
Abstract
Three different polyoxometalate (POM)-based complexes, [CoL(H2O)(α-Mo8O26)0.5]·4H2O (1), [NiL(H2O)(θ-Mo8O26)0.5]·4H2O (2) and [Ag2L2][HPMo12O40]·3H2O (3) (L = 1, 2 bis(1H-imidazole-4-carboxamido)ethyl) have been synthesized under hydrothermal conditions. The different heterometals lead to two types of polyoxomolybdates, specifically [α-Mo8O26]4− and [θ-Mo8O26]4− in complexes 1 and 2, respectively. In complex 1, the [α-Mo8O26]4− anions are linked into a 1D chain by [Co2L2(H2O)2]4+ cations. Replacing Co2+ with Ni2+, [θ-Mo8O26]4− anions were constructed in complex 2. In contrast, a discrete metal–organic cation [Ag2L2]2+ and Keggin-type [PMo12O40]3− anion resulted when Ag+ was used to prepare complex 3 in the presence of phosphoric acid. The electrochemical and photocatalytic properties of all three complexes were studied.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polyoxometalate (POM) based metal–organic complexes (POMOCs) assembled from POM anions, metal ions and organic ligands have received much attention due to not only their diverse structures, but also their many potential applications in electrochemistry, photocatalysis, magnetism, catalysis and medicine [1,2,3,4,5]. Among the known POMs, polyoxomolybdates can show different configurations in the structures of POMOCs, including [Mo6O19]2−, [Mo7O24]6−, [Mo8O26]4− and other anions [6,7,8]. Among these, the octamolybdate [Mo8O26]4− anion can exhibit different isomers, including α, β, γ, δ, ε, ζ, η, θ and ι isomers, depending on the reaction conditions, such as pH, reaction temperature and so on [9,10,11,12,13].
Selection of organic ligands plays a critical role in the synthesis of POMOCs with specific architectures. The introduction of N-donor ligands for the synthesis of a variety of POMOCs has become popular; such ligands include pyridyl- [14], triazole- [15], tetrazole-based derivatives [16] and a wide variety of POMOCs have been prepared. Recently, Cui et al. have generated two 3D Co(II) coordination polymers by using rigid or semirigid bis(imidazole) ligands [17]. In our previous work, we have studied the semirigid bis-imidazole-bis-amide ligands, 1,n-bis(1H-imidazole-4-carboxamido)metaphenylene (n = 2, 3), which can show multiple coordination modes in the structures of POMOCs. For example, three 1,3-bis(1H-imidazole-4-carboxamido)metaphenylene ligands can chelate two metal ions to give metal–organic cationic cages, or alternatively such ligands can be linked by metal ions into chains [18], etc. However, examples of POMOCs prepared by combining bis-imidazole-bis-amide ligands with isopolymolybdate have not previously been reported to our knowledge.
Considering the above and the influence of the linker between the two imidazole amide groups in such ligands, a flexible bis-imidazole-bis-amide ligand, 1,2-bis(1H-imidazole-4-carboxamido)ethyl (L), was now been prepared and utilized to construct POMOCs with different metal ions. In this way, three POMOCs, [CoL(H2O)(α-Mo8O26)0.5]·4H2O (1), [NiL(H2O)(θ-Mo8O26)0.5]·4H2O (2) and [Ag2L2][HPMo12O40]·3H2O (3) have been isolated. The heterometals show significant influence on the configurations of the polyoxoanions in the final structures. In addition, the electrochemical behaviors of all three complexes have been investigated, and the photocatalytic degradation of methylene blue by complexes 1 and 2 has been studied.
Experimental
Materials and methods
All chemicals and solvents were purchased from commercial sources and used without further purification. The proligand L was prepared according to the published method [19]. Powder X-ray diffraction (PXRD) data were collected on a D/teX Ultra diffractometer with Cu Kα radiation (λ = 0.154 nm). FTIR spectra were recorded on a Varian 640 FTIR spectrometer using KBr pellets, from 500 to 4000 cm−1. Ultraviolet–visible absorption spectra were recorded with an SP-1901 UV–Vis spectrophotometer. A CHI 760 electrochemical workstation was used for electrochemical measurements and data collection.
