
Abstract
A dinuclear copper(II) complex [Cu2L4] has been prepared by the reaction of CuCl2·2H2O and 1-[(2-iodo)benzene]-3-[benzothiazole] triazene (HL). The complex has been characterized by X-ray crystallography and by physico-chemical and spectroscopic methods. In the solid state, there is a significant antiferromagnetic coupling between the copper(II) centers with a coupling constant (J) of − 558 cm−1. In homogeneous solution, the complex shows electrocatalytic activities for hydrogen generation from both acetic acid and neutral buffer with a turnover frequency of 50 mol of H2 per mole of catalyst per hour (mol H2/mol catalyst/h) at an overpotential (OP) of 941.6 mV, and 502 mol H2/mol catalyst/h at an OP of 836.7 mV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Much current research on transition metal complexes is focussed on their potential uses as catalysts, biological agents and inorganic materials [1,2,3]. By designing appropriate ligands, the electronic properties of the metal center can be tuned to achieve specific applications [4]. Our interests have focused on triazenide ligands, which serve as bridging ligands for the assembly of binuclear and multinuclear complexes with a range of functions, for example, magnetic coupling, catalytic and luminescent performance, etc. [5,6,7,8,9,10,11]. In particular, hydrogenase enzymes based on iron and nickel can efficiently catalyze both the generation and oxidation of hydrogen [12]. Structural investigations show that the metal ligands in these enzymes facilitate the cleavage/formation of the H–H bond and the transfer of protons to and from the distal metal center [13]. Considering that triazenido ligands show greater basicity on their [N···N···N]− moieties relative to the neutral nitrogen, triazenido ligands should be more electron donating and hence amenable to binding to protons and hydrogen evolution. Hence, triazenido-metal complexes were selected as potential hydrogen evolution reaction (HER) catalysts [14, 15]. In this paper, we report the synthesis, structure, characterization and magnetic properties of a dinuclear copper(II) complex [Cu2L4], as well as its electrocatalytic properties for proton and water reduction.
Experimental
Physical measurements are given in the supplementary materials.
Synthesis of HL
A solution of 2-iodoaniline (Aladdin, Analytical, 2.19 g, 10 mmol) in water (5 mL) was mixed with 1 mol L−1 HCl 30 mL (30 mmol) at 0 °C. An aqueous solution (15%) of sodium nitrite (1.04 g, 15 mmol) was added dropwise with stirring. Once the amine was dissolved, a 15% solution of 2-aminobenzothiazole (Aladdin, Analytical, 1.5 g, 10 mmol) in ethanol (10 mL) was added at 0 °C, and the mixture was stirred for 3 h at 25 °C. The mixture was then neutralized with 15% aqueous NaCH3CO2 to give a yellow precipitate. This was filtered off, and the solid was purified by crystallization at − 4 °C from 5:1 ethyl acetate/THF to obtain yellow crystals, which were collected and dried in vacuo, Yield 2.5 g (66%). Calcd for C13H9N4SI: C, 41.06; H, 2.37; N, 14.74. Found: C, 41.57; H, 2.29; N, 14.56%. 1H NMR (400 MHz, DMSO) δ 11.50 (s, 1H), 7.91 (d, J = 12.1 Hz, 2H), 7.54 (d, J = 7.4 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.01 (d, J = 40.6 Hz, 2H). 13C NMR (400 MHz, DMSO) δ 166.14, 152.69, 139.52, 129.50, 129.24, 126.32, 125.22, 123.46, 121.81, 120.63, 118.87, 117.45, 95.70. IR (KBr): ν 3700, 3058, 1731, 1578, 1497 cm−1 (Fig. S1).
Synthesis of [Cu2L4]
To a solution of HL (0.37 g, 1 mmol) and triethylamine (0.10 g, 1 mmol) in dichloromethane/acetonitrile (20 mL, 1:1), CuCl2·2H2O (0.085 g, 0.5 mmol) was added and the mixture was stirred for 10 min. The solution was allowed to slowly evaporate, affording deep green crystals, which were collected and dried in vacuo, Yield 0.16 g (37%). Calcd for C52H32Cu2I4N16S4: C, 37.99; H, 1.97; N, 13.64. Found: C, 38.28; H, 1.94; N, 13.75%. 1H NMR (400 MHz, DMSO) 7.93 (s, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.02 (d, J = 32.6 Hz, 2H). IR (KBr): ν 3058, 1775, 1522, 1404 cm−1 (Fig. S2).
