
Abstract
A tetra-dentate ligand L [3,3′-((pyridin-2-ylmethyl)azanediyl)dipropanamide] was synthesized and characterized by spectroscopic and structural methods. The reaction of L with two different copper(II) halides [CuX2; X = Br, Cl] in a similar condition yielded two different compounds of [LCuCl]Cl, 1 and [CuLBr]2[CuBr4]·CH3OH·H2O, 2. Both compounds were characterized by several physicochemical techniques. Single-crystal X-ray studies revealed that the Cu(II) centers in the cationic complexes 1 and 2 are in a square pyramidal N2O2X (X = Cl and Br) environment. Compound 1 is chromotropic and its solvatochromism and halochromism properties were investigated. It was found that the observed positive solvatochromism is due to structural change and solvation of the vacant site of the complex. The complex demonstrated distinct reversible spectral change over the pH range 1.3–12.1 and can act as pH-induced off–on–off absorption switch through deprotonation and the Cu–O to Cu–N bond rearrangement of the coordinated amide groups in aqueous solution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Currently, ligand design becomes a crucial part of synthetic activity in coordination chemistry. This is surely due to the fine control that ligands apply to the metal center to which they are coordinated. Ligands that have considerably different functional groups, such as hard and soft donors, are named hemilabile ligands and find increasing use in molecular chemistry such as molecular activation, homogeneous catalysis, functional materials and small-molecule sensing [1,2,3,4,5,6,7,8]. Due to their ability to reversibly bind to the metal center, hemilabile coordination complexes have recently been explored for application as small-molecule chemosensors [9]. The incorporation of amide moiety in multifunctional ligands makes such ligands suitable nominees to show hemilabile properties [10, 11]. One type of such hemilabile ligand is the amide–amine class of ligands. Hemilability in this class of ligands is due to the substantially inert character of the amine group and lability of the amide moiety. Additionally, the amide group has different types of coordination modes, O-bound, through the carbonyl oxygen where metallation occurs in neutral conditions and N-bound as amidate complexes in alkaline condition. This makes such ligands good candidates to prepare chromotropic material when they are coordinated to appropriate metal ions. Among the metal complexes whose color variations are because of d–d transitions, the copper(II) is an exceptional candidate due to having various coordination numbers, diverse geometrical structure and presence of strong Jahn–Teller effect [12]. The term chromotropism refers to a change in optical characteristics (transparency or light diffraction) due to microstructural changes within the material that occur by the surrounding chemical or physical stimuli such as solvent (solvatochromism), temperature (thermochromism), pressure (pizochromism), light (photochromism), pH (halochromism), ion (ionochromism). We have recently reported a weak-link approach to the synthesis of copper(II)-based complexes using a tridentate flexible hemilabile 3-((pyridin-2-ylmethyl)amino)propanamidediazadecanediamide ligand [13] that showed solvato- and halochromism. Herein, we describe the design and synthesis of a tetra-dentate ligand with two switchable centers (amide groups) and its copper(II) complexes as illustrated in Scheme 1. In this system, the copper(II) complexes are structurally switchable in response to the pH value of the solution. The structures of the complexes were characterized by single-crystal X-ray diffraction analysis as well as standard techniques.

Complexes under study; X = Cl or Br
Experimental
Materials and methods
All reagents and solvents used in syntheses and analyses were reagent grade and purchased from Merck Chemical Company and were utilized as received. Elemental analyses were performed on a LECO CHN-600 Elemental Analyzer. Absolute metal percentages were determined by a Varian-spectra A-30/40 atomic absorption flame spectrometer. Conductance measurements were made at 25 °C with a Jenway 400 conductance meter on concentrations of 10.0 × 10−4, 6.00 × 10−4, 4.00 × 10−4 and 2.00 × 10−4 M of samples in selected solvents. Then for each solvent, a curve was plotted by drawing the molar conductance versus concentration of the sample. The curve was then extrapolated to an infinitely dilute solution to obtain the molar conductance value. Infrared spectra were recorded using potassium bromide disks and a Bruker FTIR instrument. NMR spectra were measured with a Bruker 400 DRX Fourier Transform Spectrometer at room temperature. The electronic absorption spectra were measured with a Braic2100 UV–Vis spectrophotometer. The following solvents were used in solvatochromism study: methanol (MeOH), ethanol (EtOH), water (H2O), dimethylformamide (DMF), and dimethylsulfoxide (DMSO).
