
Abstract
Interactions of O2 with the PdO(101) surface were studied using spin-dependent density-functional theory (DFT) with both the PBE and the non-local hybrid HSE exchange–correlation functional. The adsorption energies are strongly overestimated (by 40–60 kJ/mol) with PBE, whereas HSE predicts adsorption energies that are within ~5 kJ/mol of values derived from temperature programmed desorption (TPD) experiments. A detailed partial density of states analysis indicates that the band gap between the PdO d-band center and the LUMO of O2 plays an important role in determining the adsorption strength. This gap is larger for the HSE functional and leads to a decrease in the back donation of the metal d-states to the O2 LUMO orbital resulting in weaker adsorption. Based on the DFT–HSE calculations, three adsorption minima are found to be stable. The most favored configuration, with an adsorption energy of −67 kJ/mol, consists of an O2 molecule lying flat and interacting with two coordinatively unsaturated Pd (Pdcus) surface atoms. The other two configurations have weaker adsorption energies of about −25 kJ/mol and bind to a single Pdcus atom with the O2 molecule oriented away from the surface. The HSE results can be correlated with the observed TPD spectra, which shows only one type of O2 configuration at low coverages with a subsequently lower temperature (more weakly bound) peak evolving at higher coverages associated with the singly coordinated O2 adsorption configurations that start to populate when two adjacent Pdcus sites start to become unavailable.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Palladium is an important catalyst used for CO oxidation in exhaust gas remediation in automobiles and for methane combustion in lean gas turbines [1]. Normally, the catalytic oxidation of CO and methane occurs under oxygen-rich conditions where the PdO phase may develop. Indeed, catalysis experiments suggest that the PdO phase plays a key role in the activity of Pd catalysts for oxidation reactions [2, 3] but there are ongoing efforts to understand and model the role of the metal versus metal oxide for both CO and methane oxidation [4–10]. Ultrahigh vacuum (UHV) and high pressure surface science experiments have both been used to examine CO oxidation on the oxygen phases that form on Pd(100) and Pd(111). Some experiments suggest that the chemisorbed oxygen atoms on Pd are more active than oxide phases toward CO oxidation [11, 12], while others support the opposite conclusion [13]. The interactions between molecular oxygen and the oxide surface may influence the reactions on the surface and the oxidation state of the surface under reaction conditions. If the binding of O2 on the Pd oxide surfaces is sufficiently strong, the adsorbed O2 may also react with CO to form CO2 through a Langmuir–Hinshelwood mechanism. Therefore, the adsorption of O2 on PdO and its binding strength are worth further study.
For metallic Pd, the adsorption of oxygen into precursor states has been widely studied both experimentally [14–16] and theoretically [17, 18]. Imbihl et al. identified three main O2 states on Pd(111) by electron energy loss spectroscopy (EELS) and low energy electron diffraction measurements [14]. At high coverages, a superoxo state with vibrational loss of 1035 cm−1 and a peroxo state with vibrational loss of 850 cm−1 were observed at 30 K. Another peroxo state (650 cm−1) was found at 80 K. Based on the EELS results, Eichler et al. studied the structural, energetic, vibrational, and electronic properties of O2 precursors using DFT calculations [17]. The calculated vibrational frequencies of three distinct O2 configurations (top-bridge-top, top-hcp-bridge, and top-fcc-bridge) agree with the EELS peaks at 850 and 1035 cm−1 but the 650 cm−1 peak was not found in the DFT calculations for any of the O2 adsorbed configurations and might be associated with adsorption at step edges.
Contrary to the significant progress made in O2/Pd(111) studies, the fundamental understanding of O2 adsorption on PdO surfaces is limited because of difficulty in producing well-defined metal oxide surfaces [19–21]. In the past decade, Weaver and co-workers have demonstrated the growth of a high quality PdO(101) thin film on Pd(111) in UHV using an oxygen atom beam [22], and have investigated the adsorption and oxidation of several compounds on the PdO(101) surface [20, 23]. Of particular relevance is a study in which Hinojosa et al. used temperature programmed desorption (TPD) experiments to investigate the molecular adsorption of O2 on PdO(101) thin films [24]. At low O2 coverage, the O2 TPD spectrum from the PdO(101) surface exhibits one main feature (β1) centered at a peak temperature (Tp) of 250 K. As the coverage increases, the β1 peak shifts toward lower temperature and the maximum appears at 233 K once the coverage reaches 0.14 monolayer (ML) (see Sect. II for definition of ML for O2 on the PdO(101) surface). When the coverage is increased beyond about 0.14 ML, a new desorption peak (β2) appears at 117 K. Unlike the β1 peak, the β2 peak temperature does not significantly shift with increasing coverage. Both the β1 and β2 peaks further intensify with increasing coverage until the O2 layer saturates at 0.29 ML. Furthermore, experiments with co-adsorbed 16O2 and 18O2 reveal that the dissociation of O2 occurs negligibly on the PdO(101) surface under the TPD conditions examined. Based on these TPD results, Campbell and Sellers calculated prefactors and adsorption enthalpies of the two O2 states (β1 and β2) using an entropy correlation and transition state theory [19]. The estimated adsorption enthalpies are −33.4 kJ/mol at the TPD peak temperature of Tp = 117 K and −69.2 to −74.7 kJ/mol at Tp = 233–250 K. In this paper, key goals are to identify the different O2 adsorption configurations on PdO(101) associated with the β1 and β2 peaks and predict their adsorption strength using DFT. We focus on how to predict more accurate energies of the O2/PdO system using DFT with different functionals by comparison with the TPD results.