X-ray crystallography
X-ray analysis of the complex was carried out with an Xcalibur Eos Gemini X-ray diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at room temperature. Empirical absorption corrections were applied using the SADABS program [16]. The structure was solved using direct methods. Non-hydrogen atoms were all refined anisotropically. Hydrogen atoms were included in geometrically idealized positions, and thermal parameters were fixed during structural refinement. All calculations were performed using the SHELXTL-2014 computer program [17]. Table S1 gives crystallographic data for the complex, and selected bond lengths are listed in Table 1.
Results and discussion
The triazene HL was prepared by the reaction of 2-iodoaniline, sodium nitrite, and 2-aminobenzothiazole (Scheme 1 and Fig. S3). As shown in Fig. S4, the 1H NMR spectrum showed a singlet at 11.5 ppm, assigned to the triazene group hydrogen atom, and a multiplet in the range of 8.0–6.9 ppm for the aromatic protons. In the presence of Et3N, the reaction of HL with CuCl2·2H2O provided the copper complex, [Cu2(L)4].

Synthesis of HL and [Cu2(L)4]. R = NNC6H4I, (i) NaNO2, HCl; (ii) Et3N, CuCl2
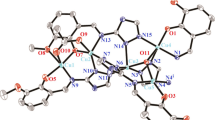
As shown in Fig. 1, [Cu2(L)4] consists of two copper centers bridged by four anionic ligands (L−). The structure is in agreement with the ESI–MS analysis. As shown in Fig. S5, the complex gave a single ion at a mass-to-charge ratio (m/z) of 1644.7788, which can be attributed to [Cu2(L)3(L–H)]+. The two copper centers share the same coordination environment, in which each copper atom is coordinated by four nitrogen atoms from four anionic ligands (L−). In contrast to previously reported bridged complexes with triazenido ligands [18, 19], L− serves as a monodentate ligand to each copper center. The bond distances between Cu and the benzothiazole N are in the range of 1.968(4)–2.050(4) Å, while the Cu–N(triazene) bond lengths are 1.964(4)–2.057(4) Å (Table 1), which are similar to those found in other copper(II) triazenide complexes 1.973–2.088 Å [20, 21]. The distance between the two copper centers (2.6925(8) Å) is longer than that found previously in the reported copper(II) complexes, Cu2(dpt)4 (dpt = 1,3-diphenyltriazenide) (2.441 Å) [21] and Cu2(OAc)4 (2.616 Å) [22].

ORTEP drawing of [Cu2(L)4] with thermal ellipsoids at the 50% probability level
Magnetic behavior of the copper complex
The magnetic properties of the complex were measured at a field of 2000 Oe over the temperature range from 2 to 300 K, with the results shown in Fig. 2. Copper(II), which has the d9 configuration, is expected to exhibit paramagnetic behavior. However, [Cu2(L)4] is diamagnetic at 300 K, because of a strong antiferromagnetic interaction between the copper atoms [23]. The magnetic analysis was carried out using the Bleaney–Bowers equation based on the Heisenberg model H = − 2JS1S2, where ρ is the fraction of monomeric impurity. The magnetism of the complex can be well reproduced by a modified Bleaney–Bowers Eq. (1) [24];

Temperature dependence of χMT at 2000 Oe for [Cu2L4]. The solid line: the fitting data
Least-squares fitting of the experimental data led to J = − 558 cm−1, Nα = 3.1 × 10−4 cm3 mol−1 and ρ = 0.00065. Hence, there is a very strong antiferromagnetic exchange interaction between the copper atoms. This result is also in agreement with the NMR analysis. According to Fig. S6, the 1H NMR spectrum of the copper complex is indicative of a single-ligand environment. 1H resonances are found in the range of 8.0–6.9 ppm for the aromatic protons.
Electrocatalytic behavior of the complex
Since triazenido complexes can act as electrocatalysts for hydrogen generation [25,26,27], we have investigated the electrochemical behavior of [Cu2(L)4]. Cyclic voltammograms (CVs) of the complex (Fig. 3a), HL (Fig. S7) and CuCl2 (Fig. S8) were measured in CH3CN with 0.10 M [(n-Bu)4N]ClO4 as the supporting electrolyte. All values are reported versus Ag/AgNO3 as a reference. As shown in Fig. S7, HL exhibited a quasireversible couple at − 1.51 V and a reduction wave at − 1.70 V. Compared to free HL, the copper complex displayed a quasireversible couple at − 1.49 V and a reduction wave at − 1.67 V (Fig. 3a). Hence, the potentials were shifted positively, and current strengths increased for the complex compared to the free ligand. According to Fig. S9, all the redox waves were characteristic of diffusion-controlled processes, as scan rate analyses of voltammograms exhibited linear dependences in plots of current versus ν1/2.

