
Abstract
The hydrodeoxygenation (HDO) of guaiacol was investigated over SiO2, ZrO2, and active carbon (AC) supported Ni2P catalysts. The physical properties of the catalysts were analyzed by temperature-programmed reduction (H2-TPR), CO-uptake chemisorption, and N2 physisorption. X-ray diffraction and extended X-ray adsorption fine structure spectroscopy were used to obtain structural properties for the supported Ni2P catalysts. The HDO was tested in a batch reactor at 573 K and 30 atm. The Ni2P/SiO2 catalyst underwent a decrease in the HDO conversion from 87 to 30 % for the first and second run of reaction. However, the Ni2P/ZrO2 and Ni2P/AC catalysts showed a little low but stable HDO conversions of 72 and 46 %, respectively. The in situ XAFS analysis revealed that differently from the cases of Ni2P/ZrO2 or and Ni2P/AC catalysts, the local structure of the Ni2P on SiO2 support underwent an oxidation to form nickel phosphate during the reaction, demonstrating that the SiO2 based Ni2P was vulnerable to the water or hydroxyl group of the reactant due to the hydrophilic nature of SiO2 support.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Depletion of petroleum reserves and stringent environmental regulation give rise to attention for alternative fuel and clean source of energy. Bio-oil produced from the pyrolysis of biomass is one of the potential alternative clean energy sources due to the CO2-neutral nature and free of SOx emission upon the combustion. However, the high amount of oxygen (45–50 %) of bio-oil can be an obstacle for directly using it as fuel [1, 2]. The oxygen can be catalytically removed via two reactions: catalytic cracking or hydrodeoxygenation (HDO). The catalytic cracking occurs at atmospheric pressure condition and high temperature (>450 °C) over acidic zeolite catalysts, but the cracking gives a lower yield of transport fuel product due to the high amount of coke formation, reaching 50–60 % [3]. Gayubo et al. studied catalytic cracking of bio-oil over ZSM-5 zeolite and showed that phenol and guaiacol had low reactivity for hydrocarbons formation; deposition of a coke formed thermally by condensation of 2-methoxyphenol was noticeable [4]. The HDO is generally conducted at high pressure and temperature and results in high product yield with high H/C ratios. As the reaction condition is similar to the traditional hydrotreating process, it can be grafted onto the existing system [5].
Traditionally the hydrotreating catalysts are composed of metal sulfides like NiMoS/Al2O3 or CoMoS/Al2O3 [6–16]. However, these catalysts can be poisoned in the lack of sulfur in the feed stream. In other words, the active sites of the catalysts can be oxidized and deactivated when the bio-oil without sulfur is applied as feedstock [12]. Therefore, alternative catalysts for HDO of bio-oil are necessary and very recently, non-sulfided metal phosphides have been studied. Among those metal phosphide catalysts, Ni2P have shown the best activity in the HDO of guaiacol [17–24]. Moreover, the type of catalyst support is also influential in the HDO. The alumina (Al2O3) is known to cause coke deposition and rapid deactivation in the HDO, particularly in the presence of water [25–29]. These drawback led to work on other supports such as silica [29, 31], carbon [15, 16], zirconia [17, 30, 31], and various mixed oxides [32].
In the present study, our attention is placed on investigating the effect of various supports like SiO2, ZrO2 and active carbon (AC) on the catalytic activity in the HDO and durability upon the exposure to water in the course of the HDO. In particular, in situ XAFS analysis was employed to elucidate the structural properties during the reaction.
2 Experimental
2.1 Synthesis of Ni2P Catalysts
The supported Ni2P catalysts were prepared by incipient wetness impregnation. The initial Ni/P molar ratio was 0.5 and the amount of Ni loading was maintained at 1.5 mmol/g of support using silica (Cabot, Cab-O-sil, M5, 200 m2 g−1), ZrO2 (Alfa Aesar), and AC (Sigma Aldrich, Darco G-60,100 mesh). The supported nickel phosphate precursor was synthesized with a solution of nickel nitrate, Ni(NO3)26H2O(Alfa Aesar, 98 %) and ammonium phosphate (NH4)HPO4 (Samchun, 99 %), followed by drying at 393 K for overnight. After drying, the supported Ni2P catalysts were calcined 673 K for 4 h in He. The resulting precursor phosphates were reduced to the corresponding phosphides at 873 K for 2 h using a hydrogen flow at 100 cm3 min−1/g of sample. After reduction, the catalysts were activated in tridecane which is used as solvent of reactant.
