact with active sites. Therefore, the reduction of this parameter clearly means the reduction of the active sites when the temperature was increased. Therefore, the metallic surface area (M.S.A.) decreased, the metallic particle size (C.C.S.) increased and, consequently, the metallic dispersion (M.D.) decreased corroborating the sintering effect suffered for this catalyst.
Being the 5.0Pt and 2.5(Pt–Pd) catalysts the ones suffering lower deactivation and showing the highest activities at all the temperatures and VHSV tested, they were selected to perform a long-term test that lasted almost 110 h (Fig. 9). Comparing the results obtained, a similar conversion decrease and deactivation tendency was observed, being the 5.0Pt catalyst slightly more active. The hydrogen yield measured for the 5.0Pt and 2.5(Pt–Pd) catalysts after 110 h was 47.3% and 36.0% consecutively.

Stability test at O2/CH4 = 0.25, S/C = 1.0, 700 °C and VHSV of 2000 LN/(h gcat) for the 5.0Pt and 2.5(Pt–Pd) catalysts
4 Conclusions
Nine bimetallic catalysts, with a total metal loading of 5.0 wt% were prepared and their activity was compared with two Pt monometallic based catalysts for the tri-reforming reaction. According to thermodynamic equilibrium calculations, the most favorable conditions for hydrogen production corresponded to O2/CH4 = 0.25 and S/C = 1.0. The activity comparison, between microreactor and FBR system, carried out using the 2.5Pt catalyst clearly demonstrated the superior performance of the microreactor system. Then, using microreactors, higher activity was measured for the two monometallic, the 2.5(Pt–Y) and the 2.5(Pt–Pd) catalysts, than for the Ni, Co, Mo, Fe, Cu, Zn y Re containing catalysts. Among the bimetallic formulations, only the 2.5(Pt–Pd) catalyst showed higher activity than the 2.5Pt catalyst. However both 2.5(Pt–Pd) and 5.0Pt catalysts showed a similar deactivation tendency during the long-term test which was attributed to the sintering effect. Therefore, the aim of the future work will be focused on the improvement of these catalytic systems using microreactors. As microreactors provide a better heat transfer, by controlling more effectively the temperature gradient, this system would allow a better improvement of the catalyst activity than the fixed bed reactor.
References
Rasi S, Veijanen A, Rintala J (2007) Trace compounds of biogas from different biogas production plants. Energy 32:1375–1380. doi:10.1016/j.energy.2006.10.018
Alves HJ, Bley Junior C, Niklevicz RR et al (2013) Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int J Hydrogen Energy 38:5215–5225. doi:10.1016/j.ijhydene.2013.02.057
Kolb G, Hessel V (2004) Micro-structured reactors for gas phase reactions. Chem Eng J 98:1–38. doi:10.1016/j.cej.2003.10.005
Kolb G, Baier T, Schürer J et al (2008) A micro-structured 5 kW complete fuel processor for iso-octane as hydrogen supply system for mobile auxiliary power units. Part II-development of water-gas shift and preferential oxidation catalysts reactors and assembly of the fuel processor. Chem Eng J 138:474–489. doi:10.1016/j.cej.2007.06.037
Germani G, Alphonse P, Courty M et al (2005) Platinum/ceria/alumina catalysts on microstructures for carbon monoxide conversion. Catal Today 110:114–120. doi:10.1016/j.cattod.2005.09.017.
