
Abstract
Genetic risk factors are important for the occurrence and prognosis of venous thromboembolism (VTE). The studies of thrombophilia families are important for dissecting the genetic background of the thrombotic disease. We conducted the systematic review of all published family-based studies on VTE genetics across all racial groups through PubMed and Embase prior to 13th April 2020. This systematic review of 287 families (including 225 Caucasian families, 52 East Asian families, and families of other ethnicities) revealed a total of 21 different genes; the five most reported mutated genes were F5 (88/287, 30.7%), SERPINC1 (67/287, 23.3%), PROC (65/287, 22.6%), F2 (40/287, 13.9%) and PROS1 (48/287, 16.7%). For Caucasian families, F5 mutations were most frequently reported at 37.8% (85/225), while PROS1 mutations were most frequently reported, at 40.4% (21/52), for East Asian families (Chinese, Japanese and Korean). Factor V Leiden was reported more frequently in Caucasians than in East Asians. Missense mutations were reported frequently in the SERPINC1, PROC and PROS1 genes. In conclusion, our study found the most likely mutated genes associated with VTE among different ethnic groups and provided indications for VTE genetic testing and research in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Highlights
-
This is the first systematic review of all published family-based studies on venous thromboembolism (VTE) genetics of all racial groups.
-
This study concluded that F2, F5, SERPINC1, PROC and PROS1 were the most frequently identified genes in VTE families.
-
For East Asian families (China, Japan and Korea), the mutations of PROS1 were the most frequently reported than other genes. For Caucasian families, however, mutations of F5 were revealed to be the most frequent compared to other genes.
-
Our review analyzed the type and prevalence of gene mutations associated with VTE among different ethnical groups, therefore, provided useful genetic information for future testing and research on VTE. Large samples may be needed for more accurate and comprehensive conclusion.
Introduction
Venous thromboembolism (VTE), comprising pulmonary thromboembolism (PTE) and deep venous thrombosis (DVT), has become a major global health problem such as stroke and myocardial infarction [1]. The global disease burden of VTE has increased steadily over the last decade, with approximately 75–269 patients per 100,000 individuals in the United States and Europe [2] and 10–30 patients per 100,000 individuals in China [3].
VTE is well-known as a multifactorial disease, but predisposing genetic factors play an important role in the occurrence and prognosis of the disease [4]. The genetic burden of VTE is characterized by a sibling relative risk of 2.5 and a strong heritability estimated from 35 to 60%, according to various studies [5].
VTE gene variations that strongly affect the VTE risk are rarely reported [5]. Notably, most causal gene mutations associated with the risk of VTE were detected in family-based studies. Moreover, family studies are generally more powerful than case–control studies as the inheritance pattern among families implies that genetics play a more important role in the pathogenesis of VTE [6].
Therefore, we conducted this comprehensive review of all published family-based studies on VTE genetics of all racial groups, aiming to identify the mutation patterns of VTE-related genes among different ethnic groups and to provide a foundation for the diagnosis of and research related to VTE. To our knowledge, our analysis is one of the most comprehensive findings on the genetics of VTE through the family-based study to date.
Method
Search strategy
We searched PubMed, Embase for articles published in English until 13th April 2020. The following search terms and their similar terms were used: “thrombosis”, “mutation” and “family”(Detailed search strategy in Supplementary material). The systematic review was conducted following the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement.
Selection criteria
Titles and abstracts of potentially relevant family studies were checked independently by two investigators (Yu Zhang and Zhu Zhang) for eligibility of full paper evaluation. For a conflicting evaluation, a third reviewer (Shi Shu) was consulted and a consensus was reached by discussion. Studies of all genetic backgrounds were included.
Identified studies satisfied the following criteria: (1) evaluating any candidate gene defect in association with VTE; (2) family studies (at least two VTE patients in one family); (3) containing sufficient information on clinical features and VTE gene mutations. The major reasons for exclusion of studies were as follows: non-English publications, reviews, duplicate publications, inherited deficiency confined to proteins levels.
Data extraction
The information was carefully extracted from all eligible literature independently by Yu Zhang and Zhu Zhang. For each selected study, information was extracted mainly on genes, genotyping method, mutation information, ethnicity, the number of families.
Result
Study selection and characteristics
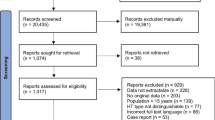
Using our search strategies, 220 potentially relevant studies were initially selected (Fig. 1). On applying the inclusion and exclusion criteria, we identified 150 studies eligible for this analysis. In addition, four more studies were manually added to our research. A total of 154 studies involving 287 families with VTE were included (225 Caucasian families, 52 East Asian families, four Arab families, two Latino families, two African families, and others; Table 1; detailed data is shown in Supplementary Table 1.1 to Supplementary Table 1.7). However, few studies have reported on clear ancestries (Supplementary Table 2). Thus, the ancestry of most families is assumed based on the country of origin. A total of 21 genes associated with VTE (Table 2) were reported among the families included in this study. The five most frequently reported genes were: F5 (88/287, 30.7%), SERPINC1 (67/287, 23.3%), PROC (65/287, 22.6%), F2 (40/287, 13.9%), PROS1 (48/287, 16.7%; Table 3). Moreover, MTHFR polymorphisms have been reported in 26 families. SERPINE1, FGA, THBD and FGB gene defects were also reported in 9, 5, 4, and 3 unrelated families, respectively. The MTHFR C677T and plasminogen activator inhibitor-1 (PAI-1) 4G/5G polymorphisms were reported to be associated with VTE only in combination with other genetic defects. The other genes had only been reported in one or two families. The mechanisms underlying the mutations that lead to the development of thrombophilia are shown in Fig. 2.