2.2 Catalyst Characterization
CO chemisorption uptake measurements were performed on reduced fresh Ni2P and on air-exposed spent samples re-reduced in hydrogen at 723 K for 2 h to examine the dispersion of Ni2P on the support. Pulses (100 μl) of CO at room temperature (300 K) were passed over the sample to measure the total dynamic gas uptake. A Micrometeritics ASAP2010 micropore size analyzer was used to measure the N2 adsorption/desorption isotherms at 77 K, and the specific surface area of the sample was calculated from the linear portion of Brunauer-Emmett-Teller (BET) plots (P/P0 = 0.01 − 0.10). The chemical composition of the samples was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (PerkinElmer, Model Optima-4300 DV). X-ray diffraction (XRD) patterns of the samples were analyzed using a diffractometer (Rigaku DMAX-2500) operated at 60 kV and 300 mA with Cu Kα radiation (λ = 0.15418 nm).
2.3 X-ray Adsorption Fine Structure (XAFS) studies
XAFS spectra at the Ni-K-edge (8.333 keV) of reference and catalyst samples were recorded in the energy range 8.23–9.283 keV using synchrotron radiation at the beamline 8C, Pohang Light Source (PLS). The X-ray ring at the PLS has a flux of 1 × 1010 photons s−1 at 100 mA and 2.5 GeV. The X-ray beamline is equipped with a Si (1 1 1) channel-cut monochromator and has an energy range capability of 4–33 keV. The samples were prepared in a kapton sealed glass cell to avoid air-exposure. The X-ray adsorption spectra were recorded at ambient temperature in transmission mode using ionization chambers for the detection of primary (I0, 100 % N2) and transmitted (IT, 100 % N2) beam intensities.
In situ XAFS analysis with H2-TPR (temperature-programmed reduction) and the HDO of phenol was conducted. The disc shaped samples were placed in an in situ reactor (quartz, 25.0 mm external diameter) using a sample holder (quartz, 15.0 mm external diameter). The holder was placed in the center of a reactor and thermocouple port was placed right above the sample disc. The catalysts were pre-reduced at 823 K with a temperature ramping rate of 5 K/min in 5 % H2/He. For in situ XAFS measurements with flowing phenol or water, the samples were exposed to a gaseous phenol flow vaporized via a bubbler (1 % phenol/hexane and distillated water) at 573 K. The obtained XAFS data were analyzed by Winxas 3.1.
2.4 Activity Test for Hydrodeoxygenation of Guaiacol
The HDO of guaiacol was measured in 150 ml autoclave batch reactor. The reduced catalyst (0.5 g) was applied with 1wt% of guaiacol (Alfa Aesar, 98+ %) in tridecane (TCI, >98 %) at 573 K and 30 atm for 8 h. After the first run of the HDO, a repetitive test was also applied to examine the stability of the catalysts, in which the catalysts were re-tested with new reactant feed of 1 wt% guaiacol after removing the previous reactant and product. The catalyst was not re-reduced after the first run but washed with tridecane two times prior to the introduction of the reactant. Moreover, the addition of water on the HDO was also examined for Ni2P catalysts by applying 1.0 wt% distillated water mixed in the reactant in the batch reactor at the same reaction condition.