Synthesis
5-(Pyridin-2-ylmethyl)-2,8-dioxo-1,5,9-triazanonane (L)
To a solution of acrylamide (1.5 g, 22 mmol) in water (3 mL) at about 5 °C was added dropwise 2-(aminomethyl)pyridine (1.0 ml, 10 mmol). After the addition was completed, the temperature was raised to 87 °C and stirred for 6 h. After cooling to room temperature, the unreacted acrylamide was removed by filtration. The thin oil residue was dissolved in dichloromethane (10 mL) and dried over anhydrous Na2SO4. Filtration and concentration under reduced pressures gave crude ligand as pale yellow oil (2.4 g, 98%). The product was suitable for use in the next reaction without further purification. Selected IR data (ν/cm−1): 3449 (br. s, N–H2 str.), 2925 and 2855 (w, C–H str. aliphatic), 1633 (s, C=O str.), 1462 (m, C–N str.), 1263 (m), 1105 (m), 1021 (m), 889 (m), 745 (m). 1H NMR (400 MHz, CHCl3), δ (ppm): 2.42 (t, J = 6.0 Hz, 4H, –NCH2CH2C(O)NH2), 2.55 (br, 4H, –CH2C(O)NH2), 2.83 (t, J = 6.0 Hz, 4H, –NCH2CH2C(O)NH2), 3.77 (s, 2H, NCH2–Py), 7.17 (m, H, Py), 7.32 (d, 1H, J = 8.0 Hz, Py), 7.67 (t, d, J = 6.0, 2.0 Hz, 1H, Py) and 8.51 (d, m, J = 5.2 Hz, 1H, Py). When one drop of D2O was added to the 1H NMR sample, the broad signal at 2.55 ppm disappeared. 13C NMR (100 MHz, CHCl3), δ (ppm): 32.17 (HNCH2CH2C(O)NH2), 49.16 (HNCH2CH2C(O)NH2),58.47, (–NH–CH2–Py), 123.12, 124.48, 138.00, 148.06, 157.01 (Py–C), 177.87(–C(O)NH2).
Preparation of chloro, 5-(pyridin-2-ylmethyl)-2,8-dioxo-1,5,9-triazanonane copper(II) chloride, [CuLCl]Cl, 1
To the solution of ligand L (0.77 g, 2 mmol) in methanol (10 mL) was slowly added a solution of CuCl2·2H2O (0.34 g, 2 mmol) in methanol (8 mL). The resultant green mixture was stirred for 2 h at room temperature and was then allowed to concentrate at room temperature. The desired compound precipitated from the solution as a greenish blue solid. The compound was recrystallized by diffusion of diethyl ether into methanol solution. Yield, 0.428 g, 56% as blue crystals. The crystals were suitable for X-ray crystallography. Anal. calcd for C12H18CuN4O2Cl2 (MW = 384.76 g mol−1): C, 37.46; H, 4.72; N, 14.56; Cu, 16.52%; Found: C, 37.2; H, 5.1; N, 14.6; Cu, 16.5%. Selected IR data (ν/cm−1 using KBr disk): 3460 (br, s, O–H str.), 3280 (br. s, N–H2 str.), 2912 and 2751 (s C–H str.), 1644 (s, C=O str.), 1452 (m, N–H bend.), 1298 (w), 1217 (m), 1116 (m), 1028 (m), 803(w), 725 (w) 573 (w), 495 (w). Ʌm (MeOH, Ω−1 cm2 mol−1): 113; the standard value for a two-ion electrolyte is 80–115.