Study flow diagram. Using our search strategies, 220 potentially relevant studies were initially identified. After applying the inclusion and exclusion criteria, 150 studies were identified. In addition, we added four studies manually. A total of 154 studies involving 287 families with VTE were included. VTE venous thromboembolism

The genes that reported in the family studies lead to thrombophilia through different pathways. Mutations in FGG, FGA and FGB genes cause dysfibrinogenemia, leading to the development of thrombophilia. Mutations in F2, F5, F12, and SERPINC1 result in the development of thrombophilia through the activation of the coagulation cascade. Mutations in of GP1BA and PEAR1 affect platelet activation, resulting in the development thrombophilia. Mutations in PROC, PROS, and THBD lead to the development of thrombophilia through the protein C system. Mutations in SERPINE1 affect fibrinolysis, leading to the development of thrombophilia.
The reported number of families with thrombophilia is smaller among the Asian populations than among the Caucasians. There are differences in the type and frequency of gene mutations between these two ethnic groups. For East Asian families (Chinese, Japanese and Korean), we found the defect of PROS1 at 40.4% (21/52), PROC at 21.2% (11/52), SERPINC1 at 19.2% (10/52), and F2 at 15.4% (8/52). Only one family from China was identified with a novel mutation of p.Glu666Asp in F5 [7]. However, for Caucasian families, the mutations in F5 were most frequently reported at 38.2% (86/225), followed by SERPNC1 at 24.0% (54/225), while PROS1 mutations were least commonly reported at 11.6% (26/225; Table 3).
F2
The gene F2 encodes prothrombin. A total of eight different mutations in six loci of F2 were reported in 40 VTE families according to our review. The F2 G20210A mutation (rs1799963, variation in the 3′ UTR region of the gene) is a well characterized mutation associated with a rather strong risk of VTE. In this review, the G20210A mutation was the most frequently reported mutation, with an incidence of 62.5% (25/40); among the families presenting this mutation, 22 families were Caucasian, one family was Colombian [8], one family was Puerto Rican [9] and one family without clear ancestry. Other missense mutations have also been reported in Caucasian families—a substitution of arginine for glutamine (p.Arg596Gln, also called prothrombin Belgrade) in four Serbian families [10,11,12], a substitution of arginine for tryptophan (p.Arg596Trp) in an Italian family [13] and a substitution of arginine for tryptophan (p.Arg173Trp) in a Dutch family [14]. Arginine at position 596 is an important site for the regulation of enzyme activity, and mutation at this site leads to a decrease in the antithrombin (AT) binding ability, resulting in the risk of thrombosis [15]. Interestingly, only eight Asian families were reported with missense mutations of F2, with G20210A has not been reported to date. Three Japanese families presented with variations at position 596; of these, two alterations were the missense mutation of p.Arg596Gln [16, 17] and one was a missense mutation of c.1787G > T, p.Arg596Leu [18]. Two rare F2 mutations—c.494C > T, p.Thr165Met and c.1274G > A, p.Arg425His—were detected in two Chinese families [19, 20]. In addition, a recent study detected a missense mutation—p.Arg541Trp—in three Chinese families [21].
F5
Factor V Leiden mutation (FVL, R506Q, rs6025) is probably the most well-known gain-of-function variation associated with VTE. It was reported by Leiden team that the majority of the patients with familial activated protein C resistance (APCR) had the same mutation, a guanine to adenine transition at nucleotide 1691 in exon 10 of F5 [22]. In our review, FVL was the most reported mutation in VTE families, with an incidence of 30.0% (86/287). Among the 86 families with FVL, 85 were of Caucasian origin, and one was of Arab origin [23]. FVL has never been reported in Asian families. Cai et al. has identified a novel mutation—heterozygous G → C transversion at base2172—changing the codon for amino acid 666 from GAG (glutamate, E) to GAC (aspartate, D) in F5, which was associated with APCR in a Chinese family with VTE, and was reported for the first time [7]. However, in the following study, the FV E666D mutation was not detectable in any of the 163 VTE patients, including 6 APCR-positive patients [24]. In a Spanish family, FVL was detected in combination with another mutation in F5 (G1091C, Arg306Thr, FV Cambridge, FVC) [25]. This mutation is rare and is associated with pathological levels of APCR. The association of the mutation with the risk of VTE is still unclear [26]. In an Italian family FVL was detected in combination with two other mutations in F5 (p.His1299Arg and p.Tyr1702Cys) [27]. FV H1299R is marked by an A → G transition at position 4070, resulting in a transition from histidine to arginine. Carriership of the FV H1299R allele is associated with mild APCR and with a relative excess of the more thrombogenic FV isoform in plasma [28]. Therefore, this mutation increases the risk of venous thrombosis in carriers of the FVL mutation [29]. The newly identified missense mutation, FV Y1702C (changing amino acid 1702 from tyrosine to cysteine), which leads to FV deficiency, may enhance APCR [27].
SERPINC1
The gene SERPINC1 encodes antithrombin. In the present review, a total of 57 different mutations in SERPINC1 were found among 67 VTE families, of which 54 were Caucasian and ten were East Asian (six Chinese [10, 30,31,32], three Japanese [33,34,35] and one Korean [36]). Among the 57 mutations, 38 are missense mutations, and others are indel, nonsense, or frameshift mutations. AT Cambridge II (p.Ala384Ser) was detected in four Caucasian VTE families, two from England [37,38,39]. This is a G → T substitution at nucleotide position 13,268 (GCA → TCA), resulting in the replacement of the normal alanine residue at position 384 by serine and the synthesis of a dysfunctional AT with normal heparin affinity but a reduction in anti-IIa activity. In addition, four families were detected with a replacement of arginase at position 393 (p.Arg393His in two families [35, 40], p.Arg393Cys [41] and p.Arg393Pro [42] in one family, respectively).
PROC
PROC encodes protein C (PC). Inherited PC deficiency is transmitted as a dominant autosomal trait. According to our review, 57 different mutations in PROC were reported among 65 families (52 Caucasian families, 11 East Asian families). In a Portuguese study, five different PROC mutations were identified in eight unrelated VTE families with reported low/borderline PC plasma levels [43]. The most frequent mutation found in this study was a nonsense mutation of p.Arg199X, detected in four families containing patients with higher numbers of VTE episodes. In a Hungarian family study, a nonsense mutation, a rare frameshift deletion, and nine different missense mutations were determined in PROC [44]. Furthermore, a Dutch family study reported that over 50% (8/15) of Dutch VTE families with PC deficiency share a founder R272C mutation [45]. Eight Chinese families [31, 46,47,48,49,50,51,52], two Japanese families [53, 54], and one Korean family [55] with PROC mutations were included in the present review.