a Cyclic voltammogram (CV) of 0.56 mM [Cu2L4] in 0.10 M of [n-Bu4N]ClO4 CH3CN solution at a glassy carbon electrode and a scan rate of 100 mV/s. b CVs of a 0.56 mM solution of [Cu2L4], with varying concentrations of acetic acid in CH3CN. Conditions: 0.10 M [n-Bu4N]ClO4 as supporting electrolyte, scan rate: 100 mV/s, glassy carbon working electrode (1 mm diameter), Pt counter electrode, Ag/AgNO3 reference electrode, Fc internal standard (*). (Color figure online)
Next, acetic acid was used as proton source to investigate the catalytic behavior of the copper complex for hydrogen evolution. According to the data plotted in Fig. 3b, with sequential increments of acetic acid concentration (from 0.00 to 4.64 mM), the potential strengths of the complex at − 1.69 and − 1.54 V were proportionately increased, which is attributed to the catalytic generation of H2 from acetic acid [28]. These results suggest that the electrocatalytic generation of hydrogen by [Cu2(L)4] involves the reduction of Cu(II) to Cu(I) or of L− to L, with associated protonations.
In order to investigate whether the triazene HL may play a role in the catalytic process, the electrochemical behavior of free HL was investigated under the same conditions. From Fig. S10, with sequential increments of acetic acid concentration from 0.00 to 4.64 mM, the peak currents at − 1.56 and − 1.71 V increased, indicating that HL can also catalyze proton reduction to H2. When the concentration of acetic acid reached 3.99 mM, a new reduction wave appeared, suggesting that new components were formed.
Based on the above-mentioned results and previous studies [29, 30], we can propose a possible catalytic H2 evolution mechanism for the present complex. As shown in Scheme 2, a putative [Cu I2 (L)4]2− species is formed by two one-electron reductions of [Cu II2 (L)4]. Then, the introduction of two protons gives a three-coordinate dinuclear copper complex, [H–L–CuI–L]2. Further addition of two protons provides a copper(III)-hydride species, {[H–L–CuIII(H)–L]2}2+. Finally, two one-electron reductions of the {[H–L–CuIII(H)–L]2}2+ species affords H2, with regeneration of the starting complex.

Possible catalytic mechanism for proton reduction by the complex. R = NNC6H4I
A series of bulk electrolysis experiments was carried out to further characterize the electrocatalytic activity of the complex, with the results listed in Fig. S11. For example, during 2 min of electrolysis at − 1.45 V, a 0.10 M [n-Bu4N]ClO4 solution of the complex exhibited a charge of 55 mC (Fig. S11a), accompanied by the generation of a gas which was confirmed as H2 by gas chromatography. According to Fig. S12, ~ 0.035 mL of H2 was produced over an electrolysis period of 1 h. Under the same conditions, this system only gave 10 mC of charge (Fig. S11b), indicating that this complex does serve as a catalyst for hydrogen production from acetic acid. The catalytic activity was estimated using Eqs. (2) and (3) [10] (Eq. S1), giving the results listed in Fig. S13. For instance, at an OP of 941.6 mV, this electrocatalytic system could produce 50 mol of hydrogen per mole of catalyst per hour (mol H2/mol catalyst/h).
where ΔC is the charge from the catalyst solution during controlled-potential electrolysis (CPE), minus the charge from an equivalent solution without catalyst during CPE; F is Faraday’s constant, n1 is the number of moles of electrons required to generate one mole of H2, n2 is the number of moles of catalyst in solution, and t is the duration of electrolysis in seconds. E⊙ +H is the standard potential for the solvated proton/dihydrogen couple, and KaHA is the dissociation constant of acetic acid.
To explore the catalytic function of the complex in aqueous media, its electrochemical behavior was investigated in aqueous buffer (CH3CN: H2O = 2:5 as solvent). As shown in Fig. S14, in the absence of the complex, the catalytic current was not apparent until a potential of − 1.84 V versus Ag/AgCl was attained. Upon addition of the complex, the onset of catalytic current was observed at about − 1.40 V, showing that the complex can efficiently reduce the potential. Moreover, the current strength increased significantly with buffer of decreasing pH values from 7.0 to 4.6 μM (Fig. S15). According to Fig. S15, the onset of this catalytic current was clearly influenced by the aqueous solution pH, such that the applied potential declined with increasing pH, which is indicative of a catalytic process [15].
