
It was shown that under the conditions of barothermal synthesis of vanadium–phosphorus oxide catalysts the individual vanadium oxide phase VOHPO4·0.5 H2O is only formed in the n-butanol medium. The duration of the synthesis has a substantial effect on the surface morphology of the samples; with increase of the synthesis time structures of “rose” type are twisted and form “plaits.” The “rose” type structures have high activity and selectivity in the oxidation of n-butane (1.5-1.7 vol.% in air). With hydrocarbon contents of 3.4 vol.% structures of the “plait” type are more effective, which makes recycling possible. Catalysts with the “plait” morphology are also more effective in the oxidative dehydrogenation of ethane.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The term barothermal synthesis, which encompasses such subjects as hydrothermal and organothermal synthesis of solids, was first used in [1] since, irrespective of the nature of the employed solvent, we felt that these processes take place under the influence of temperature and pressure and do not occur under normal conditions. The term “solvothermal synthesis” that appeared later on only covers the use of various types of solvents and does not cover the influence of the principal factors on the synthesis process. To this we can also add the fact that barothermal synthesis can also be realized without the inclusion of a solvent in the system on account, for example, of the presence of water in the initial components of the synthesis. All three methods (with water, with an organic solvent, and without solvent) were used in [1] for the synthesis of a VPO catalyst. They were developed further both in our papers [2,3,4] and in papers by other research workers [5,6,7,8,9], who used the term “VPO synthesis under the influence of pressure and temperature.”
Vanadium phosphorus oxide (VPO) catalysts are known as the main system for industrial oxidation of n-butane to maleic anhydride (MA) [10], the production of which in 2015 amounted to 2.8 million tonnes (with a growth rate of 1% a year). In recent years, however, no significant advances have been made in the activity of the catalysts in the process [11] in spite of various modifications of the traditional methods (synthesis in various types of organic solvents, synthesis in water followed by treatment in an organic solvent, etc.). Reaction mixtures with n-butane concentrations increased from 2 to 10 vol.% were proposed in order to increase the productivity with respect to MA [11,12,13,14,15]. (Traditionally a 1.5-1.7 vol.% mixture of n-C4H10 in air has been used.) This leads to the prospect of paraffin recycling [16, 17]. However, as found in [10,11,12, 14] increase of the concentration of n-C4H10 reduces the yields of the product quite sharply (to 10-20 mole %) when VPO catalysts prepared by the traditional methods are used.
On the other hand the possibility of using VPO catalysts in the oxidative dehydrogenation of ethane to ethylene was demonstrated [18,19,20,21]. However, in this case (the process was conducted with an increased C2H6 concentration of 4-8 vol.%) the yield of the olefin obtained with VPO catalysts synthesized by the traditional methods were no higher than the values obtained with other systems [19, 21] and amounted to 12-14 mole %.
In the present work we investigated the possibility of using barothermal synthesis for the production of VPO catalysts capable of working in reaction mixtures with an increased content of the hydrocarbon and producing a sufficiently high yield of the product and high productivity.
Experimental
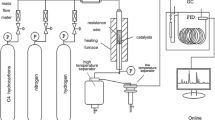
The VPO samples were synthesized in a stainless-steel laboratory autoclave with a volume of 45 mL and a teflon insert of 15 mL. In the teflon insert we placed 4 g of V2O5 (analytical grade), 3.3 mL of 85% H3PO4 (analytical), 2.34 g of oxalic acid (analytical), and 9 mL of n-butanol (analytical). The synthesis was conducted at 170 °C. The pressure formed in the autoclave was calculated by means of the Antoine equation log P=A–B(t + C) and under the given conditions amounted to 0.5 MPa. The synthesis took 5, 7, 10, and 18 h, and the samples were named accordingly as VPO-5, VPO-7, VPO-10, and VPO-18. After the synthesis the solvent was removed by filtration, and the turquoise-colored substance formed as a result of the synthesis was dried at room temperature. It was then subjected to heat treatment at a residual pressure of 0.02 MPa while the temperature was increased to 270 °C (10 °C/h).
The phase composition of the samples was determined with a DRON-4.0 diffractometer (CuKα radiation). The X-ray diffraction patterns were processed with Origin 7 software (peak fitting). The size of the crystallites was calculated by means of the Scherrer equation. The specific surface area (Ssp) of the VPO samples was determined from nitrogen adsorption–desorption isotherms obtained on a NovaWin2 instrument (Quantachrome). The surface morphology was studied with a scanning electron microscope (Hitachi S-4000, Japan) and with NanoScope E force microscope (Digital Instruments, USA).
The catalytic activity of the samples was investigated with 1 cm3 of the catalyst (0.25-0.50 mm) loaded into flow-type apparatus with full online chromatographic analysis on two chromatographs – Chrom-5 (Czech) and SELMI-Chrom-2 (Ukraine). The n-butane was oxidized with two working mixtures: 1.5 and 3.4 vol.% C4H10 in air with variation of the flow rate of the reaction mixture from 20 to 50 mL/min. For oxidative dehydrogenation of the ethane we used a reaction mixture containing 7 vol.% of C2H6 and 3 vol.% of O2 in He at flow rates from 50 to 150 mL/min.
Results and Discussion
The results of the X-ray diffraction of the synthesized samples (Table 1) show that after synthesis for 5 h the sample also contained the initial reagent V2O5 in addition to the formed active phase VOHPO4·0.5H2O, indicating that the synthesis was not complete. Increase of the synthesis time led to the result that the samples contained the vanadyl hydrophosphate phase exclusively. Table 1 also contains authentic published data on the barothermal synthesis of VPO samples. Comparison of these data shows that the VOHPO4·0.5H2O phase is only formed in the presence of the organic solvents (n- and iso-C4H9OH) or in an aqueous medium with H3PO3 as reducing agent. It should be noted that this phase is also synthesized by barothermal synthesis with reduced vanadium oxide V2O4 as initial reagent [1, 4, 6, 9].
Analysis of the presented data shows that increase of the synthesis temperature in the organic solvent has an unfavorable effect on the formation of the vanadium phosphate phase, and β-VOPO4 (a compound with low selectivity in the oxidation of n-butane) is formed [10, 11]. This may be due to oxidation of the VOHPO4·0.5H2O by the atmospheric oxygen that is present in the autoclave. If the duration of synthesis in the organic medium at moderate temperatures (170 and 145 °C, Table 1) is increased, there is some reduction in the specific surface area of the VOHPO4·0.5H2O samples (irrespective of the nature of the organic solvent) and a slight increase in the crystal size of this phase. A positive effect from the increased synthesis time is an increase in the relative content of the basal plane (I001/I220, Table 1), which is important for the selective oxidation of n-butane [10].
The SEM data presented in Fig. 1 show that the morphology of the particles of the prepared samples changes in relation to the duration of the barothermal synthesis. As seen, after synthesis for 5 h the partial formation of VOHPO4·0.5