In this work, the conversions of guaiacol and HDO are defined as below:
3 Results and Discussion
3.1 Physical Properties of Catalysts
The physical properties of the fresh and spent Ni2P catalysts are listed in Table 1. The supported Ni2P catalysts underwent a loss in the BET surface area, due to the pore occupation by the Ni2P loadings. The surface area remained unchanged during the reaction, except for Ni2P/SiO2 catalysts, indicating agglomeration of the support silica particles in the course of the HDO in the batch reactor due to the hydrophilic nature of the fumed silica [38]. Elemental analysis showed that phosphorous contents was decreased in the course of TPR for the fresh sample, compared to the initial P/Ni molar ratio of 2/1 in the oxidic precursor. During the reaction, further loss of P was observed to give the P/Ni molar ratio of 0.72, 0.68, and 0.86 for the spent samples Ni2P/SiO2, Ni2P/AC, and Ni2P/ZrO2, respectively. The extra P applied in the oxidic precursor might be removed by the formation of PH3 during the course of TPR and hydrotreating condition, as also suggested by previous reports [35, 36]. For the fresh samples the amount of CO uptake followed the order, Ni2P/SiO2 > Ni2P ZrO2 > Ni2P/AC, indicating that the particles of Ni2P are well dispersed on the SiO2 support. For the spent samples the amount of CO uptake was reduced compared to the fresh samples, especially the greatest decrease being observed for the SiO2 supported Ni2P, indicating the loss of active sites of the catalyst during the reaction.
Figure 1 showed XRD patterns for the fresh and spent Ni2P catalysts. The XRD patterns for the supported Ni2P catalysts showed three main peaks centered at 40.5, 44.8, 47.5° corresponding to the characteristic XRD peaks of Ni2P phase. For Ni2P/ZrO2 catalyst, additional peaks were observed at 30, 35, 50.1, 60, 62.9° corresponding to tetragonal zirconia (ICDD-01-072-7115). In all cases the Ni2P peaks remained even after reaction, indicating that the bulk Ni2P phase does not deteriorate during the reaction.

XRD patterns for Ni2P/SiO2, Ni2P/ZrO2 and Ni2P/AC catalyst
3.2 XAFS Analysis
The Ni K-edge XAFS spectra of the fresh and spent Ni2P catalysts were obtained as shown in Fig. 2. The X-ray adsorption fine structure (XAFS) spectra of the fresh sample of catalysts are collected after being reduced at 673 K. The analysis of X-ray adsorption near edge structure (XANES) region can provide information on the symmetry and the oxidation state of the Ni absorber. The pre-edge intensity is related to the symmetry and the occupancy of the 3d shell. The Ni K-edge XANES spectra show two region, which exhibit a pre-edge and main edge peak corresponds to the transition of 1s to 4p symmetry levels [35, 37]. For the spent samples the XANES spectra retain a rise in the absorption peak from 1s-4p transition, which is generally called white line. Notably, the spent sample of Ni2P/SiO2 showed a much stronger white line than the other Ni2P samples, indicating that a partial oxidation occurred on the surface of the Ni2P/SiO2 catalyst during the HDO.

Ni K-edge XANES, EXAFS, and Fourier transforms of fresh and spent Ni2P catalysts
Figure 2 also shows Ni K-edge extended X-ray adsorption fine structure (EXAFS) spectra and their Fourier transforms for the fresh and spent samples. The EXAFS spectra of bulk Ni2P consist of a little wider oscillation region in 30.0–80.0 nm−1 and a narrower oscillation region in 80.0–140.0 nm−1 due to the Ni–P contribution and Ni–Ni contribution, respectively. These give rise to two main peaks in the Fourier transforms, centered at 0.175 nm, corresponding to Ni–P and 0.240 nm, corresponding to Ni–Ni [35]. Also for the fresh Ni2P/SiO2, Ni2P/ZrO2, and Ni2P/AC samples, the Fourier transforms exhibit two distinct peaks in the region 0.15–0.30 nm, the shorter distance peak due to the Ni–P bond and the longer distance peak Ni–Ni bond, indicating well formation of Ni2P phase. For the spent Ni2P/ZrO2 and Ni2P/AC, the two distinct peaks located almost the same position as the fresh samples, implying the maintenance of the Ni2P phase during the reaction. However, the spectrum for Ni2P/SiO2 shows wider oscillation range at 40.0–60.0 nm than that of Ni2P reference. These results demonstrate that the phase of nickel phosphide was slightly oxidized as also observed in the XANES results.
In order to elucidate the structural properties of the Ni2P catalysts, the in situ XANES measurements were conducted as shown in Fig. 3. After the pre-reduction at 823 K, the phenol was introduced at 573 K and the spectra were collected every 15 min. For Ni2P/SiO2 samples, the white line appeared upon the introduction of phenol and developed till 30 min of the reaction. After then the spectrum was maintained after the introduction of water. In contrast, the in situ XANES spectra for Ni2P/ZrO2 samples exhibited almost no difference upon the introduction of phenol or water.