It has long been realized that the frequently used gradient-corrected approximation (GGA) functionals have several shortcomings [25, 26]. Normally GGA functionals overestimate molecular bond energies and adsorption energies of molecules on different metal surfaces [26]. This error is especially large for the O2 molecule, where the bond energy is predicted to be 5.81–6.67 eV by PBE–DFT using oxygen pseudopotentials with different 1 s core electrons [26–28] versus an experimental value of ~5.20 eV [29]. Kiejna et al. examined the O2 bond energy with the PBE functional for both all electron linear augmented plane wave and projected augmented wave (PAW) DFT calculations using different pseudopotentials for oxygen [30]. The all electron LAPW calculations result in an O2 bond energy of 6.21 eV, and this calculation can likely be taken as the most accurate value for a PBE functional. This error in the O2 bond energy is of particular importance in comparing TPD desorption energies with DFT adsorption energies since the adsorption energy is referenced to the gas phase O2 molecule. In addition, it is known that GGA functionals show errors in molecule binding, as for example with CO on metal surfaces [31, 32]. We have recently reported a significant error in DFT–PBE for CO binding on the PdO(101) surface based on a combined IRAS and DFT study [33]. In that study we found that the Heyd-Scuseria-Ernzerhof (HSE), a hybrid exchange-correlational functional, was able to capture the proper preference of CO to bind to the atop Pdcus sites of PdO(101). HSE also reproduces the bulk PdO oxidation energy and band gap [33, 34] and has been shown to provide more accurate formation energies and band structures of metal oxides [34–36]. Grönbeck and co-workers have also examined the accuracy of PBE versus PBE0, a hybrid non-local functional that introduces 25 % Hartree–Fock exact exchange, in predicting core level shifts (CLS) induced by CO adsorption on PdO(101)/Pd(100) [37]. They reported that the CLS for both the Pd 3d states for the bare PdO(101)/Pd(100) surface and the O 1 s states for the adsorbed CO is underestimated with PBE due to the self-interaction error that leads to reduced charge transfer in these systems. The PBE0 functional predicts CLS that are closer to experimental measurements. Recently, HSE has been shown to predict more accurate binding energies for O2 on Al(111) [27].
DFT–PBE accuracy issues for O2 adsorption on the PdO(101) surface have been raised in an earlier computational study of CO oxidation on PdO(101)/Pd(100) [38], but in that study the O2 adsorption energy was adjusted by destabilizing the O2 gas phase energy by about 0.45 eV (43.4 kJ/mol). This destabilization is based on an estimation of the O2 formation energy from DFT–PBE calculations of H2O and H2 gas molecules compared with the experimental formation energy for H2O from H2 and O2 [38, 39]. While this approach does reduce the overestimated O2 adsorption energies from PBE, it cannot distinguish the changes in adsorption energy on different sites on the PdO(101) surface and does not take into account details of the O2–PdO(101) surface interactions. Recently, Van den Bossche and Grönbeck reported both PBE and single point HSE06 values for elementary steps involved in methane oxidation on PdO(101) [40]. They find that HSE06 decreases the stability of adsorbed O2 on PdO(101) by 0.77 eV (74.3 kJ/mol). This change in O2 adsorption binding energy affects the reaction orders and apparent activation barrier derived from their microkinetic simulations, with the weaker O2 binding being critical to avoid poisoning of the surface at lower temperatures.
Based on these prior results we examined O2 adsorption on PdO(101) with both the PBE and HSE functionals. The adsorption energies from PBE overestimate the adsorption energies that are deduced from TPD experiments of O2 on PdO(101). This overestimation depends on the O2 adsorption configuration but can be large as 60 kJ/mol. DFT–HSE calculations predict lower O2 adsorption energies that are within 5 kJ/mol of the TPD values. The stable O2 configurations and coverage dependence from DFT calculations can be correlated qualitatively with the evolution of TPD peaks observed as a function of the O2 coverage. Analysis of the electronic density of states shows that the weaker O2 adsorption in the HSE calculations is due to an increase in the LUMO energy level of gas phase O2 combined with a downward shift of the d-band center of the coordinatively undersaturated Pd surface atoms on the PdO(101) surface. These modifications result in a reduction of the back donation from the Pd d states to the O2 LUMO, which reduces the O2 adsorption energy. This study suggests that hybrid non-local functionals, such as the HSE, are needed to accurately model molecular adsorption involving substantial π-backbonding on the PdO(101) surface and might impact ongoing work to understand the catalytic activity of Pd oxide surfaces for CO and alkane oxidation.
2 Method
The periodic plane-wave DFT calculations reported in this paper were performed using the vienna ab initio simulation package (VASP) [41, 42] with projector augmented wave (PAW) [43] pseudopotentials provided in the VASP database [44]. The soft version of the O pseudopotential was used in all calculations. Spin-polarized calculations were used and the ground state for both isolated O2 and an O atom is a triplet. Both the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [45] and the Heyd-Scuseria-Ernzerhof (HSE06) hybrid functional [36, 46] were used. A plane-wave energy cutoff of 400 eV was used and increasing these convergence criteria results in changes of total energy of less than 0.01 eV, which is sufficiently small to not affect any of the conclusions drawn in this paper.
Figure 1 illustrates the stoichiometric PdO(101) surface that is investigated in this study. Bulk crystalline PdO has a tetragonal unit cell and consists of square planar units of Pd atoms fourfold coordinated with oxygen atoms. The bulk-terminated PdO(101) surface is defined by a rectangular unit cell, where the a and b lattice vectors coincide with the \([010]\) and \([\overline{1} 01]\) directions of the PdO crystal, respectively. The stoichiometric PdO(101) surface consists of alternating rows of threefold or fourfold coordinated Pd or O atoms that run parallel to the a direction shown in Fig. 1. Thus, half of the surface O and Pd atoms are coordinatively unsaturated (cus), and we will refer to these two types of surface atoms as Ocus and Pdcus, respectively. The areal density of each type of coordinatively-distinct atom of the PdO(101) surface is equal to 35 % of the atomic density of the Pd(111) surface. Hence, the coverage of Pdcus atoms on the surface of the PdO(101) surface is equal to 0.35 ML. It is important to note that the coverage of O2 refers to O atoms in this paper and matches the definition in Ref. [24], which reports the TPD spectra for O2 on the PdO(101) surface that will be compared with the DFT results reported in this paper. Based on this definition, if there is one O2 molecule for every Pdcus site on the PdO(101) surface then we would refer to the surface as having a 0.7 ML oxygen coverage. In fact, the TPD experiments show a saturation coverage slightly below 0.35 ML suggesting that at most we can have one O2 molecule for every two Pdcus sites on the surface.

