
Abstract
Areolar activation is the most popular in vitro propagation method for cacti. Even though it is relatively simple, few established protocols exist. Acquisition of a competent state has been linked to the expression of SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) during plant organogenesis. Here, cell competence acquisition and areolar activation were investigated during Melocactus glaucescens shoot organogenesis. Degenerate PCR primers and transcriptome data enabled the characterization of MgSERK1, the first known Cactaceae SERK sequence. Phylogenetic analysis based on SERKs from 23 angiosperm species revealed elevated similarity to other SERK Dicot S1/2 sequences and identified the corresponding SERK1 orthologs. Treated explants had the areolar region punctured three times, and shoot organogenesis was induced by exposure to 17.76 µM 6-benzyladenine and 1.34 µM 1-naphthaleneacetic acid. Shoot organogenesis was analyzed by comparing treated and non-treated explants. Anatomical examination and in situ hybridization showed that shoot organogenesis occurred via areolar activation from procambial cells in the stem cortex. Wounding of the areola region activated the axillary bud and increased the number of shoots produced per explant. In situ expression revealed the association of MgSERK1 with M. glaucescens shoot organogenesis in the areola, its adjacent regions, and the roots.
Key message
This is the first characterization of a SERK1 gene in cacti. Its expression during areolar activation is associated with shoot and root organogenesis in Melocactus glaucescens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interest in ornamental cacti has witnessed a steady growth (Goettsch et al. 2015). Over the last century, cacti species have become more accessible because of more efficient propagation methods, which has facilitated the cultivation of these plants (Lema-Rumińska and Kulus 2014).
Cacti are propagated both sexually via seeds, and vegetatively via lateral branches and adventitious roots (Lema-Rumińska and Kulus 2014). However, for commercial purposes, conventional propagation may be impractical because: (i) seeds can be difficult to obtain (Godínez-Álvarez et al. 2003); (ii) their germination rate may be low (Jenkins 1993); (iii) some cacti do not form lateral branches or adventitious roots (Goettsch et al. 2015); and (iv) propagation by cuttings is difficult (Rubluo 1997). Over the past 50 years, plant tissue culture using in vitro propagation has helped overcome some of these limitations (Kim et al. 2019).
The most popular in vitro propagation method among cacti is axillary bud activation or areolar activation (Lema-Rumińska and Kulus 2014; Pérez-Molphe-Balch et al. 2015). In Cactaceae, the areola is a highly specialized axillary bud that contains meristematic tissues, from which spines, trichomes, and flowers can develop (Pérez-Molphe-Balch et al. 2015). This region can help regenerate entire plants through activation of meristematic cells following cutting or induction with plant growth regulators (PGR) (Lema-Rumińska and Kulus 2014; Pérez-Molphe-Balch et al. 2015).
Even if areolar activation is a relatively simple technique, well established in vitro propagation protocols exist solely for a few species of cacti and only a few studies have explored the anatomical and molecular aspects of in vitro regeneration (Pérez-Molphe-Balch et al. 2015). Areolar activation in cacti goes beyond the mere acquisition of competence by meristematic cells in that region. For example, Sánchez et al. (2015) observed that floral and shoot morphogenesis in Echinocereus occurred in internal regions of the buds as a result of the curling and looping of the areola meristem, caused by the periderm layer that seals and disrupts its growth.
An in vitro shoot production protocol has recently been developed for Melocactus glaucescens Buining & Brederoo, an ornamental, endemic, and endangered cactus from eastern Brazil (Torres-Silva et al. 2018). Shoots were seen to develop after areola intumescence in the fourth week of cultivation and a single shoot developed at each areola. However, in vitro propagation of M. glaucescens seemed limited by low numbers of shoots per explant, a high proportion of shoots with morphological alterations, and occurrence of somaclonal variation. Similar issues have also been reported with other species of cacti (Torres-Silva et al. 2018). Understanding the fundamental molecular events that trigger acquisition of competence during cacti areolar activation could improve Cactaceae propagation protocols.
To improve our understanding of in vitro regeneration, it is necessary to identify genetic markers of competence acquisition. SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) was first isolated from Daucus carota cell cultures, in which it triggered a single somatic cell to become competent and form an embryo (Schmidt et al. 1997). Five SERK proteins were later characterized in Arabidopsis thaliana (Hecht et al. 2001). Currently, the SERK protein family includes five homologs that share only partial functional redundancy (Podio et al. 2014; Aan den Toorn et al. 2015).
Despite ample evidence supporting a clear link between SERK gene expression and induction of somatic embryogenesis (Cueva-Agila et al. 2020), a broader role for SERK in plant morphogenesis has been suggested. First, Baudino et al. (2001) reported the expression of ZmSERK1 and ZmSERK2 in embryogenic and non-embryogenic calli of Zea mays. Further studies suggested the participation of SERK in the induction of shoot organogenesis (SO) in Medicago truncatula (Nolan et al. 2003), Helianthus annuus (Thomas et al. 2004), and Triticum aestivum (Singla et al. 2008). Sharma et al. (2008) were the first to propose SERK as a marker of pluripotency rather than embryogenesis. A robust relationship between SERK expression and in vitro organogenesis has been proposed. In situ hybridization revealed expression of CpSERK1 andCpSERK2 during somatic embryogenesis and organogenesis of Cyclamen persicum (Savona et al. 2012), whereas PeSERK1 expression was observed during the organogenesis of Passiflora edulis (Rocha et al. 2016).
Based on the suggested evidence linking competence acquisition and SERK expression during organogenic developmental programs in plants, this study investigated the role of SERK1 expression in M. glaucescens SO. The study aimed to improve existing knowledge of cell competence acquisition and areolar activation. This is the first description of a SERK gene from the Cactaceae family, as well as the first gene expression study on areolar activation.
Materials and methods
Plant material
Plant material was obtained by in vitro germination of M. glaucescens seeds collected from natural populations located in Morro do Chapéu city, Bahia State, eastern Brazil (11°29′38.4′′S; 41°20′22.5′′W) (Fig. 1A). Plant material was identified by Prof. Sheila Vitória Resende (UFBA, Bahia, Brazil). The voucher specimen was deposited at the Herbarium of Universidade Estadual de Feira de Santana (HUEFS), Bahia State (Lambert et al. 2006). Access to the genetic material (permission number A93B8DB) was granted by the Brazilian National System for the Management of Genetic Heritage and Associated Traditional Knowledge (SISGEN) in accordance with existing Brazilian biodiversity legislation. This species is endemic to Bahia State and is listed as endangered by the Convention on International Trade in Endangered Species of Wild Fauna and Flora and is on the IUCN Red List of Threatened Species (Braun et al. 2013; CITES 2021).

Melocactus glaucescens shoot production in vitro. A M. glaucescens obtained from a natural population. An explant of 3–4 mm in height was placed horizontally in culture medium: B control explant after 10 days (d) of culture; C treated explant after 10 d of culture, with the arrow pointing to de novo shoot production in the areola region; D control explant after 30 d of culture, with the arrow pointing to de novo shoot production in the areola region; E treated explant after 30 d of culture, with the arrow pointing to a shoot emerging from the areola region; F control explant after 50 d of culture, with the arrow pointing to a shoot emerging from the areola region; G treated explant after 50 d of culture, with the arrow pointing to a shoot emerging from the areola region; H control explant after 120 d of culture showing shoots with normal morphology; I treated explant after 120 d of culture showing morphologically altered shoots. Bars = 1 cm
To establish in vitro cultures, the seeds were surface-sterilized by immersion in 96% ethanol for 1 min followed by 2% commercial bleach (Super Globo®) for 10 min. The seeds were subsequently washed three times in sterile water under aseptic conditions and germinated in 350-mL flasks (AZ200; Embalagens Rio, Rio de Janeiro, RJ, Brazil) containing 50 mL MS culture medium (Murashige and Skoog 1962) at quarter-strength salt concentration, 15 g L−1 sucrose, and solidified with 7 g L−1 agar (A296 Plant TC; Phyto Technology Lab, Shawnee Mission, KS, USA). The pH was adjusted to 5.7 before autoclaving at 121 °C and 1.5 atm for 20 min. The flasks were covered with transparent polypropylene lids of 67.5 mm in diameter (TC-003-2012; Ralm, São Bernardo do Campo, SP, Brazil).