Bulk electrolyses catalyzed by [Cu2(L)4] and HL in 0.25 M aqueous buffer (pH 7.0) were then used to study the catalytic activities. From Fig. 4a, when the applied potential was − 1.45 V, the maximum charge was only 22 mC during 2 min of electrolysis in the absence of the complex. However, addition of the complex afforded 678 mC during 2 min of electrolysis (Fig. 4b), accompanied by a large amount of gas bubbles, which were confirmed as H2 by gas chromatography. According to Fig. S16a, ~ 4.26 mL of H2 was afforded over an electrolysis period of 1 h with a Faradaic efficiency of 98.6% for H2 (Fig. S16b). Turnover frequencies (TOFs) and overpotentials (OPs) were calculated from Eqs. (2) and (4) (Eq. S2) [15], giving the results listed in Fig. S17. For example, this system could afford 502 mol H2/mol catalyst/h at an OP of 837.6 mV, where

a Charge buildup versus time from electrolysis of a 0.25 M buffer (pH 7.0) under − 1.45 V versus Ag/AgCl. b Charge buildup versus time from electrolysis of a 4.18 μM [Cu2L4] in 0.25 M buffer solution (pH 7.0). All data have been deducted blank. (Color figure online)
In a similar procedure to that used for the complex, bulk electrolyses from HL was conducted in 0.25 M neutral buffer. According to Fig. S18, during 2 min of electrolysis, the addition of HL gave 178 mC of charge. The TOF was calculated as 146 mol of hydrogen per mole of catalyst per hour at an OP of 837.6 mV (Eq. S3, Fig. S19). Clearly, the complex exhibits much higher efficiency for hydrogen production than the free ligand HL.
In terms of TOF values, the results plotted in Fig. S17 show that the catalytic activity of the present complex (502 mol H2/mol catalyst/h at an OP of 869 mV) is higher than those of some previously reported molecular catalysts based on copper complexes. These include a copper complex of a tetradentate aminophenol ligand with a TOF of 300 mol H2/mol catalyst/h at an OP of 869 mV [31], a copper complex of a dicyano acetic acid methyl ester ligand giving 285 mol H2/mol catalyst/h at an OP of 787.6 mV [32], a copper(II) complex of 1,3-bis[(4-chloro)benzene]triazene ligand with a TOF of 82 mol H2/mol catalyst/h at an OP of 837.6 mV [25], and a copper(I) complex of 1-[(2-methoxy) benzene]-3-[2-(chloro)benzene] triazene (272 mol H2/mol catalyst/h at an OP of 839 mV) [26]. However, the TOF of the present complex is lower than that of [Cu I4 (L′)4] (HL′ = [1,3-bis(2-methoxy)benzene]triazene) with a TOF of 1034.31 mol H2/mol catalyst/h at an OP of 837.6 mV [10]. These findings show that the donor type and electronic properties of the ligands play a vital role in determining the structure and catalytic activity of the corresponding copper complexes.
Controlled-potential electrolysis (CPE) experiments on buffer solutions of HL, CuCl2, a mixture of HL and CuCl2 or, [Cu2(L)4] were each measured under the same conditions. According to Fig. 5, a neutral buffer solution (pH 7.0) only afforded 32 mC of charge during 2 min of electrolysis under − 1.45 V. The use of solely HL or CuCl2 as a catalyst afforded hydrogen with 157 and 187 mC of charge, respectively. However, when [Cu2(L)4] was employed as catalyst, the system provided H2 with 525 mC of charge. Thus, prior formation of the copper complex is essential for catalytic activity in this system.

Charge buildups versus time from [Cu2L4] and the related components: A neutral buffer (pH 7.0) (black), 4.18 μM HL (red), 4.18 μM CuCl2·2H2O (blue) and 4.18 μM [Cu2L4] (green) under − 1.45 V versus Ag/AgCl in a 0.25 M buffer (pH 7.0). (Color figure online)
Conclusion
The dinuclear triazenido-copper complex [Cu2L4] was formed by the reaction of CuCl2 with 1-[(2-iodo)benzene]-3-[benzothiazole] triazene. The solid copper complex shows a magnetic exchange interaction between the copper atoms mediated by triazenido bridges. In homogeneous solution, the complex can act as an electrocatalytic system for hydrogen production both from acetic acid and aqueous buffer. The present system is broadly comparable to other examples available in the literature.