Top and side views of the PdO(101) surface. The red and dark blue atoms represent O and Pd atoms, respectively. Rows of coordinatively unsaturated (cus) and fourfold-coordinated (4f) Pd or O atoms are indicated. The vertical and horizontal arrows a and b represent the \([010]\) and \([\overline{1} 01]\) crystallographic directions of PdO
The PdO(101) surface was modeled by a rectangular 4 × 1 unit cell, with a corresponding 4 × 2 × 1 Monkhorst-Pack k-point mesh [47]. To test the coverage effect, 2 × 1 and 8 × 1 unit cells were also used. Here the 2 × 1 and 8 × 1 sizes are derived by halving and doubling the 4 × 1 surface along the cus-Pd row (\(\overline{a}\)). As in our prior studies [48–51], the PdO(101) film was strained (a = 3.057 Å, b = 6.352 Å) to match the PdO(101) film structure resolved by Kan and Weaver [22, 52]. The PdO(101) slab was represented by four layers resulting in a 9 Å thick slab. The bottom layer is fixed, but other lattice atoms are allowed to relax until the forces are less than 0.03 eV/Å using the limited memory Broyden-Fletcher-Goldfarb-Shanno optimization method implemented for VASP by Sheppard et al. [53]. As in our previous work, the underlying Pd(111) surface is not included since this would require a large unit cell due to the registry of the PdO(101) film with the Pd(111) surface. We use a vacuum spacing of 20 Å, which is sufficient to eliminate spurious interactions in the surface normal direction. Vibrational frequencies were calculated with only the degrees of freedom associated with the O2 molecule included. We performed calculations for selected configurations and find that including the motions of the neighboring Pd and O atoms in the normal mode analysis has a negligible effect on the computed O2 stretch frequencies. Within this paper we use the adsorption enthalpies for O2 on PdO(101) calculated by Campbell and Sellers based on their proposed correlation [19], but in Sect S2 in the Supporting Information we evaluate the temperature-dependent prefactor from DFT calculations and compare the resulting peak temperature from DFT with the experimental values. As described in more detail in Sect. S2, the differences in the direct evaluation of the peak temperature or using the simpler correlation of Campbell and Sellers are generally small. Reported Bader charges are obtained from the electron density using the code of Henkelman and co-workers [54, 55].
3 Results and Discussion
To investigate molecular O2 adsorption on PdO(101) in DFT studies, we define the adsorption energy \(\left( {E_{\text{ads}} } \right)\) as follows:
where \(E_{{{\text{O}}_{ 2} / {\text{PdO}}}}\), \(E_{{{\text{PdO}}}}\), \(E_{{\text{O}}_{ 2}}\) denote the energy of O2 adsorbed on the PdO surface, bare PdO surface, and an isolated O2 molecule, respectively. In order to accurately predict the adsorption energy, we evaluate each term in Eq. 1 by both PBE and HSE calculations. Before turning to the adsorption configurations of O2 on the PdO(101) surface, it is worthwhile to examine the effect of HSE on both the O2 molecule and PdO since the HSE effect on the adsorption energy depends on not only the O2–PdO interactions, but also on how HSE separately affects O2 and the bare PdO surface.
3.1 O2 Molecule in the Gas Phase
Patton et al. have performed PBE calculations to estimate the bond energies of first and second row diatomic and polyatomic molecules [28]. The results show that the PBE overestimation of the O2 bond energy is the largest among the more than 20 molecules that they tested. Our calculated bond energy and bond length of an isolated O2 molecule by PBE and HSE are shown in Table 1 along with experimental values. Both the bond length and bond energy predicted by HSE agree well with the experimental results, whereas PBE overestimates the bond energy of O2 by ~150 kJ/mol and predicts a larger bond length. However, HSE significantly overestimates the vibrational frequency of O2 in comparison to experiment. Jimenez-Hoyos and coworkers have tested the accuracy of molecular vibrational frequencies for different functionals and found that PBE underestimates harmonic frequencies whereas HSE tends to more significantly overestimate them [56], so our results agree with their findings. We will use a scaling factor of 1.011 (0.905) for PBE (HSE) vibrational frequencies reported in the rest of this paper, which is the ratio of the experimental gas phase O–O vibrational frequency and the corresponding calculated DFT values. This approach to scale the DFT vibrational frequency has been shown to give relatively accurate results when studying the adsorption of CO on PdO(101) [9, 33].
We have also calculated the singlet–triplet energy difference (Table 1) and density of states (DOS) of the O2 molecule by PBE and HSE (Fig. 2). For the gas-phase O2 molecule, there are 12 valence electrons occupying the σs, σs*, π2p, σ2p, and π2p* orbitals. All molecular orbitals are fully occupied except the antibonding π2p* orbital. In the triplet O2 state, two unpaired electrons have the same spin and the antibonding π2p* orbital is partially filled by these electrons, defined as the highest occupied molecular orbital (HOMO) in Fig. 2. The unfilled π2p* states have the opposite spin and we define these states as the lowest unoccupied molecular orbital (LUMO) of O2. The DOS below the HOMO is relatively similar between the HSE and PBE functionals, but a significant difference is the energy gap between the HOMO and LUMO. PBE predicts that the HOMO–LUMO gap is about 2.3 eV, but HSE predicts that it is more than 5 eV. This difference significantly influences the adsorption of O2 on the PdO(101) surface, as discussed below.