Synthesis of complex 1
A mixture of CoCl2·6H2O (0.145 g, 0.5 mmol), L (0.0296 g, 0.1 mmol) (NH4)6Mo7O24·4H2O (0.124 g, 0.1 mmol) and H2O (10 mL) was stirred for 30 min at room temperature. The pH value was adjusted to 4.2 with 1.0 M HCl. The suspension was transferred into a Teflon-lined autoclave and kept at 120℃ for 4 days. After slow cooling to room temperature, pink block crystals of complex 1 were obtained in a yield of 0.167 g (56%) based on Co. IR (KBr pellet, cm−1): 3350 (w), 3127 (w), 2349 (s), 1616 (m), 1583 (s), 1430 (s), 1354 (m), 1246 (s), 1216 (s), 996 (s), 906 (s), 805 (s), 668 (m).
Synthesis of complex 2
The synthetic method for complex 2 was similar to that for 1, but NiCl2·6H2O (0.145 g, 0.5 mmol) was used instead of CoCl2·6H2O, and the pH was adjusted to 3.8 with 1.0 M HCl. Green crystals of complex 2 were obtained in a yield of 0.143 g (48%) based on Ni. IR (KBr pellet, cm−1): 3356 (w), 3127 (w), 2342 (s), 1615 (m), 1545 (s), 1430 (s), 1354 (m), 1246 (s), 1216 (s), 990 (s), 906 (s), 805 (s), 674 (m).
Synthesis of complex 3
The synthesis of complex 3 was also similar to that of 1, except that AgNO3 (0.145 g, 0.5 mmol) was employed instead of CoCl2·6H2O, and a drop of phosphoric acid was added (85%). The pH was adjusted to 1.0 with 1.0 M HNO3. Yellow crystals of complex 3 were obtained in a yield of 0.134 g (45%) based on Ag. IR (KBr pellet, cm−1): 3427 (w), 3140 (w), 2342 (s), 1657 (m), 1558 (m), 1430 (m), 1335 (s), 1216 (s), 1061 (s), 960 (s), 882 (m), 799 (s), 608 (m).
X-ray crystallography
A Bruker SMART APEX II CCD diffractometer was used to collect crystallographic data for complexes 1–3 using the φ and ω scan method, using Mo − Kα monochromatic radiation (λ = 0.71069 Å) at 293 K. The crystal structures were solved by direct methods, and the SHELXTL-2014 program package was used to refine the crystal structures on F2 by full-matrix least-squares techniques [20]. H atoms were placed in calculated positions and permitted to ride on their parent C atoms, while all the non-hydrogen atoms were refined with anisotropic temperature parameters. Summaries of the crystallographic data for all three complexes are given in Table 1. Selected bond lengths (Å) and angles (deg) for the complexes are provided in Table S1. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Center, with CCDC Numbers 1855843, 1855844 and 1855845.
Photocatalytic experiments
In a typical experiment, 0.15 g of the complex 1 or 2 was added to 90 mL of an aqueous solution containing MB dye at room temperature. The mixture was stirred in the dark for about 30 min to ensure the adsorption–desorption equilibrium. Then the solution was stirred under visible light. Aliquots of 5 mL volume were taken out every 30 min and analyzed with an SP-1901 UV/Vis spectrophotometer.
Result and discussion
Crystal structure of complex 1
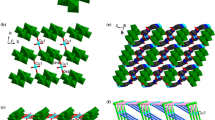
The unit cell of complex 1 contains one independent Co(II) center, half a [α-Mo8O26]4− anion (abbreviated to α-Mo8), one L ligand, one coordinated water ligand and four lattice water molecules (Fig. 1a). Bond valence sum calculations show that all of the Mo atoms are in the +VI oxidation state, while the Co atoms are in the +II oxidation state [21]. The Co1 atom has an octahedral geometry. The equatorial plane is occupied by two N atoms (N1 and N2) from the imidazolyl groups of two different L ligands, together with two O atoms (O1 and O29#1) of the carbonyl groups from two L ligands, while the axial positions are defined by one O6 atom from the α-Mo8 anion and a water ligand. The corresponding bond lengths and angles are given in Table 2.