Cultures were maintained at 25 ± 2 °C under two fluorescent lamps (110 W, T12 HO; Sylvania, Wilmington, MA, USA) with photosynthetically active radiation (PAR) of 60 µmol m−2 s−1 (assessed by a portable LI-250A Light Meter device coupled with a LI-190R Quantum Sensor; LI-COR®, Lincoln, NE, USA) and a 16/8-h light/dark photoperiod. After germination, the plants were periodically subcultured in flasks containing 50 mL MS medium at half-strength salt concentration until in vitro experiments were conducted.
Shoot organogenesis induction and tissue sampling
In vitro SO induction was performed as described by Torres-Silva et al. (2018). After 417 days of in vitro germination, the apical stem segments of the plants were removed and sectioned transversely. The resulting 3–4-mm-high explants were placed horizontally in glass tubes with 15 mL full-strength MS medium (Fig. 1).
Treated explants had the areolar regions punctured three times with 0.18 × 8 mm needles (DBC132; Dong Bang Acupuncture Inc., Chungnam, South Korea) and were placed on full-strength MS medium supplemented with 17.76 µM 6-benzyladenine (BA) and 1.34 µM α-naphthalene-1-acetic acid (NAA). Control explants did not have the areolar regions punctured and were subjected to SO induction in PGR-free medium.
Cultures were maintained at 25 ± 3 °C under two fluorescent lamps with PAR of 60 µmol m−2 s−1 and a 16/8-h light/dark photoperiod for 120 days. After this time, the percentage of responsive explants and number of shoots per explant were assessed.
Genetic material for RNA isolation, gene cloning, and probe construction was obtained from treated explants collected after 50 days of culture. Samples for anatomical analysis and in situ hybridization were collected after 0, 10, 20, 30, 40, and 50 days from both control and treated cultures.
Anatomical characterization
For anatomical characterization of SO induction, five explants of M. glaucescens from each treatment were fixed in 0.1 M Karnovsky solution (Karnovsky 1965) for 1 h and then dehydrated in an ethanol series, all under vacuum (-250 mm Hg). The samples were embedded in methacrylate resin (Historesin; Leica Instruments, Wetzlar, Germany), and transversal and longitudinal sections (average thickness of 5 µm) were prepared using a rotary microtome (RM 2155; Leica Microsystems Inc., Buffalo Grove, IL, USA). The sections were placed on slides and stained with 0.05% (v/v) toluidine blue (pH 4.4) for 10 min (O’Brien and McCully 1981).
Slides were observed under a light microscope (AX70TRF; Olympus Optical, Tokyo, Japan). Images were taken with a digital camera (SPOT Insightcolour 3.2.0; Diagnostic Instruments Inc., Sterling Heights, MI, USA) using SPOT Basic image software. Scales were projected under the same optical conditions.
RNA isolation and cDNA synthesis
Explant tissue was ground with liquid nitrogen and total RNA was extracted with Tris®-Reagent (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Next, 500 µL of Tris®-Reagent and 50 µL of chloroform:isoamyl alcohol (24:1) were added to 500 mg of frozen tissue. The mixture was vortexed, stored on ice for 5 min, and centrifuged at 12,000 × g for 15 min at 4 °C. The aqueous phase was transferred to a new microtube and an equal volume of isopropanol was added to precipitate the RNA. After incubating for 2 h at − 20 °C, the material was centrifuged again at 12,000 × g for 30 min at 4 °C. The pellet was washed in 1 mL 70% ethanol, dried, and resuspended in diethyl pyrocarbonate (DEPC)-treated water (Sigma-Aldrich). The RNA was then treated with DNase I (Thermo Scientific Technologies, Wilmington, DE, USA) to remove genomic DNA contamination. RNA integrity and quality were analyzed on a 1.5% (w/v) denaturing agarose gel, stained with gel red (Biotium Inc.), and quantifed in NanoDrop™ 2000/2000c spectrophotometer. For cDNA synthesis, 1 μg of total RNA, dNTPs, and M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA) were used according to the manufacturer’s specifications.
Single-stranded cDNA was synthesized from 3.0 μg of total RNA using the SuperScript™ II First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations.
Isolation and cloning of the MgSERK1 gene
Different combinations of degenerate primers, designed by Baudino et al. (2001) for Z. mays and based on D. carota and A. thaliana full-length transcripts, were used to amplify the coding sequence of MgSERK1 (Table 1). The cDNA of treated M. glaucescens explants subjected to SO induction and cultured for 50 days was used as template.
The PCR was conducted in a final volume of 50 μL containing 2.0 μL cDNA, 5 μL buffer 10 × , dNTPs (0.2 mM each), 2.0 mM MgSO4, primers (200 nM each), and 0.2 μL Platinum High Fidelity® Taq DNA polymerase (Invitrogen). The reaction was carried out in a C1000™ Touch thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA) with the following settings: denaturation at 94 °C for 5 min; 40 cycles at 94 °C for 1 min, 50–60 °C for 1 min for each primer pair, 68 °C for 1 min; and a final extension step at 68 °C for 5 min. The amplified fragments were cut out of the gel and purified with the Wizard® SV Gel and PCR Clean Up Kit (Promega, Madison, WI, USA) according to the manufacturer’s recommendations. The fragments were linked at a proportion of 3:1 (insert:vector) into the pGEM®-T Easy Vector System (Promega) using T4 DNA ligase (Promega) according to the manufacturer’s recommendations.
The generated recombinant plasmids were introduced into Escherichia coli DH5-alpha by thermal shock using 5 μL of insert and 100 μL of competent cells (1 × 108). After incubation at 37 °C for 1 h, the cells were plated on selective solid LB medium (Sambrook and Russel 2001) containing 100 μg mL−1 ampicillin, 20 μg mL−1 5-bromo-4-chloro-3-indolyl-β-d-galactopiranoside (X-Gal), and 0.1 mM isopropyl-beta-D-thiogalactopiranoside (IPTG), followed by overnight incubation at 37 °C.
To confirm the presence of the insert, plasmid DNA of colonies that exhibited a white color was cleaved with the EcoRI restriction enzyme and the fragments were separated by 1% agarose gel electrophoresis. Ten positive clones were sequenced using the universal M13 primers. The sequences were processed using PHRED (Ewing et al. 1998) and CAP3 software (Huang and Madan 1999). Consensus sequences were compared to public databases at NCBI (https://www.ncbi.nlm.nih.gov), Phytozome v12.0 (https://phytozome.jgi.doe.gov/pz/portal.html), and TAIR (https://www.arabidopsis.org) using the BLAST algorithm. The Beta vulgaris SERK2 gene (BvSERK2, XM_010694302.2) was used as a query against M. glaucescens transcriptome data (data not published) using the BLASTn algorithm. The identified sequence was aligned to cDNA fragments to assemble a consensus sequence.