The density of states of a free O2 molecule calculated by (a) PBE and (b) HSE. The Fermi energy is set to zero and indicated by the dashed line. The spin-up or down states are plotted accordingly
3.2 Oxidation Energy of Bulk PdO
One simple measure of the accuracy of HSE on the description of PdO is to compute the bulk oxidation energy (\(E_{\text{oxidation}}\)) defined as follows:
where \(E_{\text{PdO,bulk}}\) and \(E_{\text{Pd,bulk}}\) are the energies of bulk PdO and bulk Pd, respectively. As mentioned above, PBE overestimates the bond energy of an isolated O2 molecule, so it makes the calculated oxidation energy less negative (i.e. oxidation is less favored because the stability of the O2 molecule is overestimated in PBE) as shown in Eq. (2). Other sources of error to bulk oxidation energy can potentially come from both the \(E_{\text{PdO,bulk}}\) and \(E_{\text{Pd,bulk}}\) terms. While generally it is expected that PBE will correctly describe the electronic structure of Pd metal, it is well known that the PBE functional has errors when treating transition metal oxides due to the self-interaction issue. Wang et al. calculated the oxidation energies of several transition metal oxides and found that the effect of the self-interaction is to increase the magnitude of the oxidation energy [59].
Table 2 shows the lattice parameters, band gap, and oxidation energy of bulk PdO calculated from PBE and HSE, along with experimental data. While PBE underestimates the oxidation energy by ~29 kJ/mol, the HSE oxidation energy of PdO agrees well with the experimental result. It is interesting that errors in both the energy of bulk PdO and the O2 molecule are partially cancelled in the PBE, but this cancellation is not sufficient to give accurate values for the PdO oxidation energy. Furthermore, the lattice parameters predicted by HSE are also in a better agreement with experiments. For semiconductors and insulators, it is well-known that band gaps predicted by PBE are usually smaller than the measured results, but the HSE hybrid functional has been shown to accurately reproduce the band gap of oxides such as Cu2O, FeO, and NiO [35, 60]. Experiments show that PdO is a small band gap semiconductor but the actual band gap is uncertain and the reported values span a wide range (0–2.67 eV) depending on the measurement method [34]. From Table 2, PBE predicts no band gap of PdO, whereas HSE predicts a band gap of 0.8 eV in good agreement with that obtained from the optical transmittance measurement. We note that Grönbeck and co-workers used the PBE0 hybrid functional and reported a band gap of 1.3 eV for PdO [37]. Nevertheless, it is clear that PdO does have a band gap and only the hybrid functionals have been able to predict that PdO is in fact a semiconductor.
3.3 Adsorbed O2 Configurations on PdO(101)
Because DFT–PBE calculations are computationally less expensive, we use DFT–PBE initially to investigate the binding configurations and energetics for O2 adsorption on the pristine PdO(101) surface. On transition metal surfaces, O2 is found to bind in a variety of coordination modes, including through one or two O atoms. Recently, on the RuO2(110) surface it was shown that O2 can adsorb across two adjacent Rucus sites and form a flat-lying configuration or bind at a single Rucus site through one O atom and form an upright configuration [66]. Therefore, we initially studied several configurations of a single O2 molecule on the 4 × 1 supercell of pristine PdO(101) (equivalent to 0.175 ML O coverage) where one or both O atoms of the O2 molecule interact with surface Pd atoms (Fig. 3). Both the Pdcus and Pd4f atoms on the PdO(101) surface may interact with the adsorbed O2 molecule (\({\text{O}}_{2}^{ *}\)). For monodentate \({\text{O}}_{2}^{ *}\), we placed the upright molecule on top of and in between the Pdcus and Pd4f atoms. For bidentate \({\text{O}}_{2}^{ *}\), two O atoms may interact with one, two, or three surface Pd atoms (Pdcus and Pd4f) and the different starting geometries are shown in Fig. 3. In each of these flat-lying configurations, the molecular axis of O2 is parallel to the surface but may become tilted during the relaxation. The O2 molecule can reside on top of one surface Pd atom (Fig. 3a), between two Pd atoms (Fig. 3b), and in the triangle formed by three Pd atoms (Fig. 3c).

Twelve starting geometries of bidentate \({\text{O}}_{2}^{ *}\) on PdO(101) studied in this work. Yellow balls and sticks indicate the adsorbed O2 (a) on top of one surface Pd atom (a), between two Pd atoms and (c) in the triangle formed by three Pd atoms. In each state, there are four different configurations
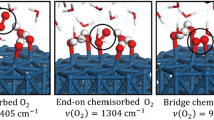
Among the sites investigated, we find four stable configurations in DFT-PBE calculations. The O2 molecule maximizes its adsorption energy when both O atoms interact with the Pdcus row of the PdO(101) surface resulting a configuration where the O2 molecule lies down flat between two Pdcus atoms (Fig. 4a). DFT–PBE identifies three additional stable configurations, all tilted, for O2 adsorbed on the bridgePdcus–Pd4f (Fig. 4b), Pdcus–O4f (Fig. 4c), and Pdcus–Ocus sites (Fig. 4d). Table 3 summarizes the adsorption energies and geometries of the four stable configurations predicted by DFT–PBE calculations.

Top and side views of the four most stable configurations for a single O2 molecule (yellow) adsorbed on the pristine PdO(101) surface predicted by DFT-PBE: (a) flat-lying-Pdcus, (b) bridgePdcus–Pd4f, (c) Pdcus–O4f, and (d) Pdcus–Ocus
Oxygen in the flat-lying-Pdcus configuration has the largest O–O bond elongation, lowest stretching frequency, and the second largest Bader charge of 0.47e among the four configurations. These features are consistent with a peroxo-like \({\text{O}}_{2}^{2 - }\) description. O2 can also bind relatively strongly on PdO(101) by interacting with two Pdcus atoms and one Pd4f atom at the bridgePdcus–Pd4f site (Fig. 4b). In this configuration, the O2 molecule is slightly tilted with respect to the PdO(101) surface plane. One O atom in the molecule resides between two Pdcus atoms (bridgePdcus) and the other leans toward the Pd4f row. The O2 molecule binds with three Pd atoms (2 Pdcus and 1 Pd4f) and has the largest Bader charge (0.54e), which also suggests a peroxo-like \({\text{O}}_{2}^{2 - }\) state. For the other two stable O2 configurations on the Pdcus–O4f and Pdcus–Ocus sites, the Bader charges (0.34 and 0.30e) and stretching frequencies (1271 and 1303 cm−1) indicate that these configurations both correspond to a superoxide (\({\text{O}}_{2}^{ - }\)) state.