Prieto PJS, Ferreira AP, Haddad PS et al (2010) Designing Pt nanoparticles supported on CeO2–Al2O3: synthesis, characterization and catalytic properties in the steam reforming and partial oxidation of methane. J Catal 276:351–359. doi:10.1016/j.jcat.2010.09.025
Duarte RB, Damyanova S, de Oliveira DC et al (2011) Study of Sm2O3-doped CeO2–Al2O3-supported Pt catalysts for partial CH4 oxidation. Appl Catal A Gen 399:134–145. doi:10.1016/j.apcata.2011.03.045
Ferreira AP, Zanchet D, Rinaldi R et al (2010) Effect of the CeO2 content on the surface and structural properties of CeO2-Al2O3 mixed oxides prepared by sol–gel method. Appl Catal A Gen 388:45–56. doi:10.1016/j.apcata.2010.08.033
Niu J, Du X, Ran J, Wang R (2016) Dry (CO2) reforming of methane over Pt catalysts studied by DFT and kinetic modeling. Appl Surf Sci 376:79–90. doi:10.1016/j.apsusc.2016.01.212
Elsayed NH, Roberts NRM, Joseph B, Kuhn JN (2015) Low temperature dry reforming of methane over Pt–Ni–Mg/ceria-zirconia catalysts. Appl Catal B Environ 179:213–219. doi:10.1016/j.apcatb.2015.05.021
Hassani Rad SJ, Haghighi M, Alizadeh Eslami A et al (2016) Sol–gel versus impregnation preparation of MgO and CeO2 doped Ni/Al2O3 nanocatalysts used in dry reforming of methane: effect of process conditions, synthesis method and support composition. Int J Hydrog Energy 41:5335–5350. doi:10.1016/j.ijhydene.2016.02.002
Luisetto I, Tuti S, Battocchio C et al (2015) Ni/CeO2–Al2O3 catalysts for the dry reforming of methane: the effect of CeAlO3 content and nickel crystallite size on catalytic activity and coke resistance. Appl Catal A Gen 500:12–22. doi:10.1016/j.apcata.2015.05.004
Levalley TL, Richard AR, Fan M (2014) The progress in water gas shift and steam reforming hydrogen production technologies—a review. Int J Hydrog Energy 39:16983–17000. doi:10.1016/j.ijhydene.2014.08.041
Cheekatamarla PK, Finnerty CM (2006) Reforming catalysts for hydrogen generation in fuel cell applications. J Power Sour 160:490–499. doi:10.1016/j.jpowsour.2006.04.078
Jiao Y, Sun D, Zhang J et al (2016) Steam reforming of n-decane toward H2 production over Ni/Ce–Al2O3 composite catalysts: effects of M (M = Fe, Co, Cu, Zn) promoters. J Anal Appl Pyrolysis 120:238–246. doi:10.1016/j.jaap.2016.05.011
Jiao Y, Zhang J, Du Y et al (2016) Steam reforming of hydrocarbon fuels over M (Fe, Co, Ni, Cu, Zn)-Ce bimetal catalysts supported on Al2O3. Int J Hydrog Energy 41:10473–10482. doi:10.1016/j.ijhydene.2015.09.151
Turco M, Cammarano C, Bagnasco G et al (2009) Oxidative methanol steam reforming on a highly dispersed CuO/CeO2/Al2O3 catalyst prepared by a single-step method. Appl Catal B Environ 91:101–107. doi:10.1016/j.apcatb.2009.05.011
Malaibari ZO, Croiset E, Amin A, Epling W (2015) Effect of interactions between Ni and Mo on catalytic properties of a bimetallic Ni-Mo/Al2O3 propane reforming catalyst. Appl Catal A Gen 490:80–92. doi:10.1016/j.apcata.2014.11.002
Huang Z, Jiang H, He F et al (2016) Evaluation of multi-cycle performance of chemical looping dry reforming using CO2 as an oxidant with Fe–Ni bimetallic oxides. J Energy Chem 25:62–70. doi:10.1016/j.jechem.2015.10.008
Rosen BA, Gileadi E, Eliaz N (2016) Electrodeposited Re-promoted Ni foams as a catalyst for the dry reforming of methane. Catal Commun 76:23–28. doi:10.1016/j.catcom.2015.12.014
Carvalho LS, Conceição KCS, Mazzieri VA et al (2012) Pt–Re–Ge/Al2O3 catalysts for n-octane reforming: influence of the order of addition of the metal precursors. Appl Catal A Gen 419–420:156–163. doi:10.1016/j.apcata.2012.01.023
Ramírez-Hernández GY, Viveros-García T, Fuentes-Ramírez R, Galindo-Esquivel IR (2016) Promoting behavior of yttrium over nickel supported on alumina-yttria catalysts in the ethanol steam reforming reaction. Int J Hydrog Energy 41:9332–9343. doi:10.1016/j.ijhydene.2016.04.080