MgSERK1 sequence analysis, domain and motif prediction, and phylogenetic inference
Open reading frame (ORF) and protein predictions were made using ORF Finder (http://www.ncbi.nlm.nih.gov/projects/gorf/). Signal peptides were predicted using SignalP 5.0 (Almagro Armenteros et al. 2019). The leucine zipper domain (PF08263) located at the N-terminus and the protein kinase domain located at the C-terminus (PF00069) were predicted using Pfam v32.0 (El-Gebali et al. 2019; https://pfam.xfam.org). The presence of conserved leucine-rich repeats (LRRs) was analyzed using the CDD Conserved Domain database (Lu et al. 2020; https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Transmembrane helices were predicted using TMHMM Server v2.0 (Krogh et al. 2001; https://services.healthtech.dtu.dk/service.php?TMHMM-2.0). Multiple sequence alignment was used to find domains and motifs (Schmidt et al. 1997; Baudino et al. 2001). Multiple sequence comparison by log-expectation (MUSCLE; https://www.ebi.ac.uk/Tools/msa/muscle/) with default settings (Edgar 2004) was used to generate a sequence identity matrix of amino acids, which was then visualized with BioEdit® software.
The B. vulgaris BvSERK2 (XP_010692604.1) protein sequence was used as a query to conduct a BLASTp search against 23 angiosperms (Table S1) available at Phytozome v12.0, Beta vulgaris Resource (http://bvseq.boku.ac.at), and SpinachBase (http://www.spinachbase.org). The 20 most frequently annotated protein sequences recovered from BLASTp were downloaded for phylogenetic analysis and annotation. The protein sequences were classified as SERK Dicot S1/2, SERK Dicot S3/4, SERK Monocot or LRRII non-SERKs, as pioneered by Aan den Toorn et al. (2015). All protein sequences classified as SERK Dicot S1/2, SERK Dicot S3/4, and SERK Monocot were kept for further bioinformatics and phylogenetic analyses, whereas LRRII non-SERKs were excluded from this work, except for Arabidopsis as an external outgroup.
All SERK and LRRII non-SERK protein sequences were aligned in MUSCLE using default settings. The alignments were fed into PhyML 3.0 (Guindon et al. 2010; http://www.atgc-montpellier.fr/phyml/) for maximum-likelihood phylogenetic analysis using automatic smart model selection to choose the right model (Lefort et al. 2017) and approximate likelihood-ratio test (aLRT) for branch support. The best selected model was JTT + G + I. The phylogenetic tree was visualized using Interactive Tree of Life (iTOL) v4 (Letunic and Bork 2019).
Protein structure analysis
To assess the conservation of the protein structure, conservation scores were determined by ConSurf (Ashkenazy et al. 2010) and plotted on the reported structure of the SERK1 protein extracellular domain (PDB: 4LSC; Santiago et al. 2013). The same multiple sequence alignment applied above for constructing distance trees (except the A. thaliana NIK1, NIK2, LRRII-RLK1, and LRRII-RLK2 sequences) was used as input together with MgSERK1 as query. Molecular graphics and analysis were performed using PyMOL Viewer version 0.99rc6.
Amplification of MgSERK1 using specific primers
To investigate the accuracy of the alignment, specific primers were designed using the NCBI primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). They bound at different positions of the consensus sequence and were employed to amplify cDNA and genomic DNA (Table 2).
Genomic DNA extraction was performed according to Doyle and Doyle (1987). The DNA samples were digested with RNAse (Thermo Scientifc) to degrade the RNA remaining from DNA extraction according to the manufacturer’s specifications. DNA integrity and quality were analyzed on a 1.2% (w/v) denaturing agarose gel, stained with gel red (Biotium Inc.), and quantifed in NanoDrop™ 2000/2000c spectrophotometer. The templates for PCR reactions included 40 ng of genomic DNA plus 100 ng of a cDNA mixture composed of control explants and test explants subjected to SO induction for 10 or 30 days. The primer sequences and combinations used are reported in Table 2. Amplification was performed using 200 nM of each primer, dNTPs (0.2 mM each), and 0.04 U/µL of Platinum™ II Hot-Start DNA polymerase (Invitrogen) according to the manufacturer’s specifications. Thermal cycling was carried out as follows: 2 min at 94 °C for enzyme activation, 15 s at 94 °C, 15 s at 64 °C, 15 s at 68 °C for 40 cycles, and a final step at 4 °C. The PCR product was analyzed using agarose gel (0.8%) and a 1-kb DNA ladder (Promega).
Sense and antisense probe construction
RNA sense and antisense probes were constructed from a cDNA clone following amplification with degenerate primers (Table 1), and were flanked by the T7 and SP6 promoters. The probe fragment was amplified from plasmid DNA using T7 and SP6 primers. Probes conjugated to digoxigenin (DIG-UTP) were synthesized by in vitro transcription using the DIG RNA Labeling Kit (SP6/T7) (Roche Applied Science, Basel, Switzerland) according to the manufacturer’s recommendations.
In situ hybridization
Plant samples were fixed in 4% paraformaldehyde pH 7 at 4 °C for 16 h, and then dehydrated in a graded series of RNAase-free ethanol. After that, the samples were embedded in paraffin (Histosec®; MERCK, Darmstadt, Germany) in a series of tert-butyl alcohol:paraffin at 50 °C. Transverse sections (average thickness of 8 µm) were made using a hand microtome (820; Reichert Jung, Seefeld, Germany) and transferred to silanated histological slides (Fisherbrand®; Thermo Fisher Scientific, Waltham, MA, USA). The slices were deparaffinized by subsequent washes in xylol, xylol:ethanol, ethanol, ethanol:DEPC water, and DEPC water.
Sense (control) and antisense DIG-labeled MgSERK1 RNA probes were used. Hybridization was performed by mixing 60 ng of probe, 60 ng of yeast tRNA, and 100 µL of hybridization buffer (Tris–HCl 10 mM pH 7.5, NaCl 300 mM, formamide 50%, EDTA 1 mM pH 8.0 in 1 × Denhardt solution), and incubating at 42 °C in a dark, moist chamber for 16 h. To visualize the hybridization signal, anti-DIG antibodies (1:2000; Roche) conjugated to alkaline phosphatase were applied for 1 h at 37 °C and the hybridization signal was detected by reaction with NBT/BCIP (Pierce, Thermo Fisher Scientific).
The hybridized slides were observed under a light microscope (AX70TRF) and images were taken with a digital camera (SPOT Insightcolour 3.2.0) using SPOT Basic image software. Scales were projected under the same optical conditions.
Statistical analyses
To detect differences between the percentage of responsive explants and the number of shoots of control and treated explants after 120 days of culture, data were subjected to a t-test at using R statistical software. For normalization, data on percentage was transformed by the arcsin √x/100 function and homogeneity of variances was checked through Shapiro–Wilk test. A confidence level of 95% was applied. The samples were randomized in a completely randomized design, with eight replicates composed of eight tubes each (1explant per tube), and the experiment was performed twice.
Results
Hormone treatment and areolar punctures promote shoot organogenesis
To test the effect of a combined hormone and wounding treatment on in vitro SO induction in M. glaucescens, we grew stem explants on MS medium (Fig. 1B). Treated explants were supplemented with auxin (1.34 µM NAA) and cytokinin (17.76 µM BA), and explant areole were punctured with a needle (Fig. 1C). Control explants were grown continuously on standard MS medium without areolar puncture. In treated explants, shoot formation started in the second week of culture (Fig. 1C); whereas in control explants, it was first detected after four weeks and the shoots appeared of a red color (Fig. 1D). After 50 days, the number of shoots stabilized (Fig. 1F, G), and was followed only by an increase in shoot size. Accordingly, histological analyses were performed only until 50 days. Shoots derived from treated explants exhibited abnormal stem shape and unusual patterns of spine production relative to control explants (Fig. 1H, I). In both control and treated plants, a single shoot emerged from or close to the areola region (Fig. 1B–I).
The percentage of explants that produced shoots was higher (P ≤ 0.01) in treated (100%) than in control explants (78.1%) (Fig. 2). The number of shoots per explant was also higher (P ≤ 0.01) in treated explants (8.9) than in controls (2.3) (Fig. 2).