To examine the effect of O2–O2 interactions, we examined the four stable O2 configurations on PdO(101) using an 8 × 1 unit cell which corresponds to a coverage of 0.088 ML. The calculated adsorption energy of each configuration does not significantly change (< 2 kJ/mol) in comparison with the results at 0.175 ML, so the coverage effect is negligible at a low coverage (≤ 0.175 ML). We also investigated how the adsorbed O2 configurations evolve at a higher coverage. On the 4 × 1 surface, a single flat-lying-Pdcus O2 species is the global energy minimum at 0.175 ML. We further added one more O2 molecule adsorbed on the adjacent vacant sites (four configurations are shown in Fig. 4) and the O atom coverage increases to 0.35 ML. The calculated energy differences (ΔE) are shown in Table 4. Here ΔE is defined as the energy difference between the final configuration at 0.35 ML and the flat-lying-Pdcus O2 at 0.175 ML plus a gas-phase O2 molecule. Compared with the adsorption energy at 0.175 ML (Table 3), the stability of each configuration is lower by 3–7 kJ/mol at 0.35 ML, which implies a weak repulsion between adjacent adsorbed O2 molecules along the Pdcus row.
We now turn our attention to comparing our results with experiments. As noted in the introduction, a single peak (β1) at Tp = 233-250 K develops in the O2 TPD spectra at coverages below about 0.14 ML. As the coverage increases from 0.14 to 0.22 ML, a second peak (β2) at Tp = 117 K appears and the β1 and β2 peaks intensify concurrently until the O2 layer saturates near 0.29 ML. Our DFT–PBE results predict that the flat-lying O2-Pdcus configuration is preferred at low coverage and we thus attribute the β1 TPD peak to this bidentate configuration. Ideally, only the flat-lying-Pdcus O2 state would reside along the Pdcus row, resulting in a saturation coverage of 0.35 ML. However, the random adsorption of O2 into the bidentate configuration would result in a theoretical saturation coverage (“jamming coverage”) of 86 % of the Pdcus sites if the O2 molecules are immobile, where 86 % corresponds to a coverage of 0.30 ML. The remaining empty Pdcus sites, so-called stranded sites, have neighboring sites that are occupied and can thus only accommodate O2 molecules in configurations that require only a single Pdcus site. We thus assert that the more weakly-bound β2 state corresponds to one of the other three O2 configurations in which only one Pdcus site is occupied by an O2 molecule, and that the β2 state populates appreciably only after the coverage of the bidentate species (β1 state) significantly lowers the concentration of empty pairs of Pdcus sites.
Interestingly, the TPD data shows that the β2 state begins to populate at total coverages (40–63 % of the Pdcus density) that are below the jamming coverage of the bidentate species, and that the total O2 coverage is also lower than the ideal maximum for a mixture of monodentate and bidentate species (0.29 vs. 0.40 ML) with the latter at the statistical jamming coverage. We assert that diffusion limitations and interactions between adsorbed O2 molecules caused the coverages of O2 species obtained in the TPD experiments to drop below the ideal values mentioned above. Despite this apparent kinetic limitation, the DFT results agree well with the experimental results as they predict that O2 can bind in multiple configurations on PdO(101). The calculations specifically suggest that O2 initially binds in the bidentate configuration and occupies neighboring Pdcus sites (β1 state) and that O2 adsorbs into a more weakly-bound configuration(s), involving only one Pdcus site, at higher O2 coverages (β2 state).
Aside from the issue of the saturation coverage, the DFT–PBE results qualitatively match and explain the features seen in the TPD experiments. However, a significant difference exists when comparing our DFT–PBE results with the TPD spectra. In the TPD experiment, the estimated adsorption energies of the two desorption features are −33.4 kJ/mol at the TPD temperature Tp = 117 K and −69.2 to −74.7 kJ/mol at Tp = 233–250 K. Clearly the adsorption energies obtained from DFT–PBE are much larger than the values estimated from TPD. To obtain more accurate adsorption energies of O2 on PdO(101), we also performed DFT–HSE calculations for the four stable configurations (Table 5). The HSE functional strongly reduces the stability of O2 adsorbed on PdO(101). For the most favored configuration, the adsorption energy drops to −67 kJ/mol compared to a PBE-DFT value of −124 kJ/mol, though it is still the most stable configuration in the DFT-HSE results. The O–O bond length decreases by 0.04 Å and the O–Pd height increases by 0.02 Å in comparison to DFT–PBE results. This decrease of 57 kJ/mol in stability of the most favored O2 adsorption configuration is smaller than that reported by the earlier single point HSE06 calculations (74.3 kJ/mol decrease) [40], but both studies indicate a large decrease in the adsorption energy with the HSE functional. While the bridgePdcus–Pd4f site was the second most stable configuration within PBE, the HSE calculation predicts that this configuration is unstable. The adsorption energy of the Pdcus–O4f configuration declines to −19 kJ/mol from a DFT–PBE value of −72 kJ/mol and the Pdcus–Ocus site becomes the second most stable site with an O2 adsorption energy of −27 kJ/mol. In these upright configurations, both the O–O and O–Pd bond lengths significantly change (HSE vs. PBE) and the O2 Bader charge drops significantly, especially for the bridgePdcus–Pd4f site.
While HSE predicts large changes in the adsorption energy compared with the PBE calculations, the HSE results suggest that the flat-lying O2-Pdcus configuration still gives rise to the β1 TPD peak. As discussed earlier based on the PBE results, because of the nonuniform population of O2 on the PdO(101) surface, adsorption into the tilted Pdcus–Ocus configuration will begin to occur at sufficiently high O2 coverage and generates the β2 peak. Both the β1 and β2 will then develop concurrently until the O2 layer saturates. The DFT–HSE predicted adsorption energies are only slightly smaller than those calculated from the TPD spectrum by ~5 kJ/mol, which is in much better agreement with the experimental values compared with DFT–PBE.