In our review, the p.Arg230Cys mutation was found in two Hungarian families [44]. This mutation was reported in the PC mutation database in 1995 and was a common genetic alteration noted among Hungarian patients [44]. The p.Thr298Met mutation was detected in two Caucasian families [56, 57] and an Arab family [23]; this mutation had previously been reported in PC deficiency families. A missense mutation, p.Arp169Trp, has also been reported in two Caucasian families [56, 58] and a Japanese family [53]. This mutation abolishes the site of thrombin cleavage that activates PC [59]. Finally, a Arg → Gln mutation was also detected in the position 169 in an English family [58].
PROS1
PROS1 encodes protein S (PS). Inherited PS deficiency is transmitted as a dominant autosomal trait. According to our review, a total of 46 different mutations in PROS1 were reported among 48 families (26 Caucasian families, 21 Asian families, and one African American family). A Chinese study identified 53 unrelated families with PS deficiency, but only 14 families with at least two VTE patients were included in the present review. The mutation p.Glu67Ala in PROS1 was reported in three families [31]. We also included nine families with at least two VTE patients from a Spanish study [60]. Among the nine families, the mutation p.Gln238X was reported in three families. The p.Met570Thr mutation in PROS1 was also identified in three families [58, 61]. The Met570Thr mutation may possibly be associated with PS deficiency [62]. Additional in vitro experiment may establish whether or not this mutation is associated with VTE. No mutation was found in either the Caucasian or Asian families.
Other genes
MTHFR
The MTHFR C677T polymorphism was identified and compounded with other gene defects in 26 Caucasian families.
SERPINE1
In a family with thrombotic disease, an 18-base pair deletion in the PAI-1 gene (SERPINE1) promoter region was identified [63]. Of the5of 11 family members experiencing thrombosis, four were heterozygous for the deletion variant. PAI-1 4G/5G polymorphism in combination with other gene defects was identified in nine VTE families.
THBD
A study reported thrombomodulin gene defects in four families from the United States with thromboembolic disease, and four heterozygous point mutations were detected [64].
FGA, FGB, FGG
Eight families were reported with fibrinogen variants [65,66,67,68,69,70,71,72]. In a Kurdish family with dysfibrinogenemia and familial venous and arterial thrombosis, a novel mutation c.221G > T in exon 2 of FGB was detected [65]. Another study used exome sequencing to identify a mutation of Arg458Cys in FGA in a family with recurrent VTE that remained undiagnosed for many years [66].
F12
F12 C67T was detected in three families with multiple genetic alterations [25, 37, 73].
GP1BA, PEAR1
Two Chinese families were detected with a mutation in GP1BA and PEAR1 respectively [74, 75]. These two genes play a role in platelet disorder, but whether these mutations increase the risk of VTE remains unclear.
Discussion
We reviewed a total of 154 published studies containing 287 VTE families (225 Caucasian families, 52 East Asian families, four Arab families, two Latino families, two African families and others) for gene defects associated with the disease. More Caucasian families (225/287) were reported to have VTE than Asian families (52/287). A reason may be that the incidence of VTE is much higher in the Caucasian population (75–269 patients per 100,000 individuals in the United States and Europe [2] vs 10–30 patients per 100,000 individuals in China [3]). Another reason may be the lack of awareness and research on VTE in Asia, leading to many Asian families having not been identified and reported in the literature. Although it has been suggested that VTE risk may be five times higher in African families than in Asian-ancestry populations [76], the present review only included two African families as some studies did not provide sufficient information on ethnicities, such as African, Hispanic, or other populations.
The prevalence of thrombophilia in Caucasian and East Asian families is shown in Table 3. FVL was detected in most Caucasian families included in the present review, while F2 G20210A was also detected in a large proportion, which should be due to the high prevalence of these two common variants in Caucasians [77, 78]. However, F5 defects were the least reported among the five genes in East Asian families. In contrast, the PROS1 defects were the most reported in East Asian families (21/52, 40.4%) while being the least identified defects in Caucasian people (26/225, 11.6%) among the five genes (Table 3). Among these genetic variants, there is a clear inverse relationship between prevalence and strength [79]. Deficiencies of natural anticoagulants are rare but increase VTE risk by up to tenfold while common variants, which include FVL and F2 G20210A, with a prevalence of several percent increasing VTE risk twofold to fivefold [80]. We collected the number of carriers with VTE in every family and calculated the penetrance as carriers with VTE/carriers. We come to a similar conclusion that the strength of natural anticoagulants deficiency (SERPINC1 [0.709], PROC [0.569], PROS1 [0.706]) is stronger than the two common mutations (FVL [0.46], G20210A [0.417]) (Supplementary Table 3). In brief, our review identifies the type and frequency of gene mutations associated with VTE among different ethnic groups; therefore, it provides useful genetic information for future testing and research on VTE in family pedigrees. Furthermore, it seems that the deficiency of anticoagulant proteins plays an important role in thrombophilia or the etiology of VTE.
There have been a number of Genome Wide Association Studies (GWAS) studies in VTE since 2009. Until now, GWAS conducted in Caucasian patients not only have confirmed known susceptible genes found in previous studies (F5, F2, ABO, FGG/FGB/FGA, PROCR), but also identified novel genes such as TSPAN15, SLC44A2, and ZFPM2. In a recent GWAS study, 14 newly reported associated loci have been found [81]. However, it should be noted that apart from the high VTE-risk SNPs (single nucleotide polymorphisms) identified by candidate gene studies, most of the genetic variants reported by GWAS only show moderate-to-low-effect sizes. Interestingly, some of the novel genes such as TSPAN15 and ZFPM2 have no role in the coagulation system, which indicates that there may be other mechanisms contributing to VTE pathogenesis.
