
Abstract
Plants produce a wide variety of secondary metabolites (SMs) of pharmacological interest that, due to their high structural complexity, are difficult to obtain from chemical synthesis. Knowledge of the enzymatic process involved in the biosynthesis of these SMs may contribute to the development of new strategies for obtaining them from plant tissues. Maytenus ilicifolia contains three main classes of bioactive compounds: sesquiterpene pyridine alkaloids, flavonoids and, primarily, quinonemethide triterpenes, which are well known for their high and broad antitumor activity. Thus, to determine the enzymatic composition in the secondary metabolism of M. ilicifolia, we carried out proteomic profiling of M. ilicifolia cell cultures with and without increased concentrations of maytenin and 22β-hydroxy-maytenin by elicitation with methyl jasmonate for 48 h. For this purpose, the analysis of soluble proteins was performed via LCMS/MS experiments using a shotgun strategy and compared to specific databases, resulting in 1319 identifications in the two types of cells. Many enzymes involved in secondary metabolism were detected, including cytochrome P450-dependent monooxygenases, which potentially catalyze the final steps in the quinonemethide triterpene biosynthesis. In conclusion, we report for the first time a proteomic study of a cell culture system that is an effective elicitor of quinonemethide triterpene production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plants produce a wide variety of secondary metabolites (SMs) of high structural complexity and pharmacological importance. As the chemical synthesis of several of these SMs is not economically feasible, their isolation from plants still represents the only option. On the other hand, producing a range of these SMs from plants, even using biotechnological techniques (e.g., in vitro tissue cultures), is still limited due to the lack of knowledge on both their biosynthetic pathways and regulation (Oksman-Caldentey and Inzé 2004; Lippmann et al. 2009).
Fortunately, advances in the field of “omics” have enabled the exploration and understanding of a variety of biological processes at the molecular level, and, among these technologies, proteomics has proven to be an efficient and powerful tool for revealing insights into plant secondary metabolism (Martínez-Esteso et al. 2015).
The first and most crucial step in proteomics-based research into plant secondary metabolism is the choice of suitable plant materials rich in the SMs of interest. Cell cultures have been considered to be the ideal biological material for these approaches. This is due to their cultivation capabilities and, mainly, their compatibility with tissues and organs specialized in the biosynthesis of particular SMs, which occurs in cell cultures or can be induced through the elicitation of stress under laboratory-controlled conditions. In fact, most proteomic studies on secondary metabolism have been carried out with cell cultures elicited by methyl jasmonate (MeJA) because is known to induce all classes of SMs (terpenes, alkaloids, phenylpropanoids) (Zhao et al. 2005; Pauwels et al. 2009; Spitzer-Rimon et al. 2010). Other elicitors, such as salicylic acid, chitosan, yeast extract also have been tried in these types of studies (Martínez-Esteso et al. 2015; Yue et al. 2016).
The medicinal plant Maytenus ilicifolia Mart ex Reissek (Celastraceae) fits perfectly into this scope. It produces various classes of bioactive SMs (Corsino et al. 2000; Gutiérrez et al. 2007; de Souza et al. 2008; Mossi et al. 2009; dos Santos et al. 2012; Jardim et al. 2015), including the antimicrotubule agent maytansine (Ahmed et al. 1981), which, when linked to the monoclonal antibody trastuzumab (as an antibody-drug conjugate), was recently approved to treat HER2-positive metastatic breast cancer (Newman and Cragg 2016).
Maytenus ilicifolia also biosynthesizes quinonemethide triterpenes (QMTs), a class of SMs that displays a range of biological activity (dos Santos et al. 2010, 2013; Gullo et al. 2012), including potent cytotoxic activity against a series of cancer cell lines (Oramas-Royo et al. 2010; Paz et al. 2013; Choi et al. 2016).
Despite the well-known pharmacological potential of QMTs, there is a considerable limitation for their studies related to their low concentration in Celastraceae species due to the QMTs showing restricted accumulation in root bark (Buffa Filho et al. 2004; Coppede et al. 2014). In addition, their syntheses require a large number of steps and still result in a low overall yield (Camelio et al. 2015). Thus, the production of this class of compounds in plant cell cultures seems to be the most viable alternative since they are richer sources of QMTs when compared to the plant material (Buffa Filho et al. 2004; Coppede et al. 2014).
In this study, we carried out proteomic profiling of M. ilicifolia cell cultures with and without increased concentrations of QMTs by elicitation with MeJA. The goals of this work were to identify the main proteins involved in the regulation of secondary metabolism in such cell cultures and generate new insights into secondary metabolism in M. ilicifolia, focusing on the biosynthesis of QMTs. We identified a variety of enzymes involved in the biosynthetic pathways of the secondary metabolites of Maytenus ilicifolia, showing that cell cultures are a promising system to explore new pathways related to some classes of natural products of interest. In addition, we aim to provide the first proteomic study of this plant species.
Materials and methods
In vitro plant materials
Callus of M. ilicifolia from the leaves (Pereira et al. 1994) was maintained in vitro for 12 years with periodic transfer to semi-solid MS culture medium (Murashige and Skoog 1962) supplemented with sucrose (30 g/l), 2,4-dichlorophenoxyacetic acid (2,4-D, 1.0 mg/l), kinetin (0.5 mg/l) and monopotassium phosphate (KH2PO4, 340 mg/l). The medium was adjusted to pH 6.0 and solidified with 2.5 g/l Phytagel® prior to autoclaving at 121 °C for 15 min. The cultures were maintained at 25 ± 2 °C and exposed to light for a 16 h photoperiod at a photosynthetic photon flux density of 20–70 mmol/m2s.
For initiating cell suspension cultures, callus of M. ilicifolia (after 60 days of growth) was transferred to 250-ml Erlenmeyer flasks (4 g/flask) containing 60 ml of same culture medium described above (without the addition of Phytagel®). The flasks were kept on an orbital shaker at 100 rpm under the same light conditions mentioned above.
Cell culture elicitation
After 7 days of incubation, the cell cultures were treated with MeJA (Sigma–Aldrich) (dissolved in EtOH/H2O, 1:1, v/v) at a final concentration of 100 µM. The controls received equivalent volumes of solvents (100 µl). After 6, 12, 24, 48 and 96 h of incubation, MeJA-elicited cells (MECs) and non-elicited cells (control cells, CCs) were filtered under reduced pressure through filter paper, washed with H2O, frozen in liquid N2 and stored at −80 °C for further analyses (phytochemical and proteomic). Experimental work was performed in triplicate with three repetitions (n = 9) of each sampling point.