To better understand how the HSE calculations influence the O2 adsorption energies we examined the electronic properties of the adsorbed system, i.e., the interaction between the electronic states of PdO(101) and the molecular orbitals of O2. In particular, we focus on explaining two key observations of the HSE versus PBE calculations. Firstly, HSE predicts a weaker bonding of all four O2 states than PBE and secondly the energy change due to the functional depends on the adsorption site with the effect on adsorption energy taking the following order: bridgePdcus–Pd4f > flat-lying-Pdcus > Pdcus–O4f > Pdcus–Ocus. We explain these two observations below by examining the electronic structure through the DOS.
The bonding of O2 on metals is due to electron transfer from the substrate to the unfilled π *2p (LUMO) orbitals [67]. Therefore, the interaction between the LUMO of O2 and the PdO(101) surface can be expected to play an important role in the determining the stability of O2 adsorbed on PdO(101). Kresse et al. studied the adsorption of CO on Pt(111) and concluded that the interaction of the LUMO of CO with Pt d orbitals is overestimated in the conventional DFT calculations and thus the predicted site preference and adsorption energy are inaccurate [68]. One difference between O2 and CO is that the antibonding orbital π2p* in O2 is half filled and splits into the HOMO and LUMO, but their bonding mechanisms on metals are similar [17, 18].
During O2 bonding on PdO(101), the d electrons in the surface Pd atoms back-donate to the O2 LUMO and stabilize the O2–PdO bonding. The interaction strength is approximately inversely proportional to the energy difference between the O2 LUMO and the surface d states for metal surfaces [68, 69]. In order to compare the energies of the O2 molecule and the PdO(101) surface predicted using the different functionals, we plot the Fermi level and the d-band center of PdO(101) and the LUMO level of O2 relative to the energy of the vacuum (Fig. 5). The details of how the reference vacuum energy is calculated within VASP are described in Sect. S3 in the SI. Based on Fig. 5, both PBE and HSE predict a similar Fermi level for the PdO(101) surface. PBE predicts that the d-band center of the clean PdO(101) surface lies below the LUMO of O2 with a gap of about 3.3 eV, while, in contrast, HSE predicts that the O2 LUMO (PdO(101) d-band center) significantly moves to a higher (lower) energy level causing the HSE gap between the O2 LUMO and PdO d-band center to increase to 5.6 eV. Consequently, HSE predicts a weaker interaction between the O2 LUMO and Pd d orbitals and less back-donation of charge leading to lower adsorption energies for all of the stable O2 configurations identified using DFT–PBE.

The energy levels relative to the vacuum energy for the LUMO of the isolated O2 molecule, the Fermi level Ef and d-band center of the PdO(101) surface calculated by PBE and HSE
The interaction between the O2 LUMO and PdO d states was also investigated by a detailed analysis of partial density of states (pDOS) of O2/PdO(101) before and after adsorption. Here we use the most favored configuration (flat-lying-Pdcus) as an example to explain the difference between the PBE and HSE results. A rotation of the orientation of the PdO(101) surface around the Pdcus row is required to match the orientation of the Pd d orbitals with the axis of the projection of the states within VASP. The details of this procedure are described in more detail in sect. S1 in the SI. The net effect of this rotation is that, for example, the Pd z2 orbital for the Pdcus atom is properly identified. For the adsorbed O2 molecule, the combination of 2s (2py) orbitals in two O atoms forms the molecular orbitals of σs and σs* (σ2p and σ2p*). The combination of 2px or 2pz orbitals forms the bonding and antibonding π orbitals (π2p and π2p*). For 2px (2pz), the formed molecular orbital is parallel (orthogonal) to the XY plane, so we use the notation π ∥ and π ∥* (π⊥ and π⊥*) to describe the O2 π states. During O2 bonding with the PdO surface, the electrons may transfer from the PdO surface to the π∥* or π⊥* orbitals, so we focus on these orbitals in the subsequent discussion.
The partial DOS of Pdcus atoms before and after adsorption and the total DOS (TDOS) of the flat-lying O2–Pdcus configuration are shown in Fig. 6 for calculations using both the PBE and HSE functionals. We also show the π states of the gas-phase O2 molecule (Fig. 6d). We first consider the PBE results in the left panel. After O2 is adsorbed on the surface, the Pdcus d bands broaden and shift down to lower energies because of the interaction with the O2 molecule. All of the d states are significantly modified. From the TDOS plot of O2, the spin splitting has completely disappeared, which corresponds to a significant stretching of the molecule (1.23 → 1.33 Å). Compared with Fig. 2, the O2 LUMO strongly interacts with the Pd d-band and shifts to lower energy resulting in more extended π2p* states (dπ) from −3 to 2 eV. Population of the newly formed dπ states results from a transfer of electrons from the surface to the originally half-filled antibonding π2p* orbital. Geometrically, the π⊥* orbital interacts with Pd dz2 states and the π∥* orbital interacts with Pd dxz states. This can be confirmed by comparing the energy levels of the two π antibonding states (Fig. 6d) with the energies of the d states (Fig. 6b). It is important to note that the π⊥*–d interaction is stronger than that of the π∥*–d, because above the Fermi level the π⊥* states are significantly modified and shift to a lower energy level whereas there is still an unpertured π∥* peak (Fig. 6d).