a View of the linking modes of metal, ligand and POM in compound 1. The hydrogen atoms are omitted. Symmetry code: #1: −1−x, 1−y, 1−z. #2: −x, −y, −z. b View of the binuclear cationic circle [Co2L2(H2O)2]4+ in 1 and the 1D chain [Co2L2(H2O)2(α-Mo8O26)]. c View of the 2D grid supramolecular layer of 1.
Complex 1 has a 2D supramolecular structure, constructed from POM-based metal–organic chains [Co2L2(H2O)2(α-Mo8O26)] linked via hydrogen bond interactions. Two L ligands utilize their imidazolyl amide groups to chelate two Co1 atoms, resulting in a binuclear cationic moiety [Co2L2(H2O)2]4+. Acting as bridging inorganic building blocks, the α-Mo8 anions link these cationic moieties into a 1D chain [Co2L2(H2O)2(α-Mo8O26)] through Co–O covalent bonds (Fig. 1b). In detail, two O6 atoms from the α-Mo8 anion coordinate with two Co1 atoms from neighboring cationic moieties. These 1D chains are linked to each other by hydrogen-bonding interactions between the α-Mo8 anions and L ligands, leading to a 2D grid supramolecular layer (Fig. 1c). The hydrogen bonds between N5-H···O10 play an important role in the formation of the supramolecular structure, and the distance between atoms N5 and O10 is 2.977 Å (Table S2).
Crystal structure of complex 2
Replacement of Co2+ with Ni2+, results in the formation of [θ-Mo8O26]4− anions in the structure of complex 2 (Fig. 2a). The cationic [Ni2L2(H2O)2]4+ moiety is similar to the analogous Co moiety seen in complex 1. The bond lengths and angles around Ni(II) are listed in Table 2. Moreover, the 1D chain [Ni2L2(H2O)2(θ-Mo8O26)] in complex 2 is similar to that formed in complex 1, being further extended into a 2D grid supramolecular layer through hydrogen bonds (Fig. 2b, Table S3).

a View of the binuclear cationic circle [Ni2L2(H2O)2]4+, the [θ-Mo8O26]4- anions and the 1D chain [Ni2L2(H2O)2(θ-Mo8O26)]. b View of 2D grid supramolecular layer of 2
Crystal structure of complex 3
When Ag+ ion and a drop of phosphoric acid were employed in the synthesis, complex 3 consisting of a discrete [Ag2L2]2+ complex cation and Keggin-type [PMo12O40]3− were generated. If the phosphoric acid was omitted, yellow crystals were obtained, which were identified as a polyoxomolybdate as confirmed by IR. However, we have not obtained the single crystals. The bond lengths around the Ag(I) atom are listed in Table 2. Complex 3 has a 3D supramolecular structure, consisting of two Ag atoms, two L ligands and one Keggin-type [PMo12O40]3− polyoxoxanion (Fig. 3a). A proton is required to balance the charge of the complex. The Ag1 atom exhibits a linear coordination mode, provided by two N atoms from the imidazole groups of two different L ligands. In turn, the L ligands connect pairs of Ag atoms, leading to a metal–organic [Ag2L2]2+ cation, which is further extended into a 3D supramolecular structure by [PMo12O40]3− polyoxoanions with the aid of O1 W molecules via hydrogen bond interactions (Fig. 3b, Table S4).

a View of the metal–organic cationic cage [Ag2L2]2+, the [PMo12O40]3- anions. b View of 1D, 2D and 3D supramolecular layer of 3
PXRD and IR analysis
The PXRD patterns of complexes 1–3 were measured at room temperature, as shown in Fig. S1. The diffraction peaks of the as-synthesized complexes are consistent with the simulated ones, confirming the phase purities of all three complexes.
The IR spectra of complexes 1–3 are shown in Fig. S2. The characteristic bands at 668–996 cm−1 for 1 and 671–990 cm−1 for 2 are attributed to the ν(Mo=Ot) and ν(Mo–O–Mo) vibrations of the polyoxomolybdates. Characteristic bands at 960, 882, 799 and 1061 cm−1 for complex 3 are attributed to ν(Mo=Ot), ν(Mo–Ob–Mo), ν(Mo–Oc–Mo) and ν(P–O), respectively [22]. In addition, bands in the regions of 1216–1616 cm−1 for 1, 1216–1615 cm−1 for 2, and 1216–1657 cm−1 for 3 can be assigned to the ligand L [23]. The bands around 3350–3450 cm−1 for complexes 1 and 2 can be ascribed to the water ligands.