Preparation of [CuLBr]2[CuBr4]·CH3OH·H2O, 2
This compound was obtained as dark green crystals in a reaction procedure similar to that of [CuLCl]Cl, except for the use of CuBr2·2H2O (0.2 mmol), instead of CuCl2·6H2O. The desired compound precipitated from the solution as a blue solid. The compound was recrystallized by diffusion of diethyl ether into methanol solution. Yield, 1.2 g, 49% as blue crystals. The crystals were suitable for X-ray crystallography. Anal. calcd for C25H42Cu3N8O6Br6 (MW = 1220.72 g mol−1): C, 24.60; H, 3.47; N, 9.18; Cu, 15.62%; Found: C, 25.2; H, 3.5; N, 9.4; Cu, 15.4%. Selected IR data (ν/cm−1 using KBr disk): 3390 (br. s, O–H str.), 3195 (br. m, N–H2 str.), 2926 (s C–H str.), 1645 (s, C=N str.), 1444 (m, N–H bend.), 1297 (w), 1108 (m), 1026 (m), 972 (w), 766 (w), 502 (w). Ʌm (DMF, Ω−1 cm2 mol−1): 157; the standard value for a three-ion electrolyte is 130–170.
Single-crystal structure determination
The X-ray data were collected with Bruker Apex-II CCD single-crystal diffractometer at room temperature in 1 and 100 K in 2 by using graphite-monochromated Mo-Kα radiation (λ = 0.71073) and the ω-scan technique. Data were integrated using the SAINT [14] program and subsequently used for the intensity corrections of the Lorentz and polarization effects. A multiscan absorption correction was applied using a SADABS program [15]. The structure was solved by conventional direct methods and refined by full-matrix least square methods using F2 data using SHELXL [15]. All non-hydrogen atoms refined as anisotropic and hydrogen atoms were placed in calculated positions. Refinement of F2 was performed against all reflections. The weighted R-factor, wR and goodness-of-fit, S are based on F2; conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2r (F2) is used only for calculating R-factors (gt) and is not relevant to the choice of reflections for refinement. Geometrical calculations were carried out with WinGX [16], and the figures were made by the use of a Olex2 [17] program. A summary of the crystal data and structure refinements for complexes is collected in Table 1.
Results and discussion
Preparation
The preparation of the ligand was accomplished in good yield by condensation of an equivalent of 2-(aminomethyl)pyridine and two equivalents of acrylamide (Scheme 2). The dinuclear copper(II) complexes were prepared in moderate yields by mixing of an equimolar copper(II) halide (Cl− and Br−) and the ligand in the solvent of methanol (Scheme 2).

Synthetic outline for preparation of complexes 1 and 2
Characterization
The IR spectra of complexes 1 and 2 are presented in Figs. S1 and S2. The presence of the characteristic bands at 1452 and 1028 cm−1 for 1, and 1444 and 1026 cm−1 for 2, suggests the v(C–N) stretching vibrations of the pyridyl ring [18]. These bands appeared in the free ligand at 1462 and 1021 cm−1. The strong bands at 1645 and 1644 cm−1 for 1 and 2, respectively, are attributed to the v(C=O) vibrations of the amide groups which appeared as a strong band at 1633 cm−1 in the free ligand. An intense and narrow band around 3260 cm−1 are associated to the v(NH2) stretching vibration of the amide moiety, which was observed to be broader in the free ligand and at the higher frequency (3450 cm−1). As the lone pair of the electrons of the donor nitrogen atom becomes involved in the metal–ligand bond, the transfer of the electron density of the donor nitrogen atom to the copper ion in the complexes and the subsequent polarization of the ligand involves electron depopulation of the N–H bond, which culminates in a shift to lower frequencies [19]. The broad band observed at around 3460 cm−1 in the complexes is identified to the stretching vibrations of -OH groups of water molecules. At lower frequency, the complexes also displayed bands around 573 and 495 cm−1 that are related to the v(Cu–O) and v(Cu–N) vibrational modes, respectively [20]. Due to the larger dipole moment change for the v(Cu–O) band compared to the v(Cu–N) band, the v(Cu–O) band usually appears at a higher frequency than the v(Cu–N) band [21].
The diffuse reflectance spectrum of complex 1 showed a broad and intense band at UV region and a broad band at about 730 nm. The band at UV region is associated with the pyridine to Cu(II) charge transfer transition (LMCT), which appear at 260 nm when the spectrum is recorded in DMF solution [22]. The broadband at 730 nm is the d–d band of Cu(II) in five-coordinated square pyramidal environments [23], which is shifted to 753 nm, when recorded in DMF solution. This band is sensitive to the solvent used and is solvatochromic, which is discussed below.