PBE (left) and HSE (right) orbital resolved electronic DOS for a Pdcus atoms on the bare PdO(101) surface, b Pdcus atoms after O2 adsorption c the TDOS for the adsorbed O2, and d π bonding and antibonding states of O2 after adsorption. The Fermi level is located at 0 eV and indicated by the dot line
We now turn our attention to the HSE result. For the bare surface, all of the d states are shifted to lower energy and the calculated d band center lies 1.03 eV lower in energy compared with the PBE result, but the overall pattern of d states is not significantly changed. The downward shift of the HSE d bands can be explained by the reduction of self-interaction error in the hybrid functional. Also, HSE shifts the occupied (unoccupied) molecular orbitals of O2 to lower (higher) energies. The combined effect is that the gap between the Pd d-band and the LUMO of O2 significantly increases and electron transfer from the d orbitals of Pd to the LUMO of O2 is hindered. From the TDOS of adsorbed O2, the spin splitting still exists in the O2 molecule due to a smaller O–O bond elongation in the HSE results. The population of O2 dπ states is reduced in the HSE results, and thus the O2-Pd bond is destabilized due to less electron transfer from the metal sites to O2. The weakening of the O2–PdO interaction can be seen clearly in the pDOS of the O2 states. For the π⊥* orbitals, the interaction with the Pd d band is not significantly changed and the corresponding dπ states become only slightly weaker. However, the π∥*-d interaction is insignificant in the HSE results. Above the Fermi level, there is a sharp unfilled π∥* band from 1 to 2 eV, which indicates that the unfilled π∥* orbital does not interact with Pd d bands and is unperturbed in the HSE calculations. Therefore, HSE reduces the adsorption energy of the flat-lying O2–Pdcus configuration because the π∥*–d interactions are weakened and the electron can only transfer from the Pd d orbitals to the O2 π⊥* orbitals.
Above we explained the overall difference in O2 adsorption energy between the PBE and HSE calculations. However, the energy difference between the HSE and PBE calculations depends on the O2 adsorption site. These site differences can be explained by the different adsorption geometries adopted by O2 on the various sites. For the bridgePdcus–Pd4f site, one O2 molecule interacts with three Pd atoms (one Pd4f and two Pdcus). The Bader analysis shows that the Pd4f–O2 bonding accounts for half of the total electron transfer. In the HSE calculation, the O2 cannot bond with Pd4f, so the bridgePdcus–Pd4f site is no longer stable. For the other two tilted configurations, Pdcus–O4f and Pdcus–Ocus, the O2 molecule binds with only one Pdcus atom, and thus the reduction of the Pd–O2 bonding strength is the least among the four sites.
4 Summary
In summary, we performed DFT calculations of molecularly adsorbed O2 on the PdO(101) surface using both the PBE and HSE functionals. The adsorption energies are strongly overestimated by PBE, whereas HSE predicts more accurate adsorption energies in comparison to TPD results. A detailed partial DOS analysis indicates that the interaction between the O2 antibonding π2p* orbital and the Pdcus d-band plays a critical role in determining the O2 adsorption energy. In the PBE calculations, the energy gap between the PdO d-band center and the LUMO of O2 is much smaller than for the HSE functional, and consequently the π2p*–d interaction and associated back-donation effects are overestimated in the PBE calculations. In contrast, HSE shifts the PdO d-band lower in energy and pushes the LUMO of O2 higher in energy. As a result, the π2p*–d interaction becomes weaker and the O2 adsorption is destabilized with the HSE functional. Using either the PBE or HSE results, we can explain the observed appearance of β1 and β2 peaks in the TPD experiment with increasing O coverage. The β1 TPD peak observed at low coverage is associated with the most stable configuration of O2 on the PdO(101) surface, where the O2 molecule lies flat and interacts with two cus-Pd sites (flat-lying-Pdcus). As the coverage increases this stable configuration cannot be accessed since two empty adjacent Pdcus sites are required. Within HSE there are two less stable O2 configurations that interact with the PdO(101) surface through one Pdcus site, and these are associated with the lower temperature β2 peak. Overall, this study demonstrates that hybrid functionals are required to properly describe O2 adsorption on the PdO(101) surface, which is a critical step in describing oxidation catalysis on these surfaces.
References
McCarty JG (1995) Catal Today 26:283–293
Datye AK, Bravo J, Nelson TR, Atanasova P, Lyubovsky M, Pfefferle L (2000) Appl Catal A 198:179–196
Carstens JN, Su SC, Bell AT (1998) J Catal 176:136–142
Hoffmann MJ, Reuter K (2014) Top Catal 57:159–170
Duan Z, Henkelman G (2014) ACS Catal 4:3435–3443
Weng X, Yuan X, Li H, Li X, Chen M, Wan H (2015) Sci China Chem 58:174–179
Martin NM, Van Den Bossche M, Grönbeck H, Hakanoglu C, Zhang F, Li T, Gustafson J, Weaver JF, Lundgren E (2014) J Phys Chem C 118:1118–1128
Hellman A, Resta A, Martin NM, Gustafson J, Trinchero A, Carlsson PA, Balmes O, Felici R, Van Rijn R, Frenken JWM, Andersen JN, Lundgren E, Grönbeck H (2012) J Phys Chem Lett 3:678–682