For most of the family studies, polymerase chain reaction (PCR) gene sequencing was used with clear targets. However, it also means sequencing was solely performed in a single selected gene, which causes other gene polymorphisms to be omitted. Whole exome sequencing (WES) has also proved its utility in VTE genetic research over the years. In WES-family studies, novel genes and novel mutations were easier to find but more difficult to prove to be causative. In WES-family studies, all polymorphisms in exons will be detected, however, introns will still be missed. None of the family studies used whole-genome sequencing. In addition, there is controversy regarding testing for thrombophilia as only some selected patients can benefit from accurate testing and individualized management, which has been well-discussed in recent reviews [82, 83]. However, it is argued that with the increasing availability of direct-to-consumer diagnostics and personalized genomic testing, we may focus on patients with identified thrombophilia rather than on the indications for undertaking tests [83].
Limitations and potential clinical implications
We have firstly made a comparison of the prevalence of five classic genes among Caucasian and East Asian VTE families. The main limitation is the lack of knowledge of ancestry of most families, so we assume ancestry based on the country of origin. Some studies that we assumed as Caucasian could be African, Hispanic, or other population in reality. Another limitation may be the underestimation of SERPINC1, PROC, and PROS1. Many VTE families with these protein deficiencies were not included in our review because of a lack of genetic research or only contain a VTE proband without symptomatic relatives, so the contribution of these three genes may be underestimated, especially in Caucasian families. Although more novel mutations have been discovered, few have undergone in-depth research. In addition, family studies may not have identified the correct mutations. The restriction of technology really limits our knowledge of how fully we have cataloged the causal mutations and their penetrance or segregation in families. In short, our results can provide indications for future VTE genetic research, but they are only based on the sequencing data of family studies. Further, it is important for clinicians to understand the genetic diversity of VTE and improve the individualized management of thrombophilia patients.
Conclusion
This review of 287 families revealed a total of 21 different genes, among which FVL was the most reported mutation in VTE families. F2, F5, SERPINC1, PROC and PROS1 were the most frequently reported genes associated with VTE. Moreover, we found that there are differences in the prevalence of individual gene mutations among different ethnic groups, providing indications for future VTE genetic testing and research. Due to the lack of genetic data from Asian countries for this study, more samples of Asian families with genetic information from increasing the application of next-generation sequencing will be needed for future investigation in order to reach a more accurate conclusion.
Abbreviations
- APCR:
-
Activated protein C resistance
- AT:
-
Antithrombin
- DVT:
-
Deep venous thrombosis
- FVL:
-
Factor V Leiden
- GWAS:
-
Genome wide association study
- PAI-1:
-
Plasminogen activator inhibitor-1
- PC:
-
Protein C
- PCR:
-
Polymerase chain reaction
- PS:
-
Protein S
- PTE:
-
Pulmonary thromboembolism
- SNPs:
-
Single nucleotide polymorphisms
- VTE:
-
Venous thromboembolism
- WES:
-
Whole exome sequencing
- IIa:
-
Activated FactorII
- Va:
-
Activated FactorV
- VIIa:
-
Activated FactorVII
- Xa:
-
Activated FactorX
References
Shahi A, Chen AF, Tan TL, Maltenfort MG, Kucukdurmaz F, Parvizi J (2017) The incidence and economic burden of in-hospital venous thromboembolism in the United States. J Arthroplast 32(4):1063–1066. https://doi.org/10.1016/j.arth.2016.10.020
Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, Ozaki Y, Wendelboe A, Weitz JI, Day ISCfWT (2014) Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol 34(11):2363–2371. https://doi.org/10.1161/ATVBAHA.114.304488
Zhang Z, Lei J, Shao X, Dong F, Wang J, Wang D, Wu S, Xie W, Wan J, Chen H, Ji Y, Yi Q, Xu X, Yang Y, Zhai Z, Wang C, China Venous Thromboembolism Study G (2019) Trends in hospitalization and in-hospital mortality from VTE, 2007 to 2016, in China. Chest 155(2):342–353. https://doi.org/10.1016/j.chest.2018.10.040
Morange PE, Suchon P, Tregouet DA (2015) Genetics of venous thrombosis: update in 2015. Thromb Haemost 114(5):910–919. https://doi.org/10.1160/TH15-05-0410
Tregouet DA, Morange PE (2018) What is currently known about the genetics of venous thromboembolism at the dawn of next generation sequencing technologies. Br J Haematol 180(3):335–345. https://doi.org/10.1111/bjh.15004
Dandine-Roulland C, Perdry H (2015) Where is the causal variant? On the advantage of the family design over the case–control design in genetic association studies. Eur J Hum Genet 23(10):1357–1363. https://doi.org/10.1038/ejhg.2014.284
Cai H, Hua B, Fan L, Wang Q, Wang S, Zhao Y (2010) A novel mutation (g2172-->c) in the factor V gene in a Chinese family with hereditary activated protein C resistance. Thromb Res 125(6):545–548. https://doi.org/10.1016/j.thromres.2010.02.009