Lenarda M, Moretti E, Storaro L et al (2006) Finely dispersed Pd-Zn catalyst supported on an organized mesoporous alumina for hydrogen production by methanol steam reforming. Appl Catal A Gen 312:220–228. doi:10.1016/j.apcata.2006.07.004
Cassinelli, Damyanova, Parizotto et al (2014) Study of the properties of supported Pd catalysts for steam and autothermal reforming of methane. Appl Catal A Gen 475:256–269. doi:10.1016/j.apcata.2014.01.006
Roy PS, Park CS, Raju ASK, Kim K (2015) Steam-biogas reforming over a metal-foam-coated (Pd-Rh)/(CeZrO2–Al2O3) catalyst compared with pellet type alumina-supported Ru and Ni catalysts. J CO2 Util 12:12–20. doi:10.1016/j.jcou.2015.09.003
https://apps.catalysts.basf.com/apps/eibprices/mp/. Accessed 10 April 2017
Izquierdo U, Barrio VL, Requies J et al (2013) Tri-reforming: a new biogas process for synthesis gas and hydrogen production. Int J Hydrog Energy 38:7623–7631. doi:10.1016/j.ijhydene.2012.09.107
O’Connell M, Kolb G, Schelhaas KP et al (2012) Towards mass production of microstructured fuel processors for application in future distributed energy generation systems: a review of recent progress at IMM. Chem Eng Res Des 90:11–18. doi:10.1016/j.cherd.2011.08.002
O’Connell M, Kolb G, Zapf R, et al (2009) Bimetallic catalysts for the catalytic combustion of methane using microreactor technology. Catal Today 144:306–311. doi:10.1016/j.cattod.2008.10.053
Izquierdo U, Wichert M, Barrio VL, Kolb G (2014) Sustainable syngas production from ethylene glycol reforming processes using Rh-based catalysts in microreactors. Appl Catal B Environ 152–153:19–27. doi:10.1016/j.apcatb.2014.01.012
Izquierdo U, Barrio VL, Lago N et al (2012) Biogas steam and oxidative reforming processes for synthesis gas and hydrogen production in conventional and microreactor reaction systems. Int J Hydrog Energy 37:13829–13842. doi:10.1016/j.ijhydene.2012.04.077
Nevanperä TK, Ojala S, Bion N et al (2016) Catalytic oxidation of dimethyl disulfide (CH3SSCH3) over monometallic Au, Pt and Cu catalysts supported on γ-Al2O3, CeO2 and CeO2–Al2O3. Appl Catal B Environ 182:611–625. doi:10.1016/j.apcatb.2015.10.012
Chen Q-Y, Li N, Luo M-F, Lu J-Q (2012) Catalytic oxidation of dichloromethane over Pt/CeO2–Al2O3 catalysts. Appl Catal B Environ 127:159–166. doi:10.1016/j.apcatb.2012.08.020
Yang H, Deng J, Liu Y et al (2016) Preparation and catalytic performance of Ag, Au, Pd or Pt nanoparticles supported on 3DOM CeO2–Al2O3 for toluene oxidation. J Mol Catal A Chem 414:9–18. doi:10.1016/j.molcata.2015.12.010
Lieske H, Lietz G, Spindler JV H (1983) Reactions of platinum in oxygen- and hydrogen-treated Ptγ-Al2O3 catalysts: I. Temperature-programmed reduction, adsorption, and redispersion of platinum. J Catal 81:8–16. doi:10.1016/0021-9517(83)90142-2
P. Ciambelli, V. Palma, A. Ruggiero, et al, (2009) Platinum catalysts for the low temperature catalytic steam reforming of ethanol. AIDIC Conf Ser 09:83–92. doi:10.3303/ACOS0909011.
Ehrfeld W, Hessel V, Lowe H (2002) Microreactors: new technology for modern chemistry. Wiley-VCH, Weinheim
Jahnisch K, Hessel V, Lowe H, Baerns M (2004) Chemistry in microstructured reactors. Angew Chem Int Ed 43:406–446. doi:10.1002/anie.200300577
Acknowledgements
The authors would like to acknowledge the University of the Basque Country (UPV/EHU) for the postdoctoral scholarship and the Central Analysis Service (SGIker) of the UPV/EHU. This research was supported by the UPV/EHU, the Spanish Ministry of Economy and Competitiveness (MINECO, ENE2014-53566-P), edp company, the European Regional Development Fund (ERDF) and the Basque Country Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Izquierdo, U., García-García, I., Barrio, V.L. et al. Hydrogen Production with a Microchannel Reactor by Tri-Reforming; Reaction System Comparison and Catalyst Development. Top Catal 60, 1210–1225 (2017). https://doi.org/10.1007/s11244-017-0798-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-017-0798-9