Quantification of Melocactus glaucescens shoot production in vitro. Number of shoots per explant and percentage of control or treated explants with shoots following 120 days of shoot organogenesis
To determine the effect of hormone treatment and areolar puncture on SO induction at the cellular level, histological analyses were carried out on explants collected after 10, 30, and 50 days of culture. In control explants, there was no indication of SO after 10 days (Fig. 3A). However, after 30 days, meristemoids in the areolar region became visible (Fig. 3B) and, by 50 days, they had become organized into an axillary meristem with associated vascular tissue (Fig. 3C). New meristemoids continued to form at 50 days (Fig. 3D).

Anatomical analysis of Melocactus glaucescens sections stained with toluidine blue. A–D Control explants cultivated in PGR-free medium for 10, 30, and 50 days (d). E–H Treated explants with a punctured areola region and cultivated in medium containing 17.76 µM BA and 1.34 µM NAA for 10, 30, and 50 days. A Transverse view, arrows indicate the vessel bundle (VB) and vascular cylinder (VC). B Transverse view, the arrow points to axillary bud activation ahead of shoot formation. C Longitudinal view, arrows point to shoot vascular bundles appearing next to the newly formed apical meristem (AM). D Transverse view, the arrow points to the meristemoid. E Longitudinal view, the arrow points to the meristemoid. F Transverse view, arrows point to the meristemoids. G Transverse view, arrows point to shoot vascular bundles appearing next to the newly formed apical meristem (AM). H Transverse view, the arrow points to shoot formation in the central part of the explant. Bars: A, D, and H = 400 µm; B, C, E, F, and G = 200 µm
Treated explants displayed meristemoid formation near the epidermis within 10 days of culture (Fig. 3E), and at a higher density than control plants at 30 days (Fig. 3F). After 50 days, apical meristems of treated explants were more fully developed than in control plants, with the associated vascular bundles, juvenile areola, spinal primordia, and phloem differentiation clearly visible (Fig. 3G). In addition to the accelerated induction of SO in areolar regions, hormone treatment and areolar puncture promoted SO within central explant tissues (Fig. 3H). Taken together, these data show that a combination of hormone treatment and areolar puncture affects the rate and patterning of de novo shoot production in M. glaucescens.
SERK gene structure is conserved in Melocactus glaucescens
SERK genes have previously been shown to regulate SO in a number of angiosperms (Sharma et al. 2008; Savona et al. 2012; Rocha et al. 2016). To test whether this role was conserved in Melocactus, we harvested RNA from explants undergoing SO. Due to the paucity of genomic information for Melocactus, we used degenerate primers designed using Z. mays (Baudino et al. 2001) to clone M. glaucescens cDNA from 50-day-old explants. We successfully cloned a 1223 bp fragment by PCR (Fig. S1). In addition, using BvSERK2 as a query, we obtained two putative sequences corresponding to MgSERK genes from a transcriptome dataset of treated explants 30 days after SO induction (data not published) (Fig. S1). Alignment of all the obtained sequences generated a consensus sequence of 2251 bp (Fig. 4).

Alignment of the Melocactus glaucescens MgSERK1 sequence. Comparison of B. vulgaris SERK2 nucleotide sequence (SERK2_B._vulgaris) with a putative M. glaucescens SERK obtained from transcriptome analysis (MG_putative_SERK1_3493.0; 4472.224, and 4472.225), a M. glaucescens clone obtained following amplification with degenerate primers (MG_clone_SERK1), the M. glaucescens probe used for in situ hybridization experiments (MG_probe_SERK1), and the contig of the putative MgSERK1 sequence (Putative contig_MgSERK1)
MgSERK1 evolutionary relationship and amino acid sequence analysis
To understand the evolutionary relationship of the consensus MgSERK1 gene, BvSERK1 (accession number XP_010692604.1) was used to find homologs in 23 angiosperm species (Table S1), including four species (Amaranthus hypochondriacus, B. vulgaris, Chenopodium quinoa, and Spinacia oleracea) from the same order (Caryophyllales) as M. glaucescens. From a set of 46 SERK amino acid sequences available from NCBI or UniProt, we were able to identify an additional 56 SERK-like proteins (Table S1), which confirmed the robustness of phylogenetic analysis and annotation. The maximum-likelihood phylogenetic tree was constructed from the predicted amino acid sequences of the consensus MgSERK1 and 102 SERK-like proteins (Fig. 5 and Fig. S2), using four A. thaliana LRRII non-SERK proteins as the outgroup. This phylogenetic analysis identified MgSERK1 as belonging to the cluster of SERK Dicot S1/2 proteins. It formed a clade containing individual SERK1 proteins from species of the same order, such as AhSERK1 (A. hypochondriacus), BvSERK1, and SoSERK1 (S. oleracea), as well as closely duplicated CqSERK1 and CqSERK2 (C. quinoa). All other 56 SERK-like proteins were clustered into SERK Dicot S1/2, SERK Dicot S3/4 or SERK Monocot (Fig. S2 and Table S1), revealing the need for renaming the genes to ensure correct interpretation. For example, BvSERK2 (XP_010692604.1) and SoSERK2 (XP_021847294.1), which presented only one candidate in the cluster SERK Dicot S1/2, were renamed as BvSERK1 and SoSERK1, respectively. Closely duplicated sequences were named based on the classification proposed by Mantelin et al. (2011); hence, CqSERK2 (XP_021714851.1) was renamed CqSERK1A and its closely duplicated sequence CqSERK1B.

Maximum-likelihood phylogeny of the MgSERK1 sequence and SERK proteins from 23 other plant species. This unrooted phylogenetic tree includes 103 putative SERK proteins that reveal a cluster of 40 SERK Dicot S1/2 sequences (expanded), 51 SERK Dicot S3/4 sequences (collapsed), 12 SERK Monocot sequences (collapsed), and four A. thaliana LRRII non-SERK proteins as an outgroup. The green star, green branches, and green SERK species names indicate MgSERK1, plant species from the same taxonomic order (Caryophyllales), and new sequences identified for the first time, respectively. The aLRT value with statistical confidence above 0.40 is indicated. Scale bars indicate the number of substitutions per site. For the sake of simplicity, the gene name, instead of the code name, was used (see Table S1)
The MgSERK1-deduced protein sequence encompassed 644 amino acids and included a putative signal peptide of 28 amino acids, five LRR domains, a serine-proline-rich domain (SPP), a transmembrane domain (TM), a varied number (I to XI) of protein kinase domains, and a C-terminal tail (Fig. 6 and Table S2). Similarity analysis among 35 protein sequences from the SERK Dicot S1/2 class revealed a high degree of identity (> 80%) between MgSERK1 and most sequences, with the highest identity corresponding to BvSERK1 (92%), which belongs to the same taxonomic order (Table S3). Interestingly, the degree of sequence conservation was not uniform along the deduced MgSERK1 protein sequence, with the signal peptide being the most divergent (Fig. 6), and was entirely absent in CqSERK2. In spite of the absence of sequence data from Melocactus, alignment of the last 12 amino acids from 35 clustered SERK Dicot S1/2 sequences showed a highly conserved region at the C-terminus (Fig. S3), suggesting that MgSERK1 shared the same last residues as its orthologs.