The molar conductance values indicate that complexes 1 and 2 are 1:1 and 1:2 electrolytes, respectively.
X-ray structure
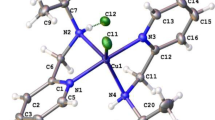
Complex 1 was crystallized in a triclinic space group P-1. A perspective view accompanied by the atom labeling scheme for the complex is shown in Fig. 1, and the selected bond parameters are collected in Table 2. The molecular structure of the complex comprises of [LCuCl]+ cation and a chloride anion. In the complex, the central copper atom is five-coordinated and the geometry around the central copper atom is defined as nearly regular square pyramidal. As stated by Addison and Rao [24], the distortion of the square pyramidal geometry toward trigonal bipyramidal can be designated by the geometrical parameter τ = |α − β|/60, where α and β are the two largest L–M–L angles of the coordination sphere. The τ values are zero for a regular square pyramid and one for a regular trigonal bipyramid. The τ value for the coordination about the copper atom is 0.055 (Fig. 2) proving the square pyramidal geometry around the copper center. The apical Cu–O(2) bond distance, 2.1862(16) Å, is normal as the Jahn–Teller elongated bond. The four coordinating atoms forming the basal plane are two nitrogen atoms of the amine moieties (N(2)) and the pyridyl group (N(3), one of the oxygen atom of the amide group (O(1)) and chlorine atom (Cl(1)), while the apical site is used by one oxygen atom of the amide group (O(2)). The basal atoms are nearly coplanar; the deviations from the least-squares plane within the CuN2OCl atoms are N(3) − 0.026, N(2) 0.027, O(1) − 0.024, Cl(1) 0.023 and Cu(1) 0.228 Å. The two oxygen atoms of the amide moieties are oriented in cis positions. The angle of the five-membered chelate ring, 81.44(7)° is nearly the same as those found for the chelate rings of 2-(methylamino)pyridine complexes [25]. The bond angles of the six-membered chelate rings are 93.69(7)° and 93.71(6)° that are very similar to those found for the chelate rings of reported amide complexes [13, 26]. Figure S3 shows the packing arrangement in the crystal of complex 1. From this figure, two inter-molecular N–H···O and N–H···Cl hydrogen bonds are noticeable (Table S1). These two types of interaction are responsible for the formation of layers of parallel molecules in a zigzag fashion along the crystallographic c axis with an angle of approximately 95°.

ORTEP view of [CuLCl]Cl, 1

Square pyramid arrangement of the coordinated atoms around the copper(II) center in complex 1
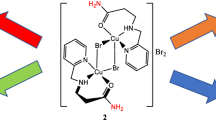
Compound 2 crystallized in the P-1 space group. In the unit cell, there are two independent cations of [LCuBr]+ (2a and 2b), an anion of [CuBr4]2−, water, and methanol molecules. These two cations are structurally identical except for a slight variation in bond angles and bond distances. An ORTEP plot of complex 2a and 2b is shown in Fig. 3 with the atom-numbering scheme. The selected bond lengths and angles are given in Table 3. In both cationic units, the copper atom adopts a five-coordinated N2O2Br environment, giving a square pyramidal geometry, where two nitrogen atoms of tertiary amine and pyridine groups, one of the oxygen atoms of amide moiety of the ligand L as well as the bromine atom located at equatorial position and the oxygen atom of another amide group occupied the axial site. The τ values for the coordination around the copper atoms in unit 2a and 2b are 0.07 and 0.02, respectively, confirming the square pyramidal geometry around the copper centers. The basal atoms are nearly coplanar; the deviations from the least-squares plane through the CuN2OBr