Roman-Gonzalez A, Cardona H, Cardona-Maya W, Alvarez L, Castaneda S, Martinez J, Torres JD, Tobón L, Bedoya G, Cadavid A (2009) The first homozygous family for prothrombin G20210A polymorphism reported in Latin America. Clin Appl Thromb Hemost 15(1):113–116. https://doi.org/10.1177/1076029608325049
Morales-Borges RH (2012) Autosomal-dominant inheritance of the prothrombin gene mutation in a Puerto Rican family: a case study. P R Health Sci J 31(4):232–234
Miljic P, Gvozdenov M, Takagi Y, Takagi A, Pruner I, Dragojevic M, Tomic B, Bodrozic J, Kojima T, Radojkovic D, Djordjevic V (2017) Clinical and biochemical characterization of the prothrombin Belgrade mutation in a large Serbian pedigree: new insights into the antithrombin resistance mechanism. J Thromb Haemost 15(4):670–677. https://doi.org/10.1111/jth.13618
Djordjevic V, Kovac M, Miljic P, Murata M, Takagi A, Pruner I, Francuski D, Kojima T, Radojkovic D (2013) A novel prothrombin mutation in two families with prominent thrombophilia—the first cases of antithrombin resistance in a Caucasian population. J Thromb Haemost 11(10):1936–1939. https://doi.org/10.1111/jth.12367
Valentina D, Kovac M, Pruner I, Francuski D, Radojkovic D (2013) A novel prothrombin c.1787G>A mutation in Serbian family with recurrent thromboembolism-another case of antithrombin resistance. J Thromb Haemost 11:374
Bulato C, Radu CM, Campello E, Gavasso S, Spiezia L, Tormene D, Simioni P (2016) New prothrombin mutation (Arg596Trp, prothrombin Padua 2) associated with venous thromboembolism. Arterioscler Thromb Vasc Biol 36(5):1022–1029. https://doi.org/10.1161/ATVBAHA.115.306914
Mulder R, Lisman T, Meijers JCM, Huntington JA, Mulder AB, Meijer K (2019) Linkage analysis combined with whole exome sequencing identifies a novel prothrombin (F2) gene mutation in a Dutch Caucasian family with unexplained thrombosis. Haematologica. https://doi.org/10.3324/haematol.2019.232504
Miyawaki Y, Suzuki A, Fujita J, Maki A, Okuyama E, Murata M, Takagi A, Murate T, Kunishima S, Sakai M, Okamoto K, Matsushita T, Naoe T, Saito H, Kojima T (2012) Thrombosis from a prothrombin mutation conveying antithrombin resistance. N Engl J Med 366(25):2390–2396. https://doi.org/10.1056/NEJMoa1201994
Yoshida R, Seki S, Hasegawa J, Koyama T, Yamazaki K, Takagi A, Kojima T, Yoshimura M (2018) Familial pulmonary thromboembolism with a prothrombin mutation and antithrombin resistance. J Cardiol Cases 17(6):197–199. https://doi.org/10.1016/j.jccase.2018.02.001
Kishimoto M, Suzuki N, Murata M, Ogawa M, Kanematsu T, Takagi A, Kiyoi H, Kojima T, Matsushita T (2016) The first case of antithrombin-resistant prothrombin Belgrade mutation in Japanese. Ann Hematol 95(3):541–542. https://doi.org/10.1007/s00277-015-2533-6
Miyawaki Y, Suzuki A, Fujimori Y, Fujita J, Maki A, Takagi A, Murate T, Sakai M, Okamoto K, Matsushita T, Kojima T (2011) A novel prothrombin gene mutation leads to an atresistant thrombin in a family with inherited thrombophilia. J Thromb Haemost 9:162. https://doi.org/10.1111/j.1538-7836.2011.04380_1.x
Sun G, Jia Y, Meng J, Ou M, Zhu P, Cong S, Luo Y, Sui W, Dai Y (2017) A genetic risk factor for thrombophilia in a Han Chinese family. Mol Med Rep 15(4):1668–1672. https://doi.org/10.3892/mmr.2017.6217
Wang L, Liu JM, Rong J, Han R, Zhao Q, Gong S, He J (2017) Thrombosis associated with congenital prothrombin deficiency: a severe procoagulant defect contrasting with thrombosis in a congenital prothrombin deficient family. Am J Respir Crit Care Med. https://doi.org/10.1164/ajrccmconference.2017.C57
Wu X, Dai J, Xu X, Li F, Li L, Lu Y, Xu Q, Ding Q, Wu W, Wang X (2020) Prothrombin Arg541Trp mutation leads to defective PC (protein C) pathway activation and constitutes a novel genetic risk factor for venous thrombosis. Arterioscler Thromb Vasc Biol 40(2):483–494. https://doi.org/10.1161/ATVBAHA.119.313373
Voorberg J, Roelse J, Koopman R, Buller H, Berends F, ten Cate JW, Mertens K, van Mourik JA (1994) Association of idiopathic venous thromboembolism with single point-mutation at Arg506 of factor V. Lancet (London, England) 343(8912):1535–1536
Brenner B, Zivelin A, Lanir N, Greengard JS, Griffin JH, Seligsohn U (1996) Venous thromboembolism associated with double heterozygosity for R506Q mutation of factor V and for T298M mutation of protein C in a large family of a previously described homozygous protein C-deficient newborn with massive thrombosis. Blood 88(3):877–880
Xin-Guang C, Yong-Qiang Z, Shu-Jie W, Lian-Kai F, Hua-Cong C (2015) Prevalence of the factor V E666D mutation and its correlation with activated protein C resistance in the Chinese population. Clin Appl Thromb Hemost 21(5):480–483. https://doi.org/10.1177/1076029613514130
Santamaría A, Soria JM, Tirado I, Mateo J, Coll I, Souto JC, Fontcuberta J (2005) Double heterozygosity for factor V Leiden and factor V Cambridge mutations associated with low levels of activated protein C resistance in a Spanish thrombophilic family. Thromb Haemost 93(6):1193–1195
Norstrom E, Thorelli E, Dahlback B (2002) Functional characterization of recombinant FV Hong Kong and FV Cambridge. Blood 100(2):524–530. https://doi.org/10.1182/blood-2002-02-0343