Alignment of predicted amino acid sequences of Melocactus glaucescens MgSERK1 and SERK-like orthologs from other plant species. Protein domains and motifs are indicated by distinct colored rectangles and include signal peptide, putative leucine zipper (ZIP), leucine-rich repeats (LRR1–LRR5), serine-proline-rich region (SPP), transmembrane domain (TM), I to XI protein kinase domains, and C-terminal domain. The 11 conserved protein kinase subdomains are fused into three regions: I to V, VI to IX, and X to XI (Schmidt et al., 1997). The SPP and C-terminal domains are marked as observed by Baudino et al. (2001). Plant species used in the alignment are A. hypochondriacus (Ah), A. thaliana (At), B. vulgaris (Bv), C. quinoa (Cq), S. oleracea (So), and V. vinifera (Vv). Accession numbers of SERK orthologs are listed in Table S1
The ConSurf algorithm (Ashkenazy et al. 2010) was used to plot the conservation score of the extracellular domain for 17 SERK1 and SERK2 sequences, including MgSERK1, on the crystal structure of AtSERK1 (PDB: 4LSC) (Fig. 7). Some motifs were consistently conserved, including residues in the extracellular domain considered essential for the interaction with BRI1 orthologs. These motifs are well conserved among SERK family members (Aan den Toorn et al. 2015). According to the conservation scores, the concave side (Fig. 7A), which interacts with the extracellular domain of BRI1, appeared more conserved than the convex side (Fig. 7B).

Melocactus glaucescens MgSERK1 protein structure showing amino acid conservation as determined by ConSurf. Data were plotted on the reported structure of the SERK1 protein extracellular domain (PDB: 4LSC), using as input 16 multiple sequences of other SERK1 and SERK2 members, plus MgSERK1 as query sequence. Highly conserved residues are plotted in magenta; whereas lower scores are depicted by other hues, with cyan corresponding to more variable residues. A Concave or solvent-exposed side turned on its right-hand side. B Convex side of the SERK extracellular domain
Amplification of the MgSERK1 transcript using specific primers
To validate the consensus sequence, PCR reactions were conducted using two combinations of specific primers (Table 2). Two bands of the expected size were obtained by amplifying cDNA: one of 1849 bp (F1 + R1) and another of 1591 bp (F2 + R1). Amplification of genomic DNA using the combination F2 + R1 yielded a product of approximately 6 kb (Fig. 8).

Amplification of the consensus sequence corresponding to MgSERK1 from cDNA and genomic DNA using specific primers
In situ hybridization
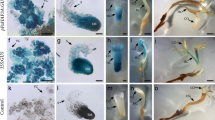
To characterize MgSERK expression during SO induction, we carried out in situ hybridization of control and hormone/areolar puncture-treated Melocactus explants at different stages of growth. To this end, we amplified a 394-bp probe based on the MgSERK sequence we cloned, which comprised the final portion of the SPP domain and the beginning of the kinase domain (Fig. S1). In control explants, no signal was detected after 10 days of growth (Fig. 9A, B). After 30 days, a weak hybridization signal was detected in parenchyma cells associated with vascular bundles (Fig. 9C, D) and, after 50 days, MgSERK expression was visible throughout newly initiated shoots (Fig. 9E, F).

In situ hybridization of Melocactus glaucescens SERK1. A, B, E, F, I, J Control explant cultivated in PGR-free medium and stained with the MgSERK1 hybridization probe (A, E, I) or negative control (B, F, J). C, D, G, H, K, L Treated explant with a punctured areola region, supplemented with 17.76 µM BA and 1.34 µM NAA, and stained with the MgSERK1 hybridization probe (C, G, K) or negative control (D, H, L). Bars = 500 µm
In treated explants, MgSERK expression was visible in epidermal structures at 10 days of growth (Fig. 9G, H). By 30 days, the hybridization signal was strong throughout the areolar and vascular bundle regions (Fig. 9I, J). Finally, after 50 days, the signal was observed at the apical meristem of the stem and throughout the areolar and vascular bundle regions (Fig. 9K, L), as well as in the roots of newly formed shoots. Taken together, these findings indicate that SERK expression is associated with SO in Melocactus.
Discussion
The areola enables the formation of three vital parts in cacti: spine clusters, flowers, and branches (Anderson 2001; Mauseth 2017). Generally, during initial formation of the areola, dividing cells form the shoot apical meristem (SAM) (Boke 1944). Once the areola SAM has produced the required number of spines’ primordia, it becomes dormant. In the Melocactus genus, all areole except the one that gives rise to the cephalium (reproductive structure) remain dormant under natural conditions (Machado 2009; Mauseth 2017).
Importantly, the SAM in each dormant areola can become active again even after decades (Mauseth 2017). For example, if the SAM is damaged, some areole below the point of injury become active and produce a branch (Machado 2009). This is possible because in the Melocactus genus the areola is a reservoir of healthy cells capable of growth (Mauseth 2017).
The dormant areole of Melocactus are activated in tissue culture by removing apical dominancy, by cutting the stem to form explants or by adding PGR to culture medium (Retes-Pruneda et al. 2007; Torres-Silva et al. 2018) (Fig. 1B, C). Explants of M. glaucescens can produce shoots even without PGR addition (Fig. 1D, F, G), but at a rate (1.3–3 shoots/explant) too low to be viable for commercial purposes (Torres-Silva et al. 2018).
In the present study, the number of shoots per explant was much higher in samples (Fig. 1E, G, I) exposed to the same PGR concentration reported previously by Torres-Silva et al. (2018) than in control samples. Specifically, an average of 2.5 shoots per explant was observed after 120 days of culture, which increased by nearly fourfold (8.3) when the areola region was punctured with a needle to stimulate the SAM. The loss of cell-to-cell communication and disruption of long-distance signaling produced by wounding can elicit changes in plasma transmembrane potential, intracellular Ca2+ concentration, and H2O2 generation (Ikeuchi et al. 2016; Xu 2018). Accordingly, wounding is believed to stimulate the production of phytohormones and is related to both shoot and root organogenesis (Ikeuchi et al. 2016, 2019, 2020; Xu 2018).
Anatomical analysis of the changes occurring during areolar activation and SO induction in M. glaucescens (Fig. 3) revealed that the shoots were produced directly from the axillary bud of the areola region, as reported by Téllez-Román et al. (2017) in Mammillaria plumosa. SO in M. glaucescens occurred both near and far from the SAM region of the areola in control and treated explants, confirming the pattern observed previously in the Echinocereus genus by Sánchez et al. (2015).
In addition to the number of shoots, the time it took to respond to SO stimuli was also different between control and treated explants. SAM activation and meristemoid formation took longer in explants placed on PGR-free medium (Fig. 3) than in treated explants, for which it happened within 10 days of starting in vitro cultures. Occurrence of meristemoids was observed in the cortex of both control and treated explants (Fig. 3D–F). The cortex is the region of the M. glaucescens stem characterized by cells with the highest level of ploidy (~ 32C) (Torres-Silva et al. 2020).
Controlled endocycles, with discrete periods of S-phase and G-phase but without cytokinesis (endocycle), give rise to cells with a single polyploid nucleus (Lee et al., 2009; Scholes and Paige, 2015). In M. glaucescens, cells with more than 15-fold DNA content (compared to 2C) were found surrounding the vascular bundles of the cortex region (Torres-Silva et al. 2020) and coincided with the area where meristemoids were observed. Propitious formation of meristemoids requires asymmetric division and consequent adoption of stem cell identity. The G2-phase might be of particular importance in preparing for this unequal cell division (Jakoby and Schnittger 2004). Therefore, in plant tissues, in which the cell cycle is modified to accommodate specific features (e.g., aquifer parenchyma), meristemoid formation may occasionally fail.
Altered meristemoid formation may give rise to shoots with unusual morphology, as observed here for M. glaucescens derived from treated explants (Fig. 1I). The transcriptional program that determines cell type-specificity relies on turning on and off temporally and spatially expressed genes to fine-tune tissue differentiation (Nagymihály et al. 2017). Failure to organize the meristemoid may change the pattern of expression regulating the differentiation of new shoot tissues.