In situ Ni K-edge XANES spectra for Ni2P/SiO2 (left), Ni2P/ZrO2 (right) catalysts
3.3 Hydrodeoxygenation of Guaiacol
Table 2 presents the product distributions for the guaiacol HDO for the Ni2P/SiO2, Ni2P/ZrO2, and Ni2P/AC catalysts. The HDO conversion followed the order: Ni2P/SiO2 > Ni2P/ZrO2 > Ni2P/AC, corresponding to the order of the amounts of CO uptake. For Ni2P/SiO2, the main products of HDO of guaiacol were benzene, cyclohexane, anisole and phenol and the conversion of the HDO was 87 %. The introduction of water in the reactor led to the decrease in the HDO conversion down to 18 % and the main product was phenol, indicating that the HDO activity of was deteriorated upon the introduction of water. For Ni2P/ZrO2, the HDO conversion was 72 % and the main product was cyclohexane, phenol, cyclohexene, and cyclohexone. The production of phenol can be explained by the side reaction occurring on the support ZrO2. Previous studies have shown that the support ZrO2 is active in the elimination of the methoxy group of guaiacol to give phenol [30, 31]. For Ni2P/AC, the HDO conversion was 46 % and the main products were cyclohexane, benzene, and cyclohexene. After the water was introduced to guaiacol, the HDO conversion of ZrO2 or AC supported Ni2P catalysts was not severely affected, demonstrating a less vulnerable nature to the exposure to water than Ni2P/SiO2 during the HDO.
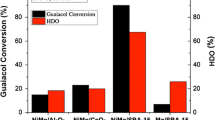
In order to examine the catalytic stability a repetitive HDO test was conducted for the catalysts as shown in Fig. 4. The product distributions are also summarized in Table 3. For the Ni2P/SiO2 catalyst, the HDO conversion was considerably decreased from 87 to 30 % in the first and second HDO tests, respectively and the main product also changed from cyclohexane to phenol, demonstrating a loss of the HDO activity, particularly in the hydrogenation activity. For Ni2P/AC, the HDO conversion was slightly decreased from 46 to 41 % with the yield of cyclohexane being decreased from 20 to 5 % and of phenol being slightly increased from 2 to 6 %. For Ni2P/ZrO2, the HDO conversion was slightly decreased from 72 to 70 % with the yield of phenol being slightly increased from 25 to 27 %, demonstrating the methoxy elimination activity ZrO2 [30, 31].

Guaiacol HDO conversion of repetitive reaction tests for Ni2P/SiO2, Ni2P/ZrO2 and Ni2P/AC catalyst
3.4 Water Durability of Ni2P Catalysts Supported on SiO2, ZrO2, and AC
From these experimental results, the schematic of the catalyst surface structure during the reaction can be proposed as shown Scheme 1. The hydrophilicity of the silica based Ni2P catalyst can facilitate the hydration of the surface of the Ni2P/SiO2, resulting in the loss of the HDO activity by the surface oxidation of Ni2P [38]. Similar results were also reported for the traditional sulfide catalysts, in which the presence of water in the HDO led to the formation of a sulfate layer covering the active phase of sulfide catalysts and reducing the catalytic activity [28, 33, 34]. For the ZrO2 or AC supported Ni2P catalysts the catalytic stability toward HDO was substantially improved, which can be explained by the nature of the support. The surface hydration behavior can be suppressed by the less hydrophilic support like ZrO2 or AC. Thus the use of hydrophobic supports could be beneficial in maintaining the HDO activity and stability of Ni2P catalysts.

Schematic of the catalyst surface structure during the HDO
4 Conclusions
The Ni2P catalysts demonstrated high activity in the HDO of guaiacol with a guaiacol conversion over 70 % in a batch reactor at 573 K and 30 atm. The characterization of structural properties of fresh and spent Ni2P catalysts made by XAFS and XRD analysis revealed that the local structure of Ni2P phase was slightly oxidized particularly over SiO2 support, but maintained over the ZrO2 or AC supports. In situ XANES analysis showed that the introduction of phenol or water led to the surface oxidation only for the Ni2P/SiO2 catalyst. All these results demonstrate that the hydrophilic nature of the support could facilitate the hydration of the catalyst surface during the HDO, while the use of less hydrophilic support like ZrO2 or AC could enhance the stability of the Ni2P catalysts.