Castoldi E, Simioni P, Kalafatis M, Lunghi B, Tormene D, Girelli D, Girolami A, Bernardi F (2000) Combinations of 4 mutations (FV R506Q, FV H1299R, FV Y1702C, PT 20210G/A) affecting the prothrombinase complex in a thrombophilic family. Blood 96(4):1443–1448
Castoldi E, Rosing J, Girelli D, Hoekema L, Lunghi B, Mingozzi F, Ferraresi P, Friso S, Corrocher R, Tans G, Bernardi F (2000) Mutations in the R2 FV gene affect the ratio between the two FV isoforms in plasma. Thromb Haemost 83(3):362–365
Faioni EM, Franchi F, Bucciarelli P, Margaglione M, De Stefano V, Castaman G, Finazzi G, Mannucci PM (1999) Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 94(9):3062–3066
Peng Y, Wang T, Zheng Y, Lian A, Zhang D, Xiong Z, Hu Z, Xia K, Shu C (2019) A novel variation of SERPINC1 caused deep venous thrombosis in a Chinese family: a case report. Medicine 98(1):e13999. https://doi.org/10.1097/MD.0000000000013999
Li L, Wu X, Wu W, Ding Q, Cai X, Wang X (2019) Clinical manifestation and mutation spectrum of 53 unrelated pedigrees with protein S deficiency in China. Thromb Haemost 119(3):449–460. https://doi.org/10.1055/s-0038-1677031
Zhu R, Cao Z, Wu W (2019) Inherited antithrombin III deficiency: a case report of familial pedigree and gene mutation screening. Eur J Vasc Endovasc Surg 58(6):e379. https://doi.org/10.1016/j.ejvs.2019.06.1011
Maruyama K, Morishita E, Karato M, Kadono T, Sekiya A, Goto Y, Sato T, Nomoto H, Omi W, Tsuzura S, Imai H, Asakura H, Ohtake S, Nakao S (2013) Antithrombin deficiency in three Japanese families: one novel and two reported point mutations in the antithrombin gene. Thromb Res 132(2):e118–e123. https://doi.org/10.1016/j.thromres.2013.06.001
Niiya K, Kiguchi T, Dansako H, Fujimura K, Fujimoto T, Iijima K, Tanimoto M, Harada M (2001) Two novel gene mutations in type I antithrombin deficiency. Int J Hematol 74(4):469–472. https://doi.org/10.1007/bf02982095
Okajima K, Abe H, Wagatsuma M, Okabe H, Takatsuki K (1995) Antithrombin III kumamoto II; a single mutation at Arg393-His increased the affinity of antithrombin III for heparin. Am J Hematol 48(1):12–18. https://doi.org/10.1002/ajh.2830480104
Jang MJ, Lee JG, Chong SY, Huh JY, Jang MA, Kim HJ, Oh D (2011) A novel splice-site mutation c.42-2A>T (IVS1-2A>T) of SERPINC1 in a Korean family with inherited antithrombin deficiency. Blood Coagul Fibrinolysis 22(8):742–745. https://doi.org/10.1097/MBC.0b013e32834a7e17
Carrasco Exposito M, Tirado Garcia I, Romero RL, Vilalta Seto N, Mateo Arranz J, Millon Caño JA, Fontcuberta Boj J (2017) Extensive molecular characterization of a family with thrombophilia: implication of multiple genetic alterations of low thrombotic risk profile. Res Pract Thromb Haemost 1:1141. https://doi.org/10.1002/rth2.12012
Orlando C, Jochmans K, Lissens W, Liebaers I, De Waele M (2009) Hereditary antithrombin deficiency caused by heterozygous Cambridge II mutation in combination with a large gene deletion. J Thromb Haemost 7(S2):376. https://doi.org/10.1111/j.1538-7836.2009.03473-2.x
Perry DJ, Daly ME, Tait RC, Walker ID, Brown K, Beauchamp NJ, Preston FE, Gyde H, Harper PL, Carrell RW (1998) Antithrombin Cambridge II (Ala384Ser): clinical, functional and haplotype analysis of 18 families. Thromb Haemost 79(2):249–253
Lane DA, Erdjument H, Flynn A, Di Marzo V, Panico M, Morris HR, Greaves M, Dolan G, Preston FE (1989) Antithrombin Sheffield: amino acid substitution at the reactive site (Arg393 to His) causing thrombosis. Br J Haematol 71(1):91–96. https://doi.org/10.1111/j.1365-2141.1989.tb06280.x
David D, Ribeiro S, Ferrao L, Gago T, Crespo F (2004) Molecular basis of inherited antithrombin deficiency in Portuguese families: identification of genetic alterations and screening for additional thrombotic risk factors. Am J Hematol 76(2):163–171. https://doi.org/10.1002/ajh.20067
Lane DA, Erdjument H, Thompson E, Panico M, Di Marzo V, Morris HR, Leone G, De Stefano V, Thein SL (1989) A novel amino acid substitution in the reactive site of a congenital variant antithrombin. Antithrombin pescara, Arg393 to Pro, caused by CGT to CCT mutation. J Biol Chem 264(17):10200–10204
Fidalgo T, Martinho P, Salvado R, Manco L, Oliveira AC, Pinto CS, Goncalves E, Marques D, Sevivas T, Martins N, Ribeiro ML (2015) Familial thrombotic risk based on the genetic background of protein C deficiency in a Portuguese study. Eur J Haematol 95(4):294–307. https://doi.org/10.1111/ejh.12488
Dávid M, Losonczy H, Sas G, Nagy A, Kutscher G, Meyer M (2000) Identification of mutations in 15 Hungarian families with hereditary protein C deficiency. Br J Haematol 111(1):129–135. https://doi.org/10.1046/j.1365-2141.2000.02324.x
Koenderman JS, Bertina RM, Reitsma PH, De Visser MCH (2011) Over 50% of Dutch VTE families with protein C deficiency share a founder R272C mutation. J Thromb Haemost 9:164. https://doi.org/10.1111/j.1538-7836.2011.04380_1.x
Liu H, Wang HF, Tang L, Yang Y, Wang QY, Zeng W, Wu YY, Cheng ZP, Hu B, Guo T, Hu Y (2015) Compound heterozygous protein C deficiency in a family with venous thrombosis: identification and in vitro study of p.Asp297His and p.Val420Leu mutations. Gene 563(1):35–40. https://doi.org/10.1016/j.gene.2015.03.002
Wu Y, Xu G, Ding Q, Xi X, Wang X, Wang H (2009) Combined occurrence of hereditary protein C and protein S deficiency in two Chinese families with venous thrombosis. J Thromb Haemost 7(S2):1043. https://doi.org/10.1111/j.1538-7836.2009.03473-2.x