Preparation of extracts
Aliquots of all frozen cells from M. ilicifolia were lyophilized, pulverized under liquid N2 and sonicated in dichloromethane (100 mg fresh weight/1 ml solvent) for 20 min (3 times) at room temperature. Next, the samples were filtered through a 0.45 μm nylon membrane under vacuum and evaporated to dryness. The extracts were resuspended in MeOH/H2O (99:1, v/v) and subjected to solid-phase extraction (SPE) with cartridges (SampliQ Agilent®) (3 ml) containing 500 mg of C18 silica gel (particle size of 40 μm) preconditioned with MeOH and MeOH/H2O (99:1, v/v). Cartridges were eluted with 4 ml of MeOH/H2O (99:1, v/v) and the eluates were evaporated to dryness. Then, the samples were resuspended in MeOH/H2O (83:17, v/v; concentrations of 0.5, 1.0 or 2.5 mg/ml), filtered through a 0.22 μm nylon membrane filter, and assayed by high-performance liquid chromatography-diode array detection (HPLC-DAD).
QMTs analysis
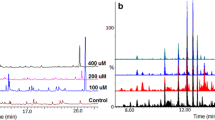
Analysis of QMTs in cell extracts of M. ilicifolia were accomplished using the method described by Paz et al. (2013) with a few modifications. The samples were analyzed on an HPLC system from Shimadzu (Kyoto, Japan), namely, an LC-10-AVP instrument equipped with an SIL-10AF autoinjector, an SPD-M20A photodiode array detector, and a Phenomenex Luna® C18 column (Torrance, CA, USA; 250 mm × 4.6 mm i.d.; particle size of 5 µm). The mobile phase was MeOH/H2O/formic acid (83:17:0.1, v/v/v), and elution was carried out in isocratic mode for 20 min at a flow rate of 1.2 ml/min. The injection volume was 20 µl for the samples and the chemical standard; detection was conducted at wavelengths of 254 and 420 nm. Maytenin (1) and 22β-hydroxy-maytenin (2) (Fig. 1) (both with HPLC purity >95%), isolated and identified by NMR and MS experiments in our laboratory from the root barks of M. ilicifolia, were employed as chemical standards. Measurements were integrated by comparison with an external standard calibration curve. Experimental work was performed in triplicate at each sampling point.

a Chromatogram of maytenin (1) and 22β-hydroxy-maytenin (2) and b concentration of both in cell cultures of Maytenus ilicifolia elicited with 100 μM methyl jasmonate (MECs) and non-elicited (control cells, CCs) in 48 h of incubation. Data are the mean ± SD for three individual experiments (n = 3)
Protein extraction
In this experiment, we used both MECs and their respective CCs that showed the greatest difference in the accumulation of QMTs [both (1) and (2)] between them after 48 h of incubation. Frozen cells (2 g) were ground in liquid N2 using a precooled mortar and pestle. Ten milliliters of extraction buffer [10 mM ammonium acetate (pH 6.7), 20% (w/v) polyvinylpolypyrrolidone (PVPP), 10 mM dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic acid (EDTA)] containing a cocktail of protease inhibitors (cOmplete Mini Protease Inhibitor Cocktail tablets, Roche Diagnostics, Mannheim, Germany) was added into each sample. The obtained solutions were manually homogenized by light shaking for 15 min at 4 °C, filtered through cheesecloth, centrifuged (10 min, 13,000 g, 4 °C) and transferred to a new tube. Total protein concentration was determined by following the Bradford (1976) method using bovine serum albumin as a standard.
In-solution protein digestion
Crude protein extracts from the M. ilicifolia cultures (250 µg) were solubilized in 50 mM ammonium bicarbonate (NH4HCO3; containing 7.5 M urea, pH 7.9) for 60 min at 37 °C for denaturation and then reduced with 10 mM DTT at 37 °C for 60 min. After this treatment, the proteins were alkylated with 40 mM iodoacetamide at 25 °C for 60 min in the dark. The samples were diluted fivefold with 100 mM NH4HCO3, pH 7.8, and sufficient amounts of calcium chloride (CaCl2) was added to the samples until a final concentration of 1 mM was reached. Non-autolytic trypsin (Promega) was added to the denatured protein solution (with trypsin/protein, 1:50, w/w) for 18 h at 37 °C. The enzymatic digestion was stopped by adding of 5 μl of formic acid and freezing the solution using liquid nitrogen. Digested samples were desalted using a Sep-Pak C18 column (Waters) conditioned with ACN, rinsed with 1 ml of 0.1% TFA, and washed with 3 ml of 0.1% TFA/H2O. Peptides were eluted from the Sep-Pak column with 1 ml of 0.1% TFA/80% ACN and concentrated using a Speed-Vac until it was completely dry. The digested samples were stored at −80 °C until analysis; the tryptic peptides were solubilized in 50% ACN and submitted to LC-IT-TOF/MS and MSn analysis.
LC-IT-TOF/MS
MS and MSn analyses were conducted on a hybrid system, where a UFLC (Prominence) was connected online to an IT/TOF MS (ion trap/time-of-flight mass spectrometer) instrument (Shimadzu, Kyoto, Japan) equipped with an electrospray ionization source. The UFLC system was composed of two LC20AD pumps, an SIL 20 AHT automatic injector, an SPD M20A diode array detector and a CTO 20A column oven. The LC system was connected to a Shim-pack XR-ODS column (100 mm × 3 mm i.d.; particle size of 2.2 µm; average pore diameter of 120 Å) and subjected to a gradient of acetonitrile [containing 0.05% (v/v) TFA] from 5% (v/v) to 90% (v/v) over 90 min at 30 °C. The elution of the tryptic peptides was monitored by UV absorption at 214 nm with a flow rate of 0.2 ml/min. Mass spectrometric analyses were carried-out using positive electrospray ionization (ESI+) mode under the conditions described as follows: the CDL temperature was adjusted to 200 °C, 4.5 kV was the voltage of capillaries, 3.5 V was set for the cone lens, the flow rate of the nebulizer gas (N2) was approximately 1.5 l h and the drying gas (N2) was at a flow rate of 100 l/h. Mass spectra were continuously acquired in the range of m/z 200–4000. The MSn spectra were obtained using the same parameters as the MS experiments. Argon (Ar) was used as the collision gas at a pressure of 100 kPa. The ions formed in the MSn experiments were trapped for 50 msec in an ion trap using a collision energy of 50% and frequency of 30 kHz. The data were acquired and analyzed using LCMS solution software (Shimadzu).