The formation of meristemoids, and consequently shoots, in regions characterized by varied cell cycle and ploidy can also be explained by the somaclonal variation observed by Torres-Silva et al. (2018) during in vitro shoot propagation of M. glaucescens. Changes in the chromatin landscape (caused by the endoreduplication process) associated with accelerated cell divisions (induced by the presence of PGR in culture medium) may cause DNA polymerase sliding and consequent pairing mistakes during DNA replication, leading to somaclonal variation (Aremu et al. 2013; Nagymihály et al. 2017).
Areolar activation in M. glaucescens was investigated here by studying the expression of the MgSERK1 gene in control and treated explants. A putative nucleotide sequence of 2251 bp was first identified based on the alignment of a cDNA clone and two sequences originating from a transcriptome database of treated explants cultured for 30 days.
Phylogenetic analysis using 107 amino acid sequences from 24 plant species with elevated sequence similarity (> 80%) against SERK Dicot S1/2 members indicated that MgSERK1 was a functional SERK1 ortholog (Figs. 4 and 5; Table S3). SERK genes comprise the largest subfamily of receptor-like kinases (RLKs) in plants and are involved in key plant development processes (Sharma et al. 2008). The general structure of SERK proteins places them in the LRR-RLK family (Walker 1994). Specifically, SERK genes belong to the LRR-RLK II group due to the presence of a small extracellular domain consisting of 4.5–5 LRRs followed by an SPP domain, a single-pass TM domain, and an intracellular kinase domain (Man et al. 2020). All conserved domains expected to be present in the established SERK orthologs were observed in MgSERK.
The structure of the MgSERK1 protein revealed conserved residues on both concave and convex sides, in agreement with other SERK members (Aan den Toorn et al. 2015; Rocha et al. 2016). The extracellular domain is involved in many different signaling processes and is important for SERK specificity (Aan den Toorn et al. 2015).
As observed in MgSERK1, the concave side is more conserved among SERK members, and presents residues that have been reported to interact with the BRI1 and FLS2 extracellular domains, and trigger downstream biological responses (Aan den Toorn et al. 2015). The convex side has not been reported to engage in specific interactions and, therefore, it may be more susceptible to evolutionary changes.
Bioinformatics analyses comparing protein sequences and structures (Figs. 6 and 7), together with the in situ hybridization results (Fig. 9), suggested that MgSERK1 played a similar role in organogenesis as CpSERK2, PeSERK1, and BnSERK2 (Brassica napus). This gene family does not mediate only embryo-specific signal transduction, but is involved also in organogenic pathways (Savona et al. 2012; Ahmadi et al. 2016; Rocha et al. 2016).
The amplification of cDNA fragments using specific primers designed to bind at different positions of the consensus sequence confirmed the presence of MgSERK1 transcripts in explants of M. glaucescens and the accuracy of the sequence generated by alignment. The specificity of MgSERK1 primers was tested also using genomic DNA, and resulted in a fragment of approximately 6 kb (Fig. 8).
According to the present in situ hybridization results, MgSERK1 transcripts were observed throughout in vitro M. glaucescens organogenesis in both control and treated explants. In particular, strong MgSERK1 expression was observed in the areola (Fig. 9), suggesting that the gene is associated with breaking its dormancy. A faint MgSERK1 hybridization signal was associated with vascular bundles of control explants during the first 30 days of culture (Fig. 9C, D). As with anatomical analysis, in situ hybridization confirmed the occurrence of SO in the inner regions of the explant. There, procambial cells possess stem cell capacity and, provided the correct signals, can become totipotent and develop into meristemoids (Podio et al. 2014; Ahmadi et al. 2016). Rocha et al. (2016) also observed a faint PeSERK1 hybridization signal in the vascular tissues of initial hypocotyl explants, which was associated with provascular tissue development.
The faster development of shoots in treated explants was observed by both anatomical and in situ hybridization assays. The MgSERK1 hybridization signal was associated with structures arising from the epidermis of treated explants within 10 days of culture (Fig. 9). The signal became stronger during SO induction (50 days of culture), and expanded from vascular tissues and the explant’s SAM to the meristematic cell populations of vascular tissues, SAM (Fig. 9), and root apical meristem of the new shoot.
Shoots formed in the inner region of treated explants generated roots. MgSERK1 expression was observed in the root apical meristem of shoots from treated explants after 50 days of culture. These results are consistent with the findings of Ahmadi et al. (2016), who observed BnSERK1 and BnSERK2 expression in both primary and developed shoots, as well as in the roots of regenerated B. napus.
The results from this study are in agreement with recent reports that advocate for a broader view of SERK function. Accordingly, SERKs may engage in the production of pluripotent cells capable of developing into several divergent cell types, a highly conserved phenomenon among land plants (Li et al. 2015; Rocha et al. 2016).
Conclusions
Anatomical and in situ hybridization analyses showed that shoot organogenesis in Melocactus glaucescens occurred by areolar activation and from parenchyma cells in the stem cortex. Shoot organogenesis was further improved by puncture of the areolar region, which activated the axillary bud and amplified the response of the explant, increasing the number of shoots produced. The MgSERK1 gene isolated in this study encodes an ortholog of other SERK Dicot S1/2 proteins. Its expression is associated with shoot organogenesis in the areola and adjacent regions, as well as with root organogenesis. These findings expand our understanding of the genetic mechanisms involved in cactus regeneration.
Availability of data materials
The raw sequence reads from next-generation sequencing of M. glaucescens transcripts were deposited at NCBI and can be accessed through the SRA accession number PRJNA663542. The Transcriptome Shotgun Assembly project where parts of the MgSERK1 were obtained has been deposited at DDBJ/EMBL/GenBank under the accession GJHH00000000. The version used in this paper is the first version, GJHH01000000.
References
Aan den Toorn M, Albrecht C, de Vries SC (2015) On the origin of SERKs: bioinformatics analysis of the somatic embryogenesis receptor kinases. Mol Plant 8:762–782. https://doi.org/10.1016/j.molp.2015.03.015
Ahmadi B, Masoomi-Aladizgeh F, Shariatpanahi ME, Azadi P, Keshavarz Alizadeh M (2016) Molecular characterization and expression analysis of SERK1 and SERK2 in Brassica napus L.: implication for microspore embryogenesis and plant regeneration. Plant Cell Rep 35:185–193. https://doi.org/10.1007/s00299-015-1878-6
Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37:420–423. https://doi.org/10.1038/s41587-019-0036-z
Anderson EF (2001) The cactus family. Timber Press, Portland
Aremu AO, Bairu MW, Szüčová L, Doležal K, Finniea JF, van Staden J (2013) Genetic fidelity in tissue-cultured ‘Williams’ bananas—the effect of high concentration of topolins and benzyladenine. Sci Hortic 161:324–327. https://doi.org/10.1016/j.scienta.2013.07.022
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533. https://doi.org/10.1093/nar/gkq399
Baudino S, Hansen S, Brettschneider R, Hecht V, Dresselhaus T, Lorz H, Dumas C, Rogowsky P (2001) Molecular characterization of two novel maize LRR receptor-like kinases, which belong to the SERK gene family. Planta 213:1–10. https://doi.org/10.1007/s004250000471
Boke NH (1944) Histogenesis of the leaf and areole in Opuntia cylindrica. Am J Bot 31:299–316. https://doi.org/10.1002/j.1537-2197.1944.tb08036.x
Braun P, Machado M, Taylor NP, Zappi D (2013) Melocactus glaucescens. The IUCN Red List of Threatened Species. e.T40923A2944067. https://doi.org/10.2305/IUCN.UK.2013-1.RLTS.T40923A2944067.en. Accessed 10 May 2020
CITES (2021) Convention on International Trade in Endangered Species of wild fauna and flora. Appendix I (valid from 22 June 2021). Available at https://cites.org/eng/app/appendices.php. Accessed on 24 June 2021).