Zhou RF, Cai XH, Xie S, Wang XF, Wang HL (2006) Molecular mechanism for hereditary protein C deficiency in two Chinese families with thrombosis. J Thromb Haemost 4(5):1154–1156. https://doi.org/10.1111/j.1538-7836.2006.01913.x
Yue Y, Liu S, Han X, Xiao L, Huang Q, Li S, Zhuang K, Yang M, Zou C, Fu Y (2019) Pathogenic variants of PROC gene caused type I activity deficiency in a familial Chinese venous thrombosis. J Cell Mol Med 23(10):7099–7104. https://doi.org/10.1111/jcmm.14563
Wu D, Zhong Z, Chen Y, Ding H, Yang M, Lian N, Huang Z, Zhang Q, Zhao J, Deng C (2019) Analysis of PROC and PROS1 single nucleotide polymorphisms in a thrombophilia family. Clin Respir J 13(8):530–537. https://doi.org/10.1111/crj.13055
Su K, Zhang H, Fang W, Zhang F, Yang L, Jin Y, Wang M (2018) Protein C deficiency (a novel mutation: ala291Thr) with systemic lupus erythematosus leads to the deep vein thrombosis. Blood Coagul Fibrinolysis 29(8):714–719. https://doi.org/10.1097/mbc.0000000000000778
Chen C, Yang L, Villoutreix BO, Wang X, Ding Q, Rezaie AR (2017) Gly74Ser mutation in protein C causes thrombosis due to a defect in protein S-dependent anticoagulant function. Thromb Haemost 117(7):1358–1369. https://doi.org/10.1160/th17-01-0043
Hoshi S, Hijikata M, Togashi Y, Aoyagi T, Kono C, Yamada Y, Amano H, Keicho N, Yamaguchi T (2007) Protein C deficiency in a family with thromboembolism and identified gene mutations. Intern Med 46(13):997–1003. https://doi.org/10.2169/internalmedicine.46.6277
Hayashida M, Yamada H, Yamazaki S, Nomura H, Yoshimura K, Kitahara O, Momose K, Kubo K, Kurihara M, Hamasaki N (2003) Combined protein C and protein S deficiency in a family with repetitive thromboembolism and segregated gene mutations. Intern Med 42(3):268–272
Park HJ, Chong SY, Oh D, Huh JY, Kim HJ, Yun-Choi HS, Park S (2011) A novel insertion mutation 718dupG in the PROC gene in a Korean thrombophilic family. Thromb Res 127(2):176–178. https://doi.org/10.1016/j.thromres.2010.07.019
Ireland HA, Boisclair MD, Taylor J, Thompson E, Swee Lay T, Girolami A, De Caterina M, Scopacasa F, De Stefano V, Leone G, Finazzi G, Cohen H, Lane DA (1996) Two novel (R(-11)C; T394D) and two repeat missense mutations in the protein C gene associated with venous thrombosis in six kindreds. Hum Mutat 7(2):176–179. https://doi.org/10.1002/(SICI)1098-1004(1996)7:2<176::AID-HUMU16>3.0.CO;2-#
Tomczak JA, Ando RA, Sobel HG, Bovill EG, Long GL (1994) Genetic analysis of a large kindred exhibiting type I protein C deficiency and associated thrombosis. Thromb Res 74(3):243–254. https://doi.org/10.1016/0049-3848(94)90112-0
Formstone CJ, Hallam PJ, Tuddenham EGD, Voke J, Layton M, Nicolaides K, Hann IM, Cooper DN (1996) Severe perinatal thrombosis in double and triple heterozygous offspring of a family segregating two independent protein S mutations and a protein C mutation. Blood 87(9):3731–3737
Esmon CT (1983) Protein-C: biochemistry, physiology, and clinical implications. Blood 62(6):1155–1158
Espinosa-Parrilla Y, Morell M, Souto JC, Tirado I, Fontcuberta J, Estivill X, Sala N (1999) Protein S gene analysis reveals the presence of a cosegregating mutation in most pedigrees with type I but not type III PS deficiency. Hum Mutat 14(1):30–39. https://doi.org/10.1002/(SICI)1098-1004(1999)14:1<30::AID-HUMU4>3.0.CO;2-X
Beauchamp NJ, Daly ME, Cooper PC, Makris M, Preston FE, Peake IR (1996) Molecular basis of protein S deficiency in three families also showing independent inheritance of factor V Leiden. Blood 88(5):1700–1707
Formstone CJ, Wacey AI, Berg LP, Rahman S, Bevan D, Rowley M, Voke J, Bernardi F, Legnani C, Simioni P, Girolami A, Tuddenham EG, Kakkar VV, Cooper DN (1995) Detection and characterization of seven novel protein S (PROS) gene lesions: evaluation of reverse transcript-polymerase chain reaction as a mutation screening strategy. Blood 86(7):2632–2641
Falk G, Sartori MT, Patrassi GM, Vettore S, Girolami A, Wiman B (1997) Identification of an 18 basepair deletion in the PAI-1 gene promoter region in a family with thrombotic disease. Fibrinolysis Proteolysis 11(5–6):239–244
Öhlin AK, Marlar RA (1999) Thrombomodulin gene defects in families with thromboembolic disease: a report on four families. Thromb Haemost 81(3):338–344
Shlebak AA, Katsarou AD, Adams G, Fernando F (2017) A novel mutation in exon 2 of FGB caused by c.221G>T† substitution, predicting the replacement of the native arginine at position 74 with a leucine (p.Arg74Leu†) in a proband from a Kurdish family with dysfibrinogenaemia and familial venous and arterial thrombosis. J Thromb Thrombolysis 43(2):263–270. https://doi.org/10.1007/s11239-016-1439-z
Fernández-Cadenas I, Penalba A, Boada C, Msc CC, Bueno SR, Quiroga A, Monasterio J, Delgado P, Anglés-Cano E, Montaner J (2016) Exome sequencing and clot lysis experiments demonstrate the R458C mutation of the alpha chain of fibrinogen to be associated with impaired fibrinolysis in a family with thrombophilia. J Atheroscler Thromb 23(4):431–440. https://doi.org/10.5551/jat.30676
Hanss MM, Ffrench PO, Mornex JF, Chabuet M, Biot F, De Mazancourt P, Dechavanne M (2003) Two novel fibrinogen variants found in patients with pulmonary embolism and their families. J Thromb Hemost 1(6):1251–1257