Protein identification
The MS and MSn data were combined to search with a taxonomic restriction of Viridiplantae (green plants) against the National Center for Biotechnology Information non-redundant protein database (NCBInr; http://www.ncbi.nlm.nih.gov, containing 2,562,600 sequences, 27 Nov 2014) and Swiss-Prot protein database (http://www.isb-sib.ch/, containing 35,737 sequences, 27 Nov 2014) using the Mascot algorithm 2.2.06 (Matrix Science, London, UK, http://www.matrixscience.com/). Search parameters were set as follows: taxonomy Viridiplantae, enzyme selected as trypsin, two maximum missing cleavage sites allowed, peptide mass tolerance of 0.5 Da for the MS and 0.5 Da for the MSn spectra, carbamidomethyl (C) specified as a fixed modification, and methionine and tryptophan oxidation were specified as variable modifications.
Bioinformatics analysis
The proteins identified by Mascot were functionally annotated according to gene ontology (GO) terms (into biological processes, molecular functions, and cellular components) and enzyme commission (EC) using Blast2GO software (v 3.0) (Götz et al. 2008) and verified manually from the Universal Protein Resource (UniProt) database (http://www.uniprot.org/). Briefly, the sequences of the proteins identified by mass spectrometry were downloaded from UniProt (saved as FASTA files) and analyzed by Blast2GO with the default parameters. Additionally, the sequences with corresponding EC numbers obtained from the annotation were mapped onto the Kyoto encyclopedia of genes and genomes (KEGG) pathway database (http://www.genome.jp/kegg/pathway.html).
Results and discussion
MeJA elicitation and QMTs accumulation
HPLC-DAD analyses confirm that the in vitro cell cultures of M. ilicifolia produce (1) and (2) (Fig. 1a) at various concentrations during all incubation periods measured (0, 6, 12, 24, 48 and 96 h) in both MECs and CCs (Supplementary Table A). However, the point of greatest difference in the accumulation of QMTs between the MECs and CCs occurred after 48 h of incubation. At this point, the concentrations of (1) and (2) in the MECs were 2.12 and 3.07 times higher, respectively, than in the CCs (Fig. 1b).
Protein profile
The proteomic profile of the in vitro cell suspension cultures of Maytenus ilicifolia changed considerably after elicitation with 100 μM MeJA. Plants subjected to stress, such as those elicited with MeJA, induce an active plant stress response, including a profound reorganization of plant proteome (Kosová et al. 2014). Using the software Mascot to search proteins with a taxonomic restriction of Viridiplantae (green plants) against the NCBInr and Swiss-Prot databases, we detected a total of 1345 proteins in M. ilicifolia cells. Among these, 26 were equally detected in the MECs and CCs, while 709 and 610 were differentially detected in the MECs and CCs, respectively.
The annotation of protein function and their cellular location is imperative to understand their role at molecular level (Shekhar et al. 2016), and therefore, the identified proteins were subjected to Blast2GO analysis. Among the proteins found in the MECs and CCs, 518 and 450 proteins, respectively, were functionally annotated (GO annotation). By predicting subcellular localization, the highest number of proteins were localized in the membrane (19.9% in CCs and 22.0% in MECs), followed by the nucleus (16.2% in CCs and 13.1% in MECs), cytoplasm (10.4% in CCs and 9.9% in MECs), plastid (9.4% in CCs and 14.5% in MECs), intracellular (10.1% in CCs and 8.1% in MECs), mitochondrion (4.7% in CCs and 3.1% in MECs) and plasma membrane (4.4 in CCs and 3.1% in MECs) (Fig. 2a).

Functional classification of proteins from the cell cultures of Maytenus ilicifolia elicited with 100 μM methyl jasmonate (MECs) and non-elicited (control cells, CCs) in 48 h of incubation. Proteins were categorized according to their gene ontology (GO) using Blast2Go software for their cellular component (a), molecular function (b) and biological process (c)
Proteins with molecular functions had the highest number of GO terms related to protein binding (17.8% in CCs and 15.9% in MECs), followed by binding (13.8% in CCs and 10.9% in MECs), nucleotide binding (13.4% in CCs and 13.9% in MECs), hydrolase activity (10.7% in CCs and 11.2% in MECs), catalytic activity (10.1% in CCs and 9.7% in MECs), DNA binding (6.9% in CCs and 5.1% in MECs) and transferase activity (5.8% in CCs and 5.1% in MECs) (Fig. 2b).
Regarding biological processes, the highest number of proteins from both cell cultures (CCs and MECs) were associated with the metabolic process (14.3% in CCs and 13.9% in MECs), followed by the cellular process (12.6% in CCs and 11.0% in MECs), biosynthetic process (11.1% in CCs and 10.0% in MECs), nucleobase-containing compound metabolic process (9.8% in CCs and 9.3% in MECs), cellular protein modification process (6.0% in CCs and 6.4% in MECs), transport (5.1% in CCs and 5.7% in MECs), carbohydrate metabolic process (4.3% in CCs and 4.6% in MECs) and response to stress (3.7% in CCs and 3.3% in MECs) (Fig. 2c).
Proteins with catalytic features were further classified, and the classes of the enzymes present in the cell cultures could be revealed. The enzyme distribution (Fig. 3) shows that hydrolases accounted for the largest proportion in both the CCs and MECs of M. ilicifolia enzymes, followed by transferases, oxidoreductases, lyases, isomerases and ligases.

General classification of enzyme from the cell cultures of Maytenus ilicifolia elicited with 100 μM methyl jasmonate (MECs) and non-elicited (control cells, CCs) in 48 h of incubation. Enzymes were classified according to enzyme commission (EC) by searching against the Kyoto encyclopedia of genes and genomes pathway database (KEGG), using Blast2Go software. Oxidoreductases (EC 1.x.x), Transferases (EC 2.x.x), Hydrolases (EC 3.x.x), Lyases (EC 4.x.x), Isomerases (EC: 5.x.x), and Ligases (EC 6.x.x)
Enzymes involved in the biosynthesis of SMs
By focusing on the biosynthetic pathways directly involved with secondary metabolites, some enzymes considered to be the manufacturers of terpenes were identified (Fig. 4), including isopentenyl-diphosphate delta-isomerase I (EC 5.3.3.2), which is involved in the isomerization of isopentenyl pyrophosphate (IPP) to dimethylallyl pyrophosphate (DMAPP) and is considered the key enzyme in the construction of all terpenes (Dewick 2009). Interestingly, IPP and DMAPP are synthesized by two well-characterized metabolic pathways: the mevalonate (MVA) pathway, which is generally involved in the biosynthesis of sesqui- and triterpenes, and the 2-C-methyl-d-erythritol 4-phosphate (MEP) pathway, which is involved in the biosynthesis of mono- and diterpenes that are compartmentalized in the cytosol and plastids, respectively (Van Cutsem et al. 2011). Among these, only the MVA pathway was shown to be active in the in vitro system in proteomic analysis (Fig. 4). In this regard, recent research reported by our group has shown that QMTs are biosynthesized by the MVA pathway (Pina et al. 2016).