Electrochemical properties
The electrochemical behaviors of complexes 1–3 were investigated in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution by means of bulk-modified carbon paste electrodes (1-, 2- and 3-CPE), prepared according to the literature [24]. As shown in Figs. 4a–6a, three pairs of redox peaks with mean peak potentials E1/2 = (Epc + Epa)/2 (scan rate: 200 mV s−1) at +273 (I–I′), +91 (II–II′), −143(III–III′) mV for 1, +207 (I–I′), +87 (II–II′), −136 (III–III′) mV for 2 and +280 (I–I′), +89 (II–II′), −125(III–III′) mV for 3 were observed in the potential range of 700 ~ −300 mV, which can be assigned to three consecutive two-electron redox processes of the Mo centers of the polyoxoanions [25].

Cyclic voltammograms of 1-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates (a) and electrocatalytic behaviors of 1 toward the reduction of H2O2 (b), bromate (c), and nitrite (0-8.0 mM) (d). Scan rate: 200 mV·s−1.
The electrocatalytic behaviors of all three complexes toward the reduction of H2O2, bromate and nitrite were measured in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution. When the amount of H2O2, bromate and nitrite was gradually increased, the reduction peak currents all three CPEs gradually increased in the potential range of +700 to −300 mV, while the corresponding oxidation peak currents were decreased (Figs. 4, 5, 6). The results suggest that all three CPEs have good electrocatalytic activities for the reduction of H2O2, bromate and nitrite [26]. Additionally, the electrochemical behavior of 3-CPE under different pH conditions was investigated. As shown in Fig. 7, upon increasing of pH from 1.35 to 4.0, the peak potentials shift gradually in the negative direction, and the peak currents also decrease [27]. These observations confirm that the electrochemical behavior of 3-CPE is pH-dependent, suggesting that complex 3 may be used as a potential pH sensor.

Cyclic voltammograms of 2-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates (a) and electrocatalytic behaviors of 1 toward the reduction of H2O2 (b), bromate (c), and nitrite (0-8.0 mM) (d). Scan rate: 200 mV·s−1

Cyclic voltammograms of 3-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates (a) and electrocatalytic behaviors of 1 toward the reduction of H2O2 (b), bromate (c), and nitrite (0-8.0 mM) (d). Scan rate: 200 mV·s−1

The cyclic voltammograms for 3-CPE in 0.1M H2SO4 + 0.05M Na2SO4 solutions with different pH (from 1.35 to 4.0)
Photocatalytic activity
Many POM-based hybrids exhibit good photocatalytic activities for the degradation of organic dyes under UV irradiation, including methylene blue (MB), rhodamine B (RhB), methyl orange (MO) and so on [28]. Hence, the photocatalytic activities of complexes 1 and 2 for degradation of MB have been investigated in this study. These experiments were carried out according to the literature procedure [29]. The results are shown in Fig. 8. The absorption peak of MB decreased gradually for both complexes with increasing UV irradiation time, giving MB conversions of 67.6% for 1 after 180 min and 65.3% for 2 after 150 min. Hence, complexes 1 and 2 both show photocatalytic activities for the degradation of MB under UV irradiation.

Photocatalytic decomposition of MB solution under UV light irradiation with the use of complexes 1 and 2
Conclusions
In summary, three POM-based complexes have been obtained by combining different metal ions with a flexible bis-imidazole-bis-amide ligand under hydrothermal conditions. The structural analysis of the complexes reveals that each metal ion plays a key role in determining the final structures of the polyoxoanions. All three complexes show good electrocatalytic properties for the reduction of H2O2, bromate and nitrite. The electrochemical behavior of complex 3 is pH-dependent, indicating that it may have potential applications in pH sensing. Complexes 1 and 2 show photocatalytic activities for the degradation of MB under UV irradiation.