Brennan SO, Hammonds B, Spearing R, George PM (1997) Electrospray ionisation mass spectrometry facilitates detection of fibrinogen (Bβ14 Arg→Cys) mutation in a family with thrombosis. Thromb Haemost 78(6):1484–1487
Salomon O, Barel O, Eyal E, Ganor RS, Kleinbaum Y, Shohat M (2019) C.259A>c in the fibrinogen gene of alpha chain (FGA) is a fibrinogen with thrombotic phenotype. Appl Clin Genet 12:27–33. https://doi.org/10.2147/TACG.S190599
Siebenlist KR, Mosesson MW, Meh DA, DiOrio JP, Albrecht RM, Olson JD (2000) Coexisting dysfibrinogenemia (gammaR275C) and factor V Leiden deficiency associated with thromboembolic disease (fibrinogen Cedar Rapids). Blood Coagul Fibrinolysis 11(3):293–304
Cheah CY, Brennan SO, Kennedy H, Januszewicz EH, Maxwell E, Burbury K (2012) Fibrinogen Melbourne: a novel congenital hypodysfibrinogenemia caused by γ326Cys-Phe in the fibrinogen γ chain, presenting as massive splanchnic venous thrombosis. Blood Coagul Fibrinolysis 23(6):563–565. https://doi.org/10.1097/MBC.0b013e328354a23b
Koopman J, Haverkate F, Grimbergen J, Lord ST, Mosesson MW, DiOrio JP, Siebenlist KS, Legrand C, Soria J, Soria C et al (1993) Molecular basis for fibrinogen Dusart (A alpha 554 Arg-->Cys) and its association with abnormal fibrin polymerization and thrombophilia. J Clin Invest 91(4):1637–1643. https://doi.org/10.1172/jci116371
Ortiz AS (2005) Complexity of the genetic contribution of different thrombotic risk factors in a Spanish thrombophilic family. Thromb Haemost 93(5):997–998
Chang WA, Sheu CC, Liu KT, Shen JH, Yen MC, Kuo PL (2018) Identification of mutations in SLC4A1, GP1BA and HFE in a family with venous thrombosis of unknown cause by next-generation sequencing. Exp Ther Med 16(5):4172–4180. https://doi.org/10.3892/etm.2018.6693
Fu Y, Sun S, Liang J, Liu S, Jiang Y, Xu L, Mei J (2016) PEAR1 gene polymorphism in a Chinese pedigree with pulmonary thromboembolism. Medicine (United States) 95(51):e5687. https://doi.org/10.1097/MD.0000000000005687
Zakai NA, McClure LA (2011) Racial differences in venous thromboembolism. J Thromb Haemost 9(10):1877–1882. https://doi.org/10.1111/j.1538-7836.2011.04443.x
Rees DC, Cox M, Clegg JB (1995) World distribution of factor V Leiden. Lancet 346(8983):1133–1134. https://doi.org/10.1016/s0140-6736(95)91803-5
Rosendaal FR, Doggen CJ, Zivelin A, Arruda VR, Aiach M, Siscovick DS, Hillarp A, Watzke HH, Bernardi F, Cumming AM, Preston FE, Reitsma PH (1998) Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 79(4):706–708
Rosendaal FR, Reitsma PH (2009) Genetics of venous thrombosis. J Thromb Haemost 7(Suppl 1):301–304. https://doi.org/10.1111/j.1538-7836.2009.03394.x
Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T, Mackman N (2015) Venous thrombosis. Nat Rev Dis Primers 1:15006. https://doi.org/10.1038/nrdp.2015.6
Lindstrom S, Wang L, Smith EN, Gordon W, van Hylckama VA, de Andrade M, Brody JA, Pattee JW, Haessler J, Brumpton BM, Chasman DI, Suchon P, Chen MH, Turman C, Germain M, Wiggins KL, MacDonald J, Braekkan SK, Armasu SM, Pankratz N, Jackson RD, Nielsen JB, Giulianini F, Puurunen MK, Ibrahim M, Heckbert SR, Damrauer SM, Natarajan P, Klarin D, Million Veteran P, de Vries PS, Sabater-Lleal M, Huffman JE, Group CHW, Bammler TK, Frazer KA, McCauley BM, Taylor K, Pankow JS, Reiner AP, Gabrielsen ME, Deleuze JF, O'Donnell CJ, Kim J, McKnight B, Kraft P, Hansen JB, Rosendaal FR, Heit JA, Psaty BM, Tang W, Kooperberg C, Hveem K, Ridker PM, Morange PE, Johnson AD, Kabrhel C, Tregouet DA, Smith NL (2019) Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood 134(19):1645–1657. https://doi.org/10.1182/blood.2019000435
Connors JM (2017) Thrombophilia testing and venous thrombosis. N Engl J Med 377(12):1177–1187. https://doi.org/10.1056/NEJMra1700365
Moran J, Bauer KA (2020) Managing thromboembolic risk in patients with hereditary and acquired thrombophilias. Blood 135(5):344–350. https://doi.org/10.1182/blood.2019000917
Funding
This work was supported by the Fund of The National Key Research and Development Program of China (No. 2016YFC0905600), the Fundamental Research Funds for the Central Universities (NO. 3332018184), CAMS Innovation Fund for Medical Sciences (CIFMS) (2018-I2M-1-003).
Author information
Authors and Affiliations
Contributions
CW and ZZ conceived and designed the study, having full access to all of the data in the study and taking responsibility for the content of the manuscript. YZ, ZZ and SS analyzed the data, took responsibility for the accuracy of the data analysis and wrote the first draft of the manuscript. WN, WJ and WX contributed to the interpretation of the data and clinical inputs. All authors were involved in the revision of the manuscript for important intellectual content and approved the final version to be published.
Corresponding authors
Ethics declarations
Conflict of interest
No conflicts of interest are involved in this manuscript, and manuscript is approved by all authors for publication.
Ethical approval
No additional ethical approval needs to be obtained.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Y., Zhang, Z., Shu, S. et al. The genetics of venous thromboembolism: a systematic review of thrombophilia families. J Thromb Thrombolysis 51, 359–369 (2021). https://doi.org/10.1007/s11239-020-02203-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-020-02203-7