Summary of metabolic pathways predicted from of proteomic analysis in cell cultures of Maytenus ilicifolia. Enzymes related to enzyme commission numbers are shown in Table 1. Enzymes coded green were detected in both, 100 μM methyl jasmonate-elicited cells (MECs) and non-elicited (control cells, CCs); those coded blue detected only in CCs; those coded red detected only in MECs. CYP450s cytochrome P450-dependent monooxygenases, OSC oxidosqualene cyclase; *Enzyme was not found in cell cultures of Maytenus ilicifolia. (Color figure online)
The bifunctional enzyme farnesyl pyrophosphate synthase 2 (FPS2, EC 2.5.1.1 and 2.5.1.10) and squalene monooxygenase-like (EC 1.14.13.132, transferred in 2015 to EC 1.14.14.17) were detected in MECs and CCs, respectively. These enzymes are involved in the biosynthetic pathway sequences to produce 2,3-oxidosqualene, which corroborated the possible biosynthesis of triterpenes regarding cyclization steps catalyzed by oxidosqualene cyclases (OSCs), showing their possible involvement in the biosynthesis of QMTs in the in vitro system (Fig. 4).
The cyclization of oxidosqualene in plants is one of the most complex reactions known for terpene biosynthesis (Wendt 2005; Phillips et al. 2006; Abe 2007). However, we have not identified any OSC in the cell cultures of M. ilicifolia. Investigations conducted by our research group with the species M. aquifolium and Salacia campestris (both Celastraceae) showed that friedelin (a friedelane triterpenoid) is the precursor of the QMTs (Corsino et al. 2000). Thus, we expected to find a cyclase enzyme that could form (directly or indirectly) a friedelane triterpene, such as, for example, friedelin synthase (FS, EC 5.4.99.50). FS is an OSC cloned from the plant Kalanchoe daigremontiana that is responsible for the cyclization of 2,3-oxidosqualene (at various stages) to friedelin (Su et al. 2010). Once synthesized, friedelin undergoes sequential oxidation steps and is converted into QMTs. Cytochrome P450-dependent monooxygenases (CYP450s) are good candidates for the oxidative processes. They are known to perform this type of modification of secondary metabolites. We found nine CYPs of yet unknown function (Supplementary Table S1) that could be responsible for the last stages of the formation of the QMTs.
The fact that we have found only a small number of enzymes involved in the biosynthesis of QMTs is related to the gaps remaining in this pathway for which the catalysis is unknown. Furthermore, a large percentage of proteins found in this study had an unknown function in GO analysis. This highlights the limit of proteomic approaches that rely on databases having many non-annotated proteins or enzymes with very general annotations (e.g., oxidoreductases), which can act upon more differentiated metabolic pathways. This can be further aggravated in the case of secondary metabolism, which is much less well characterized than primary metabolism (Champagne et al. 2012).
In addition, the results lead to the prediction of several enzymes involved in important metabolic pathways of secondary metabolism (Fig. 4). The shikimate pathway, which links the metabolism of carbohydrates to the biosynthesis of aromatic compounds (Herrmann and Weaver 1999) in plants, is important for both protein biosynthesis and for secondary metabolism (Guo et al. 2014). The end products and many intermediates of this pathway serve as precursors for plant hormones and for a large variety of secondary metabolites (e.g., alkaloids, benzenoids, and phenylpropanoids, including lignin). This seven-step metabolic route leads from phosphoenolpyruvate and erythrose 4-phosphate to chorismate, the common precursor for the biosynthesis of folic acid, ubiquinone, aromatic amino acids (l-phenylalanine, l-tyrosine and l-tryptophan) and other metabolites (Dewick 1995; Weaver and Herrmann 1997; Herrmann and Weaver 1999). Our results demonstrate that this pathway is active in M. ilicifolia. In the CCs was found a bifunctional enzyme with 3-dehydroquinate dehydratase/shikimate dehydrogenase (DQD/SDH, EC 1.1.1.25 and 4.2.1.10) activity, which catalyzes the third and fourth steps of this pathway (the conversion of shikimate from 3-dehydroquinate via 3-dehydroshikimate). In the MECs, we detected isochorismate synthase (ICS, EC 5.4.4.2), an enzyme responsible for the isomerization of chorismate to isochorismate (an SN2′-type of reaction), which is the immediate precursor of the well-known salicylic acid (Wildermuth et al. 2001). Also in the MECs, we identified one enzyme with o-succinylbenzoate-CoA ligase (EC 6.2.1.26) activity, which converts o-succinylbenzoate (OSB), an intermediate formed from chorismate, to OSB-CoA after four enzymatic steps. These intermediaries have been implicated in the biosynthesis of a wide range of plant SMs (Dewick 2009; Yamazaki et al. 2013).
Related to phenylpropanoid pathway, we have found in the MECs phenylalanine ammonia-lyase (PAL, EC 4.3.1.24), the first key enzyme in this pathway; it catalyzes the deamination of l-phenylalanine to form trans-cinnamic acid (Vogt 2010). The phenylpropanoid pathway is a secondary metabolism pathway responsible for producing flavonoids (as well as a variety of other important metabolites, including lignins, coumarins, phytoalexins, and stilbenes) (Harakava 2005). In the CCs was detected the cinnamyl-alcohol dehydrogenase 1 (CAD1, EC 1.1.1.195), which catalyzes the NAPDH-dependent reduction of cinnamyl aldehydes (p-coumaryl, coniferyl and sinapyl) to form their corresponding cinnamyl alcohols (monolignols) (Kutsuki et al. 1982; Deng et al. 2013). From oxidative coupling reactions governed by class III peroxidases (plant peroxidases), monolignols are polymerized to form lignins (Ralph et al. 2004). In cultures of M. ilicifolia (in both the CCs and MECs) we have found ten peroxidases of this group (EC 1.11.1.7) capable of polymerizing the cinnamyl alcohols (p-coumaryl, coniferyl and sinapyl) to form their corresponding lignins (p-hydroxyphenyl, guaiacyl and syringyl lignin). Several works have reported the direct implication of CAD and POD enzymes in lignin biosynthesis in the cell suspension cultures of many plant species (Messner and Boll 1993; Negrel and Javelle 1995; Koutaniemi et al. 2005; Sasaki et al. 2006). Lignin biosynthesis occurs during the differentiation of distinct cell types in different tissues/organs of a plant and, beyond ensuring proper cellular function, also occurs in response to environmental changes (Weng and Chapple 2010; Barros et al. 2015).