atoms are N(2) − 0.118, N(3) 0.032, Br(1) − 0.098, O(1) 0.033 and Cu(1) 0.152 Å in 2a and N(7) − 0.076, N(8) 0.005, Br(2) − 0.066, O(1) 0.008 and Cu(1) 0.155 Å in 2b. The bond distances around the copper center are typical [27,28,29]. The tetra-dentate ligand offers N2O2-donor sets chelated to the copper center, thereby forming three fused metallocyclic (6,6,5)-membered rings with O,N and N,N bite angles. The six-membered bite angles vary in the range of 90.4(3)–94.5(3)° and the five-membered bite angles are 82.9(3)° and 82.6(3)° in 2a and 2b, respectively. The five-membered chelate rings are puckered, so that the torsion angles of N(2)–C(4)–C(5)–N(3) and N(7)–C(19)–C(20)–N(8) are 26.71(7) and 24.40(7)°, respectively. The six-membered chelate rings have boat conformations. The oxygen atoms O(2) in 2a and O(3) in 2b that are located in the apical site of the basal planes with Cu(1)–O(2) distance of 2.202(6) Å and Cu(2)–O(3) distance of 2.194(6) Å are coordinated loosely to the copper atom because of a strong Jahn–Teller effect. The copper(II) in the anionic complex, [CuBr4]2−, has a distorted tetrahedral geometry with Br–Cu–Br angles between 94.70° and 134.43°, the resulting cis-angle, the average of the four smallest angles, is 98.93°. The water and methanol molecules in the unit cell are free from coordination. The crystal lattice is stabilized by the intra-H-bonding system between two cationic units through N(4)–H(4)···O(3) and N(5)–H(5)···O(2), a cationic dimeric entity [LCuBr] 2+2 is formed (dotted lines in Fig. S4), which is further linked to water molecule N(6)–H(6)···O(6), cationic complex N(1)–H(1)···Br(4) and methanol O(5)–H(5)···Br(3). Details of the hydrogen bonding are given in Table S2.

ORTEP views of cations [CuLBr]+, 2a (right) and 2b (left)
Thermal study
The thermal stabilities of compounds 1 and 2 were studied under nitrogen and air atmospheres, respectively, as shown in Figs. S1 and S2. The thermal analysis measurements show that the decomposition of compounds 1 and 2 continues in several stages. TG–DTA thermogram of compound 1 displays an endothermic process at 195 °C, along with a mass loss of 5.52% (calc. 4.48%), allocated to the elimination of a water molecule. This stage proceeds by three decomposition events as evidenced by the endothermic and exothermic processes at 275, 373, and 470 °C, respectively. Two amide moieties (CH2CH2C(O)NH2) of the ligand molecules (calc. 36.30, found 37.31) as well as a pyridyl moiety (Py–CH2N) (calc. 59.16%; found 57.00%) were lost in the second and third events and the remnant was lost in the final stage of the decomposition, forming a metallic copper as final solid product (10.16%).
As shown in Figure S2, the pyrolysis of compound 2 shows an endothermic process at 170 °C, together with a mass loss of 4.90% (calc. 4.30%), assigned to the elimination of the latticed water and methanol molecule. This event proceeds by four decomposition stages as proved by the strong endothermic and exothermic processes at 243, 341, 573, and 772 °C, respectively, because of the exclusion of the ligand parts, forming a CuO as final solid products (Calc. 6.51%; Found 6.76%).
Chromotropism study
The complexes are chromotropic in solution and show distinct color changes. Their color change is due to a d–d transition of copper(II) with d9 electron configuration. However, the chromotropism property of complex 2 was not taken into consideration due to the presence of the [CuBr4]2− species with a high intensity d–d band that interferes with the d–d band of [LCuBr]+ and makes complicated visible spectra in different solvents and was problematic to be resolved.