Cueva-Agila AY, Alberca-Jaramillo N, Cella N, Concia L (2020) Isolation, phylogenetic analysis, and expression of a Somatic Embryogenesis Receptor-like Kinase (SERK) gene in Cattleya maxima Lindl. Curr Plant Biol 21:100139. https://doi.org/10.1016/j.cpb.2020.100139
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
El-Gebali S, Mistry J, Bateman A et al (2019) The Pfam protein families database in 2019. Nucleic Acids Res 47:D427–D432. https://doi.org/10.1093/nar/gky995
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using Phred. I. Accuracy Assessment. Genom Res 8:175–185. https://doi.org/10.1101/gr.8.3.175
Godínez-Álvarez H, Valverde T, Ortega-Baes P (2003) Demographic trends in the Cactaceae. Bot Rev 69:173–203. https://doi.org/10.1663/0006-810168
Goettsch B, Hilton-Taylor C, Cruz-Piñón G et al (2015) High proportion of cactus species threatened with extinction. Nat Plants 1:15142. https://doi.org/10.1038/NPLANTS.2015.142
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010
Hecht V, Vielle-Calzada J-P, Hartog MV, Schmidt EDL, Boutilier K, Grossniklaus U, de Vries SC (2001) The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE 1 gene is expressed in developing ovules and embryos and enhances embryogenic competence in culture. Plant Physiol 127:803–816. https://doi.org/10.1104/pp.010324
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome 9:868–877. https://doi.org/10.1101/gr.9.9.868
Ikeuchi M, Ogawa Y, Iwase A, Sugimoto K (2016) Plant regeneration: cellular origins and molecular mechanisms. Development 143:1442–1451. https://doi.org/10.1242/dev.134668
Ikeuchi M, Favero DS, Sakamoto Y, Iwase A, Coleman D, Rymen B, Sugimoto K (2019) Molecular mechanisms of plant regeneration. Annu Rev Plant Biol 70:377–406. https://doi.org/10.1146/annurev-arplant-050718-100434
Ikeuchi M, Rymen B, Sugimoto K (2020) How do plants transduce wound signals to induce tissue repair and organ regeneration? Curr Opin Plant Biol 57:72–77
Jakoby M, Schnittger A (2004) Cell cycle and differentiation. Curr Opin Plant Biol 7:661–669. https://doi.org/10.1016/j.pbi.2004.09.015
Jenkins M (1993) The wild plant trade in Europe—results of a traffic Europe survey of European nurseries. Traffic Europe, Cambridge, pp 3–17
Karnovsky MJ (1965) A formaldehyde-glutaraldehyde fixative of high osmolality foruse in electron microscopy. J Cell Biol 27:1-149A
Kim DG, Enkhtaivan G, Saini RK, Keum Y-S, Kang KW, Sivanesan I (2019) Production of bioactive compounds in cladode culture of Turbinicarpus valdezianus (H. Moeller) Glass & R. C. Foster. Ind Crops Prod 138:111491. https://doi.org/10.1016/j.indcrop.2019.111491
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. https://doi.org/10.1006/jmbi.2000.4315
Lambert SM, Borba EL, Machado MC (2006) Allozyme diversity and morphometrics of the endangered Melocactus glaucescens (Cactaceae), and investigation of the putative hybrid origin of Melocactus x albicephalus (Melocactus ernestii x M. glaucescens) in north-eastern Brazil. Plant Species Biol 21:93–108. https://doi.org/10.1111/j.1442-1984.2006.00155.x
Lee HO, Davidson JM, Duronio RJ (2009) Endoreplication: polyploidy with purpose. Genes Dev 23:2461–2477. https://doi.org/10.1101/gad.1829209
Lefort V, Longueville JE, Gascuel O (2017) SMS: smart model selection in PhyML. Mol Biol Evol 34:2422–2424. https://doi.org/10.1093/molbev/msx149
Lema-Rumińska J, Kulus D (2014) Micropropagation of cacti—a review. Haseltonia 18:46–63. https://doi.org/10.2985/026.019.0107
Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. https://doi.org/10.1093/nar/gkz239
Li W, Fang Y-H, Han J-D, Bai S-N, Rao G-Y (2015) Isolation and characterization of a novel SOMATIC EMBRYOGENESIS RECEPTOR KINASE gene expressed in the fern Adiantum capillus-veneris during shoot regeneration in vitro. Plant Mol Biol Rep 33:638–647. https://doi.org/10.1007/s11105-014-0769-2
Lu S, Wang J, Chita F, Derbyshire MK et al (2020) CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268. https://doi.org/10.1093/nar/gkz991
Machado MC (2009) The genus Melocactus in eastern Brazil: part I—an introduction to Melocactus. Brit Cac Succ J 27:1–16
Man J, Gallagher JP, Bartlett M (2020) Structural evolution drives diversification of the large LRR-RLK gene family. New Phytol 226:1492–1505. https://doi.org/10.1111/nph.16455
Mantelin S, PengH-C LiB, Atamian HS, Takken FLW, Kaloshian I (2011) The receptor-like kinase SlSERK1 is required for Mi-1-mediated resistance to potato aphids in tomato. Plant J 67:459–471. https://doi.org/10.1111/j.1365-313X.2011.04609.x
Mauseth JD (2017) An introduction to cactus areoles, part II. Brit Cac Succ J 89:219–229. https://doi.org/10.2985/015.089.0503
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497. https://doi.org/10.1111/j.13993054.1962.tb08052.x
Nagymihály M, Veluchamy A, Györgypál Z, Ariel F, Jégu T, Benhamed M, Szűcs A, Kereszt A, Mergaert P, Kondorosi E (2017) Ploidy-dependent changes in the epigenome of symbiotic cells correlate with specific patterns of gene expression. Proc Nat Acad Sci USA 114:4543–4547. https://doi.org/10.1073/pnas.1704211114
Nolan KE, Irwanto RR, Rose RJ (2003) Auxin up-regulates MtSERK1 expression in both Medicago truncatula root-forming and embryogenic cultures. Plant Physiol 133:218–230. https://doi.org/10.1104/pp.103.020917
O’Brien TP, McCully ME (1981) The study of plant structure: principles and selected methods. Termacarphi Pty Ltd, Melbourne
Pérez-Molphe-Balch E, Santos-Díaz MS, Ramírez-Malagón R, Ochoa-Alejo N (2015) Tissue culture of ornamental cacti. Sci Agric 72:540–561. https://doi.org/10.1590/0103-9016-2015-0012
Podio M, Felitti SA, Siena LA, Delgado L, Mancini M, Seijo JG, González AM, Pessino SC, Ortiz JPA (2014) Characterization and expression analysis of SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) genes in sexual and apomictic Paspalum notatum. Plant Mol Biol 84:479–495. https://doi.org/10.1007/s11103-013-0146-9
Retes-Pruneda JL, Valadez-Aguilar ML, Pérez-Reyes ME (2007) Propagación in vitro de especies de Echinocereus, Escontria, Mammillaria, Melocactus y Polaskia (Cactaceae). Bol Soc Bot Méx 81:9–16. https://doi.org/10.17129/botsci.1761
Rocha DI, Monte-Bello CC, Aizza LCB, Dornelas MC (2016) A passion fruit putative ortholog of the SOMATIC EMBRYOGENESIS RECEPTOR KINASE1 gene is expressed throughout the in vitro de novo shoot organogenesis developmental program. Plant Cell Tissue Organ Cult 25:107–117. https://doi.org/10.1007/s11240-015-0933-x
Rubluo A (1997) Micropropagation of Mammillaria species (Cactaceae). In: Bajaj YPS (ed) Biotechnology in agriculture and forestry 40. Springer-Verlag, Berlin, pp 193–205
Sambrook J, Russell DW (2001) Molecular Cloning, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sánchez D, Grego-Valencia D, Terrazas T, Arias S (2015) How and why does the areole meristem move in Echinocereus (Cactaceae)? Ann Bot 115:19–26. https://doi.org/10.1093/aob/mcu208