In addition to being a precursor of the phenylpropanoids, as discussed above, l-tyrosine can follow a different pathway and be oxidized to 3,4-dihydroxy-l-phenylalanine (l-Dopa) by polyphenol oxidase (PPO, EC 1.10.3.1), which was found in the CCs of M. ilicifolia. Transcriptomic studies of different tissues of Panax notoginseng have demonstrated that the gene encoding PPO is involved in isoquinoline alkaloid biosynthesis (Liu et al. 2015). Another enzyme found in the MECs, arginine decarboxylase (ADC, EC 4.1.1.19), has been shown to be associated with tropane alkaloid biosynthesis in plant in vitro cultures (Georgiev et al. 2013).
Also related to amino acid metabolism, we have found in the CCs branched-chain-amino-acid aminotransferase (BCAT, EC 2.6.1.42). BCATs play a crucial role in the metabolism of leucine, isoleucine, and valine (branched-chain amino acids, BCAA) (Diebold et al. 2002), which are important compounds not only for their function as the building blocks of proteins but also because they serves as precursors of several SMs in plants (Rizhsky et al. 2004).
To summarize, the proteome showed enzymes involved in pathways that lead to the biosynthesis of the major metabolites of the Maytenus ilicifolia species: enzymes involved in the MVA pathway, which lead to the formation of the QMTs and sesquiterpene moiety of the sesquiterpene pyridine alkaloids; enzymes involved in the amino acid pathway, which lead to the formation of the nitrogenous portion of the pyridine alkaloids; and enzymes involved in the acetyl-coenzyme A and phenylpropanoid pathways, which lead to the formation of polyphenols (catechins, flavonoids and tannins).
Enzymes involved in the biosynthesis of precursors of secondary metabolites
In the central metabolic pathway that provides energy and generates precursors for the synthesis of primary and secondary metabolites (Plaxton 1996), i.e., glycolysis, we have detected fructose-bisphosphate cytoplasmic (EC 4.1.2.13) in both cells (the CCs and MECs) and hypothetical protein ZEAMMB73_453652 (EC 2.7.1.11) in the CCs. Among the proteins in the MECs, we have detected the enzyme that catalyzes the reversible dehydration of 2-phosphoglycerate to phosphoenolpyruvate (PEP) in glycolysis, enolase (2-phospho- d -glycerate hydratase, EC 4.2.1.11). Genes encoding plant enolases have been cloned from diverse plant species (Blakeley et al. 1994; Forsthoefel et al. 1995; Fox et al. 1995; Lal et al. 1998). PEP formed through the enolase reaction represents a central metabolite in plant primary and secondary metabolism and has different possible fates (Voll et al. 2009). It not only can serve as precursor for the biosynthesis of aromatic amino acids in the shikimate pathway but can also be converted to pyruvate by pyruvate kinase (PK, EC 2.7.1.40), another enzyme detected in the MECs. In addition to these fates, PEP can undergo a β-carboxylation in the presence of HCO3− to yield inorganic phosphate (Pi) and oxaloacetate using Mg2+ as a cofactor. An enzyme able to govern this irreversible reaction, phosphoenolpyruvate carboxylase (EC 4.1.1.31), was detected in the CCs of M. ilicifolia. It is part of an essential anaplerotic reaction and is needed to replenish the tricarboxylic acid (TCA) cycle when organic acids are being used as building blocks for the synthesis other biomolecules (Voll et al. 2009). Another enzyme also found in this study (in the CCs), malate dehydrogenase (EC 1.1.1.37), also enables the formation of oxaloacetate, this time using (S)-malate as the substrate. In addition to malate dehydrogenase, in the CCs was detected another enzyme that also acts in the TCA cycle, a predicted protein with isocitric dehydrogenase (EC 1.1.1.41) activity.
In terms of the acetyl-CoA pathway, an enzyme capable of catalyzing one of the more important reactions within this route, chloroplast carboxyltransferase alpha subunit (acetyl-CoA carboxylase activity, EC 6.4.1.2) was found in the MECs. This enzyme catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, which is considered the first committed step of fatty acid biosynthesis in many organisms (Radakovits et al. 2010). Two more enzymes involved in fatty acids biosynthesis were detected in the MECs, delta(12) fatty acid dehydrogenase (EC 1.14.99.33) and stearoyl-[acyl-carrier-protein] 9-desaturase 4, chloroplastic (EC 1.14.19.2). The latter effects the induction of double bonds at specific positions in fatty acids of defined chain lengths (Cahoon et al. 1997).
Many other important enzymes for primary and secondary metabolism were detected in the cell cultures of M. ilicifolia in the present work, including difunctional riboflavin kinase/phosphatase FMN (EC 2.7.1.26 and EC 2.7.7.2), NADH dehydrogenase subunit F (EC 1.6.99.3) and alcohol dehydrogenase I (EC 1.1.1.1). Beyond them, several enzymes involved in other essential functions, such as the starch and sucrose metabolisms and synthesis of proteins, nucleotides, primordial amino acids, hormones and others.
Proteins related to stress
The proteome plays an important role in plant stress responses since proteins are directly involved in virtually all types of biotic or abiotic stress. Generally, stress factors induce profound alterations in the protein network covering signaling, energy metabolism, protein metabolism and several other biosynthetic pathways (Kosová et al. 2014). In the present work, according to the functional annotation of the two protein datasets of cell cultures of M. ilicifolia (CCs and MECs), 50 proteins were categorized into the GO term “response to stress” (Supplementary Table S1 and S2). For instance, in the CCs were detected CBL-interacting serine/threonine-protein kinase 7 (CIPK7) and 11 (CIPK11). Transcriptomic analysis have shown that the genes CIPK7 and CIPK11 are involved in the response to low temperature (cold stress) in several plants (Huang et al. 2011; Ren et al. 2014; Yang et al. 2015).