References
Furimsky E (2000) Catalytic hydrodeoxygenation. Appl Catal B 199:147–190
Bridgwater AV (1996) Production of high grade fuels and chemicals from catalytic pyrolysis of biomass. Catal Today 29:285–295
Bulushev DA, Ross JRH (2011) Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal Today 171:1–13
Gayubo AG, Aguayo AT, Atuxtxa A, Aguado R, Olazar M, Bilbao J (2004) Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. II. Aldehydes, ketones, and acids. Ind Eng Chem Res 43:2619–2626
Huber GW, Iborra S, Corma A (2006) Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem Rev 106:4044–4098
Bui VN, Laurenti D, Afanasiev P, Geantet C (2011) Hydrodeoxygenation of guaiacol with CoMo Catalysts. Part I: promoting effect of cobalt on HDO selectivity and activity. Appl Catal B 101:239–245
Bui VN, Laurenti D, Afanasiev P, Geantet C (2011) Hydrodeoxygenation of guaiacol. Part II: support effect for CoMoS catalysts on HDO activity and selectivity. Appl Catal B 101:246–255
Gevert BS, Otterstedt JE, Massoth FE (1987) Kinetics of the HDO of methyl-substituted phenols. Appl Catal 31:119–131
Dupont C, Lemeur R, Daudin A, Raybaud P (2011) Hydrodeoxygenation pathways catalyzed by MoS2 and NiMoS active phases: a DFT study. J Catal 279:276–286
Popov A, Kondratieva E, Mariey L, Goupil JM, El Fallah J, Gilson JP, Travert A, Maugé F (2013) Bio-oil hydrodeoxygenation: adsorption of phenolic compounds on sulfided (Co)Mo catalysts. J Catal 297:176–186
Şenol OI, Ryymin EM, Viljava TR, Krause AOI (2007) Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts. J Mol Catal A-Chem 277:107–112
Romero Y, Richard F, Brunet S (2010) Hydrodeoxygenation of 2-ethylphenol as a model compound of bio-crude over sulfided Mo-based catalysts: promoting effect and reaction mechanism. Appl Catal B 98:213–223
Romero Y, Richard F, Renème Y, Brunet S (2009) Hydrodeoxygenation of benzofuran and its oxygenated derivatives (2,3-dihydrobenzofuran and 2-ethylphenol) over NiMoP/Al2O3 catalyst. Appl Catal A 353:46–53
Ferrari M, Maggi R, Delmon B, Grange P (2001) Influences of the hydrogen sulfide partial pressure and of a nitrogen compound on the hydrodeoxygenation activity of a CoMo/carbon catalyst. J Catal 198:47–55
Ferrari M, Delmon B, Grange P (2002) Influence of the impregnation order of molybdenum and cobalt in carbon-supported catalysts for hydrodeoxygenation reactions. Carbon 40:497–511
Gutierrez A, Kaila RK, Honkela M, Slioor R, Krause AOI (2009) Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal Today 147:239–246
Ardiyanti AR, Gutierrez A, Honkela ML, Krause AOI, Heeres HJ (2011) Hydrotreatment of wood-based pyrolysis oil using zirconia-supported mono- and bimetallic (Pt, Pd, Rh) catalysts. Appl Catal A 407:56–66
Wang W, Yang Y, Luo H, Hu T, Liu W (2011) Amorphous Co-Mo-B catalyst with high activity for the hydrodeoxygenation of bio-oil. Catal Comm 12:436–440
Sun J, Karim AM, Zhang H, Kovarik L, Li XS, Hensley AJ, McEwen JS, Wang Y (2013) Carbon-supported bimetallic Pd-Fe catalysts for vapor-phase hydrodeoxygenation of guaiacol. J Catal 306:47–57
Zhao C, He J, Lemonidou AA, Li X, Lercher JA (2011) Aqueous-phase hydrodeoxygenation of bio-derived phenols to cycloalkanes. J Catal 280:8–16
Zhu X, Lobban LL, Mallinson RG, Resasco DE (2011) Bifunctional transalkylation and hydrodeoxygenation of anisole over a Pt/HBeta catalys. J Catal 281:21–29