Martin NM, Van Den Bossche M, Hellman A, Grönbeck H, Hakanoglu C, Gustafson J, Blomberg S, Johansson N, Liu Z, Axnanda S, Weaver JF, Lundgren E (2014) ACS Catal 4:3330–3334
Blomberg S, Hoffmann MJ, Gustafson J, Martin NM, Fernandes VR, Borg A, Liu Z, Chang R, Matera S, Reuter K, Lundgren E (2013) Phys Rev Lett 110:117601
Kolasinski KW, Cemic F, Demeijere A, Hasselbrink E (1995) Surf Sci 334:19–28
Gabasch H, Knop-Gericke A, Schlögl R, Borasio M, Weilach C, Rupprechter G, Penner S, Jenewein B, Hayek K, Klötzer B (2007) Phys Chem Chem Phys 9:533–540
Toyoshima R, Yoshida M, Monya Y, Kousa Y, Suzuki K, Abe H, Mun BS, Mase K, Amemiya K, Kondoh H (2012) J Phys Chem C 116:18691–18697
Imbihl R, Demuth JE (1986) Surf Sci 173:395–410
Guo X, Hoffman A, Yates JT (1989) J Chem Phys 90:5787
Sjovall P, Uvdal P (1998) J Vac Sci Technol A 16:943–947
Eichler A, Mittendorfer F, Hafner J (2000) Phys Rev B 62:4744–4755
Honkala K, Laasonen K (2001) J Chem Phys 115:2297–2302
Campbell CT, Sellers JRV (2013) Chem Rev 113:4106–4135
Weaver JF (2013) Chem Rev 113:4164–4215
Lundgren E, Mikkelsen A, Andersen JN, Kresse G, Schmid M, Varga P (2006) J Phys 18:R481–R499
Kan HH, Weaver JF (2008) Surf Sci 602:L53–L57
Weaver JF, Hakanoglu C, Antony A, Asthagiri A (2014) Chem Soc Rev 43:7536–7547
Hinojosa JA, Kan HH, Weaver JF (2008) J Phys Chem C 112:8324–8331
Zygmunt SA, Curtiss LA (2005) Quantum-chemical studies of molecular reactivity in nanoporous materials. In: Curtiss LA, Gordon MS (eds) Computational materials chemistry. Kluwer, Dordrecht, pp 191–245
Hammer B, Hansen L, Nørskov J (1999) Phys Rev B 59:7413–7421
Liu H-R, Xiang H, Gong XG (2011) J Chem Phys 135:214702
Patton DC, Porezag DV, Pederson MR (1997) Phys Rev B 55:7454–7459
Lide DR (2013) CRC Handbook of chemistry and physics, 94th Edition, 2013–2014. CRC Press, Boca raton
Kiejna A, Kresse G, Rogal J, De Sarkar A, Reuter K, Scheffler M (2006) Phys Rev B 73:35404
Stroppa A, Termentzidis K, Paier J, Kresse G, Hafner J (2007) Phys Rev B 76:195440
Gajdos M, Eichler A, Hafner J (2004) J Phys 1141:16
Zhang F, Pan L, Li T, Diulus JT, Asthagiri A, Weaver JF (2014) J Phys Chem C 118:28647–28661
Bruska MK, Czekaj I, Delley B, Mantzaras J, Wokaun A (2011) Phys Chem Chem Phys 13:15947–15954
Marsman M, Paier J, Stroppa A, Kresse G (2008) J Phys 20:064201
Paier J, Marsman M, Hummer K, Kresse G, Gerber IC, Angyán JG (2006) J Chem Phys 124:154709
Van Den Bossche M, Martin NM, Gustafson J, Hakanoglu C, Weaver JF, Lundgren E, Grönbeck H (2014) J Chem Phys 141(3):034706
Hirvi JT, Kinnunen T-JJ, Suvanto M, Pakkanen TA, Nørskov JK (2010) J Chem Phys 133:084704
Nørskov JK, Rossmeisl J, Logadottir A, Lindqvist L, Kitchin JR, Bligaard T, Jónsson H (2004) J Phys Chem B 108:17886–17892
Van den Bossche M, Grönbeck H (2015) J Am Chem Soc 137:12035–12044
Kresse G, Hafner J (1993) Phys Rev B 47:558–561
Kresse G, Hafner J (1993) J Non Cryst Solids 156–158:956–960
Blöchl PE (1994) Phys Rev B 50:17953–17979
Kresse G, Joubert D (1999) Phys Rev B 59:1758–1775
Perdew J, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865–3868
Heyd J, Scuseria GE, Ernzerhof M (2003) J Chem Phys 118:8207
Monkhorst H, Pack J (1976) Phys Rev B 13:5188–5192
Weaver JF, Hakanoglu C, Hawkins JM, Asthagiri A (2010) J Chem Phys 132:024709
Hakanoglu C, Hawkins JM, Asthagiri A, Weaver JF (2010) J Phys Chem C 114:11485–11497
Weaver JF, Hakanoglu C, Antony A, Asthagiri A (2011) J Am Chem Soc 133:16196–16200
Antony A, Hakanoglu C, Asthagiri A, Weaver JF (2012) J Chem Phys 136:054702
Kan HH, Weaver JF (2009) Surf Sci 603:2671–2682
Sheppard D, Terrell R, Henkelman G (2008) J Chem Phys 128:134106
Tang W, Sanville E, Henkelman G (2009) J Phys 21:084204
Henkelman G, Arnaldsson A, Jónsson H (2006) Comput Mater Sci 36:354–360
Jiménez-Hoyos CA, Janesko BG, Scuseria GE (2008) Phys Chem Chem Phys 10:6621–6629
Irikura KK (2007) J Phys Chem Ref Data 36:389–397
Schweitzer C, Schmidt R (2003) Chem Rev 103:1685–1758
Wang L, Maxisch T, Ceder G (2006) Phys Rev B 73:195107
Scanlon DO, Morgan BJ, Watson GW, Walsh A (2009) Phys Rev Lett 103:096405
Rogers DB, Shannon RD, Gillson JL (1971) J Solid State Chem 3:314–316
Nilsson PO (1979) J Phys C 12:1423
Okamoto H, Asô T (1967) Jpn J Appl Phys 6:779
Rey E, Kamal MR, Miles RB, Royce BSH (1978) J Mater Sci 13:812–816
Pawlas-Foryst E, Zabdyr L (2008) Arch Metall Mater 53:1173–1175
Wang H, Schneider WF, Schmidt D (2009) J Phys Chem C 113:15266–15273
Finlay RJ, Her T, Wu C, Mazur E (1997) Surface femtochemistry of oxygen and coadsorbates on Pt(111). In: Sundstrom V (ed) Femtochemistry femtobiology ultrafast react. Imperial College, London, pp 629–659
Kresse G, Gil A, Sautet P (2003) Phys Rev B 68:3–6
Hammer B, Nørskov JK (1995) Surf Sci 343:211–220
Acknowledgments
We acknowledge the Ohio Supercomputing Center for providing computational resources. We gratefully acknowledge financial support for this work provided by the Department of Energy, Office of Basic Energy Sciences, Catalysis Science Division through Grant DE-FG02-03ER15478.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pan, L., Weaver, J.F. & Asthagiri, A. First Principles Study of Molecular O2 Adsorption on the PdO(101) Surface. Top Catal 60, 401–412 (2017). https://doi.org/10.1007/s11244-016-0705-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-016-0705-9