We also have found ten peroxidases (PODs, EC 1.11.1.7), nine and one in the CCs and MECs, respectively, which are included in the list of the most important antioxidant enzymes reported to respond to oxidative stress (Demidchik 2015). Even under normal environmental conditions, plants generate reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), superoxide anion radical (O2 ·−), hydroxyl radical (·OH) and singlet oxygen (1O2), and virtually all environmental stresses induce over-production and accumulation of these molecules. Plants have developed a sophisticated defense mechanism of enzymatic antioxidants to fight against elevated reactive oxygen species (ROS) levels and scavenge them from cells (Demidchik 2015). Other proteins that play important roles in plant stress also were found in the M. ilicifolia cells, including beta-insoluble isoenzyme cwinv1, nodulin 25 family protein, bifunctional riboflavin, kinase fmn hydrolase, acidic 27 kDa endochitinase precursor, alpha-partial, abc transporter g family member 22, peroxidase 70-like, and others (Supplementary Table S1 and S2).
Conclusion
This study showed the production in high concentration of the QMTs maytenin (1) and 22β-hydroxy-maytenin (2) in the cell culture of Maytenus ilicifolia, which was elicited using methyl jasmonate. In addition, the first proteomic report on the secondary metabolism of a Maytenus ilicifolia cell culture was obtained. The elicitation caused a large variation in the protein composition while still not fundamentally changing its profile at a functional level. In addition, we also observed that the secondary metabolic pathways of the cells were preserved after elicitation. Analyses of both cells, elicited and non-elicited, were complementary in the detection of enzymes involved in the QMT pathway and in the discovery of uncharacterized CYP450 enzymes that potentially catalyze the ultimate steps in the biosynthesis of the QMTs maytenin (1) and 22β-hydroxy-maytenin (2). In addition, a significant amount of information at the proteome level has been gathered and, taken together, offers interesting possibilities to empower natural product drug discovery through biotechnology techniques, including metabolic engineering.
References
Abe I (2007) Enzymatic synthesis of cyclic triterpenes. Nat Prod Rep 24:1311–1331. doi:10.1039/b616857b
Ahmed MS, Fong HHS, Soejarto DD et al (1981) High-performance liquid chromatographic separation and quantitation of maytansinoids in Maytenus ilicifolia. J Chromatogr A 213:340–344. doi:10.1016/S0021-9673(00)81919-6
Barros J, Serk H, Granlund I, Pesquet E (2015) The cell biology of lignification in higher plants. Ann Bot 115:1053–1074. doi:10.1093/aob/mcv046
Blakeley SD, Dekroon C, Cole KP et al (1994) Isolation of a full-length cDNA encoding cytosolic enolase from Ricinus communis. Plant Physiol 105:455–456. doi:10.1104/pp.105.1.455
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Buffa Filho W, Bolzani V da S, Furlan M et al (2004) In vitro propagation of Maytenus ilicifolia (Celastraceae) as potential source for antitumoral and antioxidant quinomethide triterpenes production. A rapid quantitative method for their analysis by reverse-phase high-performance liquid chromatograp. Arkivoc 2004:137–146. doi:10.3998/ark.5550190.0005.617
Cahoon EB, Lindqvist Y, Schneider G, Shanklin J (1997) Redesign of soluble fatty acid desaturases from plants for altered substrate specificity and double bond position. Proc Natl Acad Sci 94:4872–4877. doi:10.1073/pnas.94.10.4872
Camelio AM, Johnson TC, Siegel D (2015) Total synthesis of celastrol, development of a platform to access celastroid natural products. J Am Chem Soc 137:11864–11867. doi:10.1021/jacs.5b06261
Champagne A, Rischer H, Oksman-Caldentey K-M, Boutry M (2012) In-depth proteome mining of cultured Catharanthus roseus cells identifies candidate proteins involved in the synthesis and transport of secondary metabolites. Proteomics 12:3536–3547. doi:10.1002/pmic.201200218
Choi JY, Ramasamy T, Kim SY et al (2016) PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy. Acta Biomater 39:94–105. doi:10.1016/j.actbio.2016.05.012
Coppede J da S, Pina ES, Paz TA et al (2014) Cell cultures of Maytenus ilicifolia Mart. are richer sources of quinone-methide triterpenoids than plant roots in natura. Plant Cell Tissue Organ Cult 118:33–43. doi:10.1007/s11240-014-0459-7
Corsino J, De Carvalho PRF, Kato MJ et al (2000) Biosynthesis of friedelane and quinonemethide triterpenoids is compartmentalized in Maytenus aquifolium and Salacia campestris. Phytochemistry 55:741–748. doi:10.1016/S0031-9422(00)00285-5
de Souza LM, Cipriani TR, Iacomini M et al (2008) HPLC/ESI-MS and NMR analysis of flavonoids and tannins in bioactive extract from leaves of Maytenus ilicifolia. J Pharm Biomed Anal 47:59–67. doi:10.1016/j.jpba.2007.12.008
Demidchik V (2015) Mechanisms of oxidative stress in plants: from classical chemistry to cell biology. Environ Exp Bot 109:212–228. doi:10.1016/j.envexpbot.2014.06.021
Deng W-W, Zhang M, Wu J-Q et al (2013) Molecular cloning, functional analysis of three cinnamyl alcohol dehydrogenase (CAD) genes in the leaves of tea plant, Camellia sinensis. J Plant Physiol 170:272–282. doi:10.1016/j.jplph.2012.10.010
Dewick PM (1995) The biosynthesis of shikimate metabolites. Nat Prod Rep 12:579–607
Dewick PM (2009) The shikimate pathway: aromatic amino acids and phenylpropanoids. In: Medicinal natural products: a biosynthetic approach, 3rd edn. Wiley, pp 137–186
Diebold R, Schuster J, Däschner K, Binder S (2002) The branched-chain amino acid transaminase gene family in Arabidopsis encodes plastid and mitochondrial proteins. Plant Physiol 129:540–550. doi:10.1104/pp.001602