Santiago J, Henzler C, Hothorn M (2013) Molecular mechanism for plant steroid receptor activation by somatic embryogenesis coreceptor kinases. Science 341:889–892. https://doi.org/10.1126/science.1242468
Savona M, Mattioli R, Nigro S, Falasca G, Rovere FD, Costantino P, de Vries SC, Ruffoni B, Trovato M, Altamura MM (2012) Two SERK genes are markers of pluripotency in Cyclamen persicum Mill. J Exp Bot 63:471–488. https://doi.org/10.1093/jxb/err295
Schmidt EDL, Guzzo F, Toonen FAJ, de Vries SC (1997) A leucine-rich repeat containing receptor-like kinase marks somatic plant cells competent to form embryos. Development 124:2049–2062
Scholes DR, Paige KN (2015) Plasticity in ploidy: a generalized response to stress. Trends Plant Sci 20:165–175. https://doi.org/10.1016/j.tplants.2014.11.007
Sharma SK, Millam S, Hein I, Bryan GJ (2008) Cloning and molecular characterization of a potato SERK gene transcriptionally induced during initiation of somatic embryogenesis. Planta 228:319–330. https://doi.org/10.1007/s00425-008-0739-8
Singla B, Khurana JP, Khurana P (2008) Characterization of three somatic embryogenesis receptor kinase genes from wheat, Triticum aestivum. Plant Cell Rep 27:833–843. https://doi.org/10.1007/s00299-008-0505-1
Téllez-Román J, López-Peralta MCG, Hernández-Meneses E, Estrada-Luna AA, Mancera HAZ, Muñoz ML (2017) In vitro morphogenesis of Mammillaria plumosa Weber. Rev Mex Cien Agric 8:863–876
Thomas C, Meyer D, Himber C, Steinmetz A (2004) Spatial expression of a sunflower SERK gene during induction of somatic embryogenesis and shoot organogenesis. Plant Physiol Biochem 42:35–42. https://doi.org/10.1016/j.plaphy.2003.10.008
Torres-Silva G, Resende SV, Lima-Brito A, Bezerra HB, Santana JRF, Schnadelbach AS (2018) In vitro shoot production, morphological alterations and genetic instability of Melocactus glaucescens (Cactaceae), an endangered species endemic to eastern Brazil. S Afr J Bot 15:100–107. https://doi.org/10.1016/j.sajb.2018.01.001
Torres-Silva G, Matos EM, Correia LNF, Fortini EA, Soares WS, Batista DB, Otoni CG, Azevedo AA, Viccini LF, Koehler AD, Resende SV, Specht SD, Otoni WC (2020) Anatomy, flow cytometry, and X-ray tomography reveal tissue organization and ploidy distribution in long-term in vitro cultures of Melocactus species. Front Plant Sci 11:1314. https://doi.org/10.3389/fpls.2020.01314
Walker JC (1994) Structure and function of the receptor-like protein kinases of higher plants. Plant Mol Biol 26:1599–1609. https://doi.org/10.1007/BF00016492
Xu L (2018) De novo root regeneration from leaf explants: wounding, auxin, and cell fate transition. Curr Opin Plant Biol 41:39–45. https://doi.org/10.1016/j.pbi.2017.08.004
Acknowledgements
We thank Delmar Lopes Alvim (in memoriam) for help during fieldwork, Evandro S. B. Oliveira and Jessé de Andrade Ribeiro for the fruitful discussion on acupuncture in plants, and Susan Strickler for help with transcriptome data mining. Editage (www.editage.com) is also acknowledged for English language editing.
Funding
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, DF, Brazil), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, Belo Horizonte, Brazil; Grant APQ-00772-19), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília, DF, Brazil; Finance Code 001 and Internship Grant PDSE 88881.132727/2016-01 to GTS).
Author information
Authors and Affiliations
Contributions
GTS, ADK, DSB, SVR, SS, CDS, and WCO conceived and designed the study; GTS performed the experiments; GTS and LNFC performed the histological and in situ analyses; GTS and DSB performed statistical analysis; ADK, DVF, SS, CDS, and ER performed phylogenetic and bioinformatics analyses; GTS, ADK, DSB,SVR, JF, CDS, ER, and WCO wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interests.
Additional information
Communicated by T. Winkelmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.



11240_2021_2137_MOESM4_ESM.xlsx
Supplementary Table S1. SERK and non-SERK-like genes identified in 24 angiosperm species. Taxonomic classification, species name, sequence phylogenetic classification, gene name, accession number for protein ID, references, gene code, updated gene name, and library source consulted in this study are provided. Accession numbers for previously studied proteins are indicated. The library source for all sequences is Phytozome v12.0, Beta vulgaris Resource or SpinachBase. The original gene name was kept when it did not interfere with the classification. Eudicot and monocot protein IDs not explored previously received a gene name based on their phylogenetic relationship, clustering with SERK Dicot S1/2 and SERK Dicot S3/4, and meaningful classification according to Mantelin et al. (2011) and Aan den Toorn et al. (2015). SERK Dicot S1/2 closely duplicated sequences were named SERK#A and SERK#B, as proposed by Mantelin et al. (2011). The gene name used in the phylogenetic tree (Fig. 4 and Fig. S1) includes the species initials, such as Ah (Amaranthus hypochondriacus), At (Arabidopsis thaliana), Bd (Brachypodium distachyon), Bv (Beta vulgaris), Cp (Carica papaya), Cq (Chenopodium quinoa), Gh (Gossypium hirsutum), Gm (Glycine max), Ha (Helianthus annuus), Ls (Lactuca sativa), Md (Malus domestica), Me (Manihot esculenta), Mg (Melocactus glaucescens), Mt (Medicago truncatula), Oe (Olea europaea), Os (Oryza sativa), Pt (Populus trichocarpa), Rc (Ricinus communis), Sv (Setaria viridis), Sl (Solanum lycopersicum), So (Spinacia oleracea), Tc (Theobroma cacao), Vv (Vitis vinifera), and Zm (Zea mays). (XLSX 20 KB)
11240_2021_2137_MOESM5_ESM.xlsx
Supplementary Table S2. Conserved SERK protein domains and motifs. Software-predicted protein domains and motif regions in SERK proteins include: signal peptide, putative leucine zipper (ZIP), leucine-rich repeats (LRR1–LRR5), transmembrane domain (TM), and protein kinase domain. Alignment (ALN) was used to predict the serine-proline-rich region (SPP) and C-terminal domain, as observed for A. thaliana by Baudino et al. (2001). If no such region was identified, the spot is marked with (-); however, if the alignment confirmed the presence or absence of the region (Fig. 5), this is indicated as (Yes) or (No), respectively. Accession numbers of SERK orthologs are listed in Table S1. (XLSX 10 KB)
11240_2021_2137_MOESM6_ESM.xlsx
Supplementary Table S3. Identity matrix, and percentage (%) of identical residues between SERK-like proteins in 23 plant species. All SERK Dicot S1/2 and one clade from SERK Monocot sequences were analyzed. MgSERK1 identity compared with other plant species is highlighted in green. The gene name and corresponding species are described in Table S1. (XLSX 13 KB)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Torres-Silva, G., Correia, L.N.F., Koehler, A.D. et al. Expression of Melocactus glaucescens SERK1 sheds new light on the mechanism of areolar activation in cacti. Plant Cell Tiss Organ Cult 147, 437–451 (2021). https://doi.org/10.1007/s11240-021-02137-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-021-02137-9