Kim YT, Dumesic JA, Huber GW (2013) Aqueous-phase hydrodeoxygenation of sorbitol: a comparative study of Pt/Zr phosphate and PtReOx/C. J Catal 304:72–85
Bui P, Cecilia JA, Oyama ST, Takagaki A, Infantes-Molina A, Zhao H, Li D, Rodríguez-Castellón E, Jiménez López A (2012) Studies of the synthesis of transition metal phosphides and their activity in the hydrodeoxygenation of a biofuel model compound. J Catal 294:184–198
De La Puente G, Gil A, Pis JJ, Grange P (1999) Effects of support surface chemistry in hydrodeoxygenation reactions over CoMo/activated carbon sulfided catalysts. Langmuir 15:5800–5806
Laurent E, Delmon B (1994) Study of the hydrodeoxygenation of carbonyl, carboxylic and guaiacyl groups over sulfided CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalysts. I. Catalytic reaction schemes. Appl Catal A 109:77–96
Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM (2010) The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev 110:3552–3599
Laurent E, Delmon B (1994) Study of the hydrodeoxygenation of carbonyl, carboxylic and guaiacyl groups over sulfided CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalyst. II. Influence of water, ammonia and hydrogen sulfide. Appl Catal A 109:97–115
Laurent E, Delmon B (1994) Influence of water in the deactivation of a sulfided NiMo γ-Al2O3 catalyst during hydrodeoxygenation. J Catal 146(281–285):288–291
Moon JS, Kim EG, Lee YK (2014) Active sites of Ni2P/SiO2 catalyst for hydrodeoxygenation of guaiacol: a joint XAFS and DFT study. J Catal 311:144–152
Centeno A, Laurent E, Delmon B (1995) Influence of the Support of CoMo Sulfide Catalysts and of the Addition of Potassium and Platinum on the Catalytic Performances for the Hydrodeoxygenation of Carbonyl, Carboxyl, and Guaiacol-Type Molecules. J Catal 154:288–298
Wu SK, Lai PC, Lin YC, Wan HP, Lee HT, Chang YH (2013) Atmospheric hydrodeoxygenation of guaiacol over alumina-, zirconia-, and silica-supported nickel phosphide catalysts. ACS Sustainable Chem Eng 1:349–358
Nimmanwudipong T, Aydin C, Lu J, Runnebaum RC, Brodwater KC, Browning ND, Block DE, Gates BC (2012) Selective hydrodeoxygenation of guaiacol catalyzed by platinum supported on magnesium oxide. Catal Lett 142:1190–1196
Gevert BS, Otterstedt JE, Massoth FE (1987) Kinetics of the HDO of methyl-substituted phenols. Appl Catal 31:119–131
Kresse G, Hafner J (1993) Ab initio molecular dynamics for liquid metals. Phys Rev B: Condens Matter 47:558–561
Lee YK, Oyama ST (2006) Bifunctional nature of a SiO2-supported Ni2P catalyst for hydrotreating: EXAFS and FTIR studies. J Catal 239:376–389
Oyama ST, Wang X, Lee YK, Chun WJ (2004) Active phase of Ni2P/SiO2 in hydroprocessing reactions. J Catal 221:263–273
Seo HR, Cho KS, Lee YK (2011) Formation mechanisms of Ni2P nanocrystals using XANES and EXAFS spectroscopy. Mater. Sci. Eng B 176:132–140
Zhuravelev LT (2000) The surface chemistry of amorphous silica Zhuravlev model. Colloids Surf B 173:1–38
Acknowledgments
This research was funded by NRF (2012R1A1A2008651).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Moon, JS., Lee, YK. Support Effects of Ni2P Catalysts on the Hydrodeoxygenation of Guaiacol: In Situ XAFS Studies. Top Catal 58, 211–218 (2015). https://doi.org/10.1007/s11244-015-0362-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-015-0362-4