dos Santos VADFFM, Dos Santos DP, Castro-Gamboa I et al (2010) Evaluation of antioxidant capacity and synergistic associations of quinonemethide triterpenes and phenolic substances from Maytenus ilicifolia (celastraceae). Molecules 15:6956–6973. doi:10.3390/molecules15106956
dos Santos VFFM, Regasini LO, Nogueira CR et al (2012) Antiprotozoal sesquiterpene pyridine alkaloids from Maytenus ilicifolia. J Nat Prod 75:991–995. doi:10.1021/np300077r
dos Santos VAFFM, Leite KM, Da Costa Siqueira M et al (2013) Antiprotozoal activity of quinonemethide triterpenes from Maytenus ilicifolia (Celastraceae). Molecules 18:1053–1062. doi:10.3390/molecules18011053
Forsthoefel NR, Cushman MA, Cushman JC (1995) Posttranscriptional and post translational control of enolase expression in the facultative crassulacean acid metabolism plant Mesembryanthemum Crystallinum L. Plant Physiol 108:1185–1195. doi:10.1104/pp.108.3.1185
Fox TC, Mujer CV, Andrews DL et al (1995) Identification and gene expression of anaerobically induced enolase in Echinochloa phyllopogon and Echinochloa crus-pavonis. Plant Physiol 109:433–443. doi:10.1104/pp.109.2.433
Georgiev V, Marchev A, Berkov S, Pavlov A (2013) Plant in vitro systems as sources of tropane alkaloids. In: Ramawat GK, Mérillon J-M (eds) Natural products: phytochemistry, botany and metabolism of alkaloids, phenolics and terpenes. Springer, Berlin, pp 173–211
Götz S, García-Gómez JM, Terol J et al (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435. doi:10.1093/nar/gkn176
Gullo FP, Sardi JCO, Santos VAFFM et al (2012) Antifungal activity of maytenin and pristimerin. Evidence-Based Complement Altern Med 2012:1–6. doi:10.1155/2012/340787
Guo J, Carrington Y, Alber A, Ehlting J (2014) Molecular characterization of quinate and shikimate metabolism in Populus trichocarpa. J Biol Chem 289:23846–23858. doi:10.1074/jbc.M114.558536
Gutiérrez F, Estévez-Braun A, Ravelo ÁG et al (2007) Terpenoids from the medicinal plant Maytenus ilicifolia. J Nat Prod 70:1049–1052. doi:10.1021/np070019g
Harakava R (2005) Genes encoding enzymes of the lignin biosynthesis pathway in Eucalyptus Genet. Mol Biol 28:601–607. doi:10.1590/S1415-47572005000400015
Herrmann KM, Weaver LM (1999) The shikimate pathway. Annu Rev Plant Physiol Plant Mol Biol 50:473–503. doi:10.1146/annurev.arplant.50.1.473
Huang C, Ding S, Zhang H et al (2011) CIPK7 is involved in cold response by interacting with CBL1 in Arabidopsis thaliana. Plant Sci 181:57–64. doi:10.1016/j.plantsci.2011.03.011
Jardim ACG, Igloi Z, Shimizu JF et al (2015) Natural compounds isolated from Brazilian plants are potent inhibitors of hepatitis C virus replication in vitro. Antiviral Res 115:39–47. doi:10.1016/j.antiviral.2014.12.018
Kosová K, Vítámvás P, Prášil IT (2014) Proteomics of stress responses in wheat and barley—search for potential protein markers of stress tolerance. Front Plant Sci 5:14. doi:10.3389/fpls.2014.00711
Koutaniemi S, Toikka MM, Kärkönen A et al (2005) Characterization of basic p-coumaryl and coniferyl alcohol oxidizing peroxidases from a lignin-forming Picea abies suspension culture. Plant Mol Biol 58:141–157. doi:10.1007/s11103-005-5345-6
Kutsuki H, Shimada M, Higuchi T (1982) Regulatory role of cinnamyl alcohol dehydrogenase in the formation of guaiacyl and syringyl lignins. Phytochemistry 21:19–23. doi:10.1016/0031-9422(82)80006-X
Lal SK, Lee C, Sachs MM (1998) Differential regulation of enolase during anaerobiosis in maize. Plant Physiol 118:1285–1293. doi:10.1104/pp.118.4.1285
Lippmann R, Kaspar S, Rutten T et al (2009) Protein and metabolite analysis reveals permanent induction of stress defense and cell regeneration processes in a tobacco cell suspension culture. Int J Mol Sci 10:3012–3032. doi:10.3390/ijms10073012
Liu M-H, Yang B-R, Cheung W-F et al (2015) Transcriptome analysis of leaves, roots and flowers of Panax notoginseng identifies genes involved in ginsenoside and alkaloid biosynthesis. BMC Genomics 16:265. doi:10.1186/s12864-015-1477-5
Martínez-Esteso MJ, Martínez-Márquez A, Sellés-Marchart S et al (2015) The role of proteomics in progressing insights into plant secondary metabolism. Front Plant Sci 6:504. doi:10.3389/fpls.2015.00504
Messner B, Boll M (1993) Elicitor-mediated induction of enzymes of lignin biosynthesis and formation of lignin-like material in a cell suspension culture of spruce (Picea abies). Plant Cell Tissue Organ Cult 34:261–269. doi:10.1007/BF00029715
Mossi AJ, Mazutti M, Paroul N et al (2009) Chemical variation of tannins and triterpenes in Brazilian populations of Maytenus ilicifolia Mart. Ex Reiss. Braz J Biol 69:339–345. doi:10.1590/S1519-69842009000200015
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497. doi:10.1021/jf9040386
Negrel J, Javelle F (1995) Induction of phenylpropanoid and tyramine metabolism in pectinase- or pronase-elicited cell suspension cultures of tobacco (Nicotiana tabacum). Physiol Plant 95:569–574. doi:10.1111/j.1399-3054.1995.tb05524.x
Newman DJ, Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79:629–661. doi:10.1021/acs.jnatprod.5b01055
Oksman-Caldentey K-M, Inzé D (2004) Plant cell factories in the post-genomic era: new ways to produce designer secondary metabolites. Trends Plant Sci 9:433–440. doi:10.1016/j.tplants.2004.07.006
Oramas-Royo SM, Chávez H, Martín-Rodríguez P et al (2010) Cytotoxic triterpenoids from Maytenus retusa. J Nat Prod 73:2029–2034. doi:10.1021/np100517u
Pauwels L, Inzé D, Goossens A (2009) Jasmonate-inducible gene: what does it mean? Trends Plant Sci 14:87–91. doi:10.1016/j.tplants.2008.11.005
Paz TA, dos Santos VAFFM, Inácio MC et al (2013) Production of the quinone-methide triterpene maytenin by in vitro adventitious roots of Peritassa campestris (Cambess.) A.C. Sm. (Celastraceae) and rapid detection and identification by APCI-IT-MS/MS. Biomed Res Int 2013:485837. doi:10.1155/2013/485837