Solvatochromism
The visible absorption spectrum of compound 1 was provided in solid state (powder) and in the solution state in water and some organic solvents with various donor numbers (DN): the donor number is defined as a measure of coordinating ability of solvent on the standard of dichloromethane [30]. The solid-state spectrum of compound 1 demonstrates a broad maximum at around 740 nm which tails to the longer wavelength that is characteristic of five-coordinated copper(II) complexes with a square pyramidal geometry [31], that is in accord with its stereochemistry in the crystal structure. In solution, the complexes exhibited similar spectral pattern but their absorption maxima are changed in different solvents, i.e., the complexes are solvatochromic. The visible spectra of the complexes in different solvents are presented in Fig. 4. The positions of the absorption maxima of the complexes with their molar absorptivity values are gathered in Table 4. As can be seen, all the solution spectra are also different from the solid-state spectrum. This suggests that in solutions the core (CuN2O2Cl) geometry of 1 is substantially altered from the square-pyramidal-based structure in the solid state. Regression analysis of the band maxima of compound 1 versus the donor number of solvents is illustrated in Fig. 5 and signifies good correlation and confirms the solvatochromic behavior of these complexes. The vacancy of one of the apical sites of the complexes and also the facile elimination of the coordinated amide group on the other site of the chelate plane cause the approach of the solvents molecules to the metal center with no difficulty, but each solvent has its own basicity strength attributable to its electron donating power. As donor number of the solvent increases its power and ability to coordinate to the copper(II) center increases. The approach of the polar solvent molecules to the apical site of the complexes triggers an intense repulsion between the electron in the \({\text{d}}_{{z^{2} }}\) orbital of the copper(II) ions and the electron pair of the solvents that causes the required energy for transferring the electrons to \({\text{d}}_{{x^{2} - y^{2} }}\) orbital decreases. Therefore, the location of this band declines almost linearly with the increase of the electron pair donating power of the solvents that cause a redshift.

Solvent-dependent electronic absorption spectra of complex 1 in different solvents and powder (arbitrary unit)

Dependence of the λmax values of compound 1 on the solvent donor number value
Halochromism
Complex 1 is halochromic and its halochromism was studied in different pH (1–12). However, it seems that in aqueous solution the Cu-halide bonds in [LCuX]+ are broken and replaced by water molecules as shown in Scheme 3. This was further established by an increase in the molar conductance of the compound to the standard value of 2:1 electrolyte. The original blue color of the aqueous solution turns to green as the pH of the solution was increased from 6.5 to 12.0 by the addition of NaOH (0.1 M). Also, the absorption band at 688 nm moved to a lower wavelength (580 nm) with the development of three isosbestic points (at 595, 618, and 633 nm) that became detectable in different pH scales (Fig. 6). These spectral changes might be due to the ionization of coordinated water molecule and the amide protons followed by the Cu–O to Cu–N bond exchange as the pH of the solution increased to 12.0. It appears that the first and the second isosbestic points are due to the ionization of the coordinated water and the amide protons, respectively, and the last isosbestic point is attributed to the reversible interconversion of the Cu–O to Cu–N coordination modes as displayed in Scheme 3. The observed hypsochromic shift in the absorption maxima of the complex with escalating the pH of the solution discloses that the deprotonated amide group is a stronger donor than neutral amide. Such behaviors were observed before in amide derivative ligands [25, 32]. Spectrophotometric titration of the compounds with sodium hydroxide at λmax = 688 nm (inset of Fig. 6) at pH range 4.8–12.0 correspond to the utilization of three equivalent protons that is perhaps related to deprotonation of the coordinated water and amide moieties.

Interconversion of complex in acid and base aqueous solution (pH = 1.15–12.26)

Visible absorption spectra of the complex 1 in alkaline solution (5.0 × 10−3 mol dm−3) at 25 °C. The pH values are denoted on the figure. The upper graphs show the decrease of 688 nm absorbance on titration of 1, NaOH (0.10 M) and the isosbestic points observed in the pH range 4.8–12.0
The complex is almost stable in acidic solution so that the original blue color of its aqueous solution (λmax = 688 nm) becomes unchanged upon addition of perchloric acid (0.1 M). (Fig. S4).
Conclusion
In summary, the present paper includes the synthesis, structural and spectroscopic studies of two copper(II) complexes with a tetra-dentate N2O2 ligand that contain two amide groups. The complexes were synthesized by a similar method. The X-ray structures of the complexes indicate the copper(II) ion in 1 has the square pyramidal geometry but in compound 2 copper(II) appeared as square pyramidal and tetrahedral geometries. The results of chromotropism studies of 1 indicate that the complex is solvato- and halochromic. Its halochromism property is due to hemilability of the amide moieties of the coordinated ligand and presence of a vacant axial site in the compound. Complex 1 can act as pH-induced off–on–off absorption switch through deprotonation and the Cu–O to Cu–N bond rearrangement of the coordinated amide groups in aqueous solution.