Supporting information
CCDC 860769 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Gan L, Groy TL, Tarakeshwar P, Mazinani SKS, Shearer J, Mujica V, Jones AK (2015) J Am Chem Soc 137:1109–1115
Tang H, Hall MB (2017) J Am Chem Soc 139:18065–18070
Tarai A, Baruah JB (2018) Cryst Growth Des 18:456–465
Mohamed AA, Abdou HE, Fackler JP Jr (2006) Inorg Chem 45:11–13
Nuricumbo-Escobar JJ, Campos-Alvarado C, Ríos-Moreno G, Morales-Morales D, Walsh PJ, Parra-Hake M (2007) Inorg Chem 46:6182–6189
Barrett AGM, Crimmin MR, Hill MS, Hitchcock PB, Kociok-Kohn G, Procopiou PA (2008) Inorg Chem 47:7366–7376
Rofouei MK, Hematyar M, Ghoulipour V, Gharamaleki JA (2009) Inorg Chim Acta 362:3777–3784
Li W, Chen JY, Xu W-Q, He E-X, Zhan SZ, Cao DR (2011) Inorg Chem Commun 14:916–919
Fang T, Zhou LL, Fu LZ, Zhan SZ, Lv QY (2015) Polyhedron 85:355–360
Xue D, Luo SP, Chen YY, Zhang ZX, Zhan SZ (2017) Polyhedron 132:105–111
Luo SP, Lei JM, Zhan SZ (2018) Inorg Chem Commun 90:45–50
Reijerse EJ, Pham CC, Pelmenschikov V, Gilbert-Wilson R, Adamska-Venkatesh A, Siebel JF, Gee LB, Yoda Y, Tamasaku K, Lubitz W, Rauchfuss TB, Cramer SP (2017) J Am Chem Soc 139:4306–4309
Ezzaher S, Capon JF, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J (2007) Inorg Chem 46:9863–9872
Zhang YX, Lin CN, Zhan SZ (2016) J Coord Chem 69:2832–2844
Xue D, Luo SP, Zhan SZ (2017) Chem Sel 2:8673–8678
Sheldrick GM (1996) SADABS: program for empirical absorption correction of area detector data. University of Götingen, Götingen
Sheldrick GM (2015) Acta Cryst C71:3–8
Rios-Moreno G, Aguirre G, Parra-Hake M, Walsh PJ (2003) Polyhedron 22:563–568
Johnson AL, Willcocks AM, Richards SP (2009) Inorg Chem 48:8613–8622
Rodriguez JG, Parra-Hake M, Aguirre G, Ortega F, Walsh PJ (1999) Polyhedron 18:3051–3055
Corbett M, Hoskins BF, McLeod NJ, O’Day BP (1975) Aust J Chem 28:2377–2392
de Meester P, Fletcher SR, Skapski AC (1973) Dalton Trans 2575–2578
Cotton FA, Wilkinson G (1988) Advanced inorganic chemistry. Wiley, New York
Bleaney B, Bowers KD (1952) Proc R Soc Lond Ser A 214:451–465
Cao JP, Fang T, Wang ZQ, Ren YW, Zhan SZ (2014) J Mol Catal A Chem 391:191–197
Fang T, Lu HX, Zhao JX, Zhan SZ, Lv QY (2015) J Mol Catal A Chem 396:304–309
Zhou LL, Fu LZ, Tang LZ, Zhang YX, Zhan SZ (2015) Int J Hydrog Energy 40:5099–5105
Karunadasa HI, Chang CJ, Long JR (2010) Nature 464:1329–1333
Zhang YX, Lin CN, Zhan SZ (2016) J Coord Chem 69:2832–2844
Li D, Lin CN, Zhan SZ, Ni CL (2017) Chin Chem Lett 28:1424–1428
Fang T, Li W, Zhan SZ, Wei XL (2015) J Coord Chem 68:573–585
Lin CN, Tang LZ, Ren ST, Ye LP, Chen CH, Zhan SZ (2017) Polyhedron 121:13–18
Acknowledgements
This work was supported by the National Science Foundation of China (Nos. 20971045 and 21271073).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Luo, Sp., Jiang, Wx. & Zhan, Sz. Synthesis, structure, magnetic and catalytic behavior of a dinuclear copper(II) complex with triazendio ligands. Transit Met Chem 43, 431–437 (2018). https://doi.org/10.1007/s11243-018-0230-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0230-8