Pereira AMS, Moro JR, Cerdeira RMM, França SC (1994) Micropropagation of maytenus aquifolium martius. J Herbs Spices Med Plants 2:11–18. doi:10.1300/J044v02n03_03
Phillips DR, Rasbery JM, Bartel B, Matsuda SPT (2006) Biosynthetic diversity in plant triterpene cyclization. Curr Opin Plant Biol 9:305–314. doi:10.1016/j.pbi.2006.03.004
Pina ES, Silva DB, Teixeira SP et al (2016) Mevalonate-derived quinonemethide triterpenoid from in vitro roots of Peritassa laevigata and their localization in root tissue by MALDI imaging. Sci Rep 6:22627. doi:10.1038/srep22627
Plaxton WC (1996) The regulation and organization of plant glycolysis. Annu Rev Plant Physiol Plant Mol Biol 47:185–214. doi:10.1146/annurev.arplant.47.1.185
Radakovits R, Jinkerson RE, Darzins A, Posewitz MC (2010) Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell 9:486–501. doi:10.1128/EC.00364-09
Ralph J, Lundquist K, Brunow G et al (2004) Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem Rev 3:29–60. doi:10.1023/B:PHYT.0000047809.65444.a4
Ren L, Sun J, Chen S et al (2014) A transcriptomic analysis of Chrysanthemum nankingense provides insights into the basis of low temperature tolerance. BMC Genomics 15:844. doi:10.1186/1471-2164-15-844
Rizhsky L, Liang H, Shuman J et al (2004) When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol 134:1683–1696. doi:10.1104/pp.103.033431.1
Sasaki S, Baba K, Nishida T et al (2006) The cationic cell-wall-peroxidase having oxidation ability for polymeric substrate participates in the late stage of lignification of Populus alba L. Plant Mol Biol 62:797–807. doi:10.1007/s11103-006-9057-3
Shekhar S, Mishra D, Gayali S et al (2016) Comparison of proteomic and metabolomic profiles of two contrasting ecotypes of sweetpotato (Ipomoea batata L.). J Proteomics 143:306–317. doi:10.1016/j.jprot.2016.03.028
Spitzer-Rimon B, Marhevka E, Barkai O et al (2010) EOBII, a gene encoding a flower-specific regulator of phenylpropanoid volatiles’ biosynthesis in petunia. Plant Cell 22:1961–1976. doi:10.1105/tpc.109.067280
Su CF, Wang YC, Hsieh TH et al (2010) A novel MYBS3-dependent pathway confers cold tolerance in rice. Plant Physiol. doi:10.1104/pp.110.153015
Van Cutsem E, Simonart G, Degand H et al (2011) Gel-based and gel-free proteomic analysis of Nicotiana tabacum trichomes identifies proteins involved in secondary metabolism and in the (a)biotic stress response. Proteomics 11:440–454. doi:10.1002/pmic.201000356
Vogt T (2010) Phenylpropanoid biosynthesis. Mol Plant 3:2–20. doi:10.1093/mp/ssp106
Voll LM, Hajirezaei MR, Czogalla-Peter C et al (2009) Antisense inhibition of enolase strongly limits the metabolism of aromatic amino acids, but has only minor effects on respiration in leaves of transgenic tobacco plants. New Phytol 184:607–618. doi:10.1111/j.1469-8137.2009.02998.x
Weaver LM, Herrmann KM (1997) Dynamics of the shikimate pathway in plants. Trends Plant Sci 2:346–351. doi:10.1016/S1360-1385(97)84622-5
Wendt KU (2005) Enzyme mechanisms for triterpene cyclization: new pieces of the puzzle. Angew Chemie Int Ed 44:3966–3971. doi:10.1002/anie.200500804
Weng J-K, Chapple C (2010) The origin and evolution of lignin biosynthesis. New Phytol 187:273–285. doi:10.1111/j.1469-8137.2010.03327.x
Wildermuth MC, Dewdney J, Wu G, Ausubel FM (2001) Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 414:562–565. doi:10.1038/417571a
Yamazaki M, Mochida K, Asano T et al (2013) Coupling deep transcriptome analysis with untargeted metabolic profiling in Ophiorrhiza pumila to further the understanding of the biosynthesis of the anti-cancer alkaloid camptothecin and anthraquinones. Plant Cell Physiol 54:686–696. doi:10.1093/pcp/pct040
Yang Q-S, Gao J, He W-D et al (2015) Comparative transcriptomics analysis reveals difference of key gene expression between banana and plantain in response to cold stress. BMC Genomics 16:446. doi:10.1186/s12864-015-1551-z
Yue W, Ming Q-L, Lin B et al (2016) Medicinal plant cell suspension cultures: pharmaceutical applications and high-yielding strategies for the desired secondary metabolites. Crit Rev Biotechnol 36:215–232. doi:10.3109/07388551.2014.923986
Zhao J, Davis LC, Verpoorte R (2005) Elicitor signal transduction leading to production of plant secondary metabolites. Biotechnol Adv 23:283–333. doi:10.1016/j.biotechadv.2005.01.003
Acknowledgements
We thank the São Paulo Research Foundation (FAPESP) for the CIBFar-2013/07600-3 grant. T. A. Paz thanks CAPES for the provision of a scholarship. V. A. F. F. M. dos Santos and M. C. Inácio thank FAPESP for their fellowships (2011/01544-9 and 2014/19362-2, respectively). V. A. F. F. M. dos Santos is also grateful to the National Council for Scientific and Technological Development (CNPq) for her fellowship. M. Furlan and M. S. Palma would also like to thank CNPq for their research fellowships.
Author contributions
TAP was responsible for the conception and design of all experiments, data analysis, drafting and editing of the manuscript. MCI contributed with plant cell experiments and revision of the manuscript. VAFFMS contributed with proteomic experiments. NBD and MSP contributed with proteomic experiments and revised the manuscript. AMSP supervised part of experiments and contributed to data interpretation and final revision of the manuscript. MF contributed to the experimental design, to data interpretation, drafting and final revision of the manuscript, and supervised all experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Ali R. Alan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Paz, T.A., dos Santos, V.A.F.F.M., Inácio, M.C. et al. Proteome profiling reveals insights into secondary metabolism in Maytenus ilicifolia (Celastraceae) cell cultures producing quinonemethide triterpenes. Plant Cell Tiss Organ Cult 130, 405–416 (2017). https://doi.org/10.1007/s11240-017-1236-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-017-1236-1