Supporting materials
Supporting materials include Tables of selected bond lengths and angles, Tables of hydrogen-bonding geometries, Figures PXRD patterns and IR spectra of complexes 1–3.
References
Hirano T, Uehara K, Kamata K, Mizuno N (2012) J Am Chem Soc 134:6425–6433
Wang XL, Li N, Tian AX, Ying J, Lin XL, Luan J, Yang Y (2014) Inorg Chem 53:7118–7129
Sartzi H, Miras HN, Nadal L, Long DL, Cronin L (2015) Angew Chem Int Ed 127:15708–15492
Wang XL, Zhang R, Wang X, Lin HY, Liu GC (2016) Inorg Chem 55:6384–6393
Liu YP, Zhao SF, Guo SX, Bond AM, Zhang J (2016) J Am Chem Soc 138:2617–2628
Korenev VS, Boulay AG, Dolbecq A, Sokolov MN, Hijazi A, Floquet S, Fedin VP, Cadot E (2010) Inorg Chem 49:9740–9742
Hao XL, Ma YY, Wang YH, Zang HY, Li YG (2014) Cryst Eng Commun 16:10017–10027
Du HJ, Wang CH, Li Y, Niu YY, Hou HW (2015) RSC Adv 5:74065–74074
Lan YQ, Li SL, Wang XL, Shao KZ, Du DY, Zang HY, Su ZM (2008) Inorg Chem 47:8179–8187
Lan YQ, Li S, Wang XL, Shao KZ, Su ZM, Wang EB (2008) Inorg Chem 47:529–534
Du XD, Li CH, Zhang Y, Liu S, Ma Y, You XZ (2011) Cryst Eng Commun 13:2350–2357
Kan WQ, Yang J, Liu YY, Ma JF (2012) Inorg Chem 51:11266–11278
Xu N, Zhang JW, Wang XL, Liu GC, Li TJ (2016) Dalton Trans 45:760–767
Zhao J, Wang YN, Dong WW, Wu YP, Li DS, Zhang QC (2016) Inorg Chem 55:3265–3271
Sha JQ, Li MT, Sun JW, Zhang YN, Yan PF, Li GM (2013) Dalton Trans 42:7803–7809
Wang LD, Liu TT (2018) Chin J Catal 39:327–333
Cui JW, Hou SX, Heckeb KV, Cui GH (2017) Dalton Trans 46:2892–2903
Wang XL, Zhang S, Wang X, Liu GC, Lin HY, Zhang HX (2017) Dalton Trans 46:16580–16588
Sarkar M, Biradha K (2006) Cryst Growth Des 6:202–208
Sheldrick GM (2008) Acta Crystallogr Sect A Found Crystallogr 64:112–122
Brown ID, Altermatt D (1985) Acta Crystallogr Sect B Struct Sci 41:244–247
Tian AX, Ning YL, Wang XL (2015) Cryst Eng Commun 44:10499–10507
Saeed A, Ashraf Z, Erben MF, Simpson J (2017) J Mol Struct 1129:283–291
Wang XL, Xu C, Lin HY, Liu GC, Luan J, Chang ZH (2013) RSC Adv 3:3592–3598
Fernandes DM, Teixeira A, Freire C (2015) Langmuir 31:1855–1865
Dolbecq A, Mialane P, Keita B, Nadjo L (2012) J Mater Chem 22:24509–24521
Wang P, Wang XP, Yuan Y, Zhu GY (2000) J NonCryst Solids 277:22–29
Liu B, Yu ZT, Yang J, Hua W, Liu YY, Ma JF (2011) Inorg Chem 50:8967–8972
Tang J, Zou Z, Yin J, Ye J (2003) Chem Phys Lett Elsevier 382:175–179
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 21671025, 21471021, 201401010 and YJC20170021) and Program for Distinguished Professor of Liaoning Province (No. 2015399).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, X., Pan, X., Wang, X. et al. Polyoxometalate-based complexes with a flexible bis-imidazole-bis-amide ligand: structures, electrochemical and photocatalytic properties. Transit Met Chem 44, 207–217 (2019). https://doi.org/10.1007/s11243-018-0284-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0284-7