Supplementary data
CCDC 1896321 and 1896322 contain the supplementary crystallographic data for the complex 1 and 2, respectively. The data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033 or e-mail: deposit@ccdc.cam.uk.
References
Adams GM, Weller AS (2018) Coord Chem Rev 355:150–172
Braunstein P, Naud F (2001) Angew Chem Int Ed 40:680–699
Bader A, Lindner E (1991) Coord Chem Rev 108:27–110
Lindner E, Pautz S, Haustein M (1996) Coord Chem Rev 155:145–162
Hesp KD, Wechsler D, Cipot J, Myers A, McDonald R, Ferguson MJ, Schatte G, Stradiotto M (2007) Organometallics 26:5430–5437
Duran J, Oliver D, Polo A, Real J, Benet-Buchholz J, Fontrodona X (2003) Tetrahedron Asymmetry 14:2529–2538
Uh YS, Boyd A, Little VR, Jessop PG, Hesp KD, Cipot-Wechsler J, Stradiotto M, McDonald R (2010) J Organomet Chem 695:1869–1872
Wiester MJ, Mirkin CA (2009) Inorg Chem 48:8054–8056
Angell SE, Rogers CW, Zhang Y, Wolf MO, Jones WE Jr (2006) Coord Chem Rev 250:1829–1841
Chao M-S, Lu H-H, Tsai M-L, Huang S-L, Hsieh T-H (2009) Inorg Chim Acta 362:3835–3839
Golchoubian H, Moayyedi G, Reisi N (2015) Spectrochim Acta Part A 138:913–924
Camard A, Ihara Y, Murata F, Mereiter K, Fukuda Y, Linert W (2005) Inorg Chim Acta 358:409–414
Rostami L, Golchoubian H (2017) J Coord Chem 70:3660–3676
SAINT (2005) Version 7.06A. Bruker AXS Inc, Madison
Sheldrick GM (2015) Acta Cryst A 71:3–8
Farrugia LJ (2012) J Appl Cryst 45:849
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339–341
Tsiamis C, Themeli M (1993) Inorg Chim Acta 206:105–115
Golchoubian H, Rezaee E (2009) J Mol Struct 927:91–95
Raman N, Esthar S, Thangaraja CA (2004) J Chem Sci 116:209–213
Nakamoto K, Ohkaku N (1971) Inorg Chem 10:798–805
Ciampolini M, Nardi N (1966) Inorg Chem 5:41
Mautner FA, Soileau JB, Bankole PK, Gallo AA, Massoud SS (2008) J Mol Struct 889:271
Addison AW, Rao TN, Reedijk J, Rijn JV, Verschoor GC (1984) J Chem Soc Dalton Trans 7:1349–1356
Golchoubian H, Tarahomi M, Rezaee E, Bruno G (2015) Polyhedron 85:635–642
Golchoubian H, Rostami L (2017) Inorg Chim Acta 462:215–222
Du M, Guo YM, Bu XH (2002) Inorg Chim Acta 335:136–140
Bu XH, Liu H, Du M, Wong KMC, Yam VWW (2002) Inorg Chim Acta 333:32–40
Bu XH, Du M, Shang ZL, Zhang RH, Liao DZ, Shionoya M, Clifford T (2000) Inorg Chem 39:4190–4199
Cataldo F (2015) Eur Chem Bull 4:92–97
Murakami T, Hatakeyama S, Igarashi S, Yukawa Y (2000) Inorg Chim Acta 310:96–102
Golchoubian H, Heidarian A, Rezaee E, Nicolò F (2014) Days Pigments 104:175–184
Acknowledgements
We are grateful for the financial support of the University of Mazandaran of the Islamic Republic of Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Golchoubian, H., Mehrbanian, D., Rezaee, E. et al. Structural and chromotropism properties of copper(II) complexes with 3,3′-((pyridin-2-ylmethyl)azanediyl)dipropanamide ligand. Transit Met Chem 44, 671–680 (2019). https://doi.org/10.1007/s11243-019-00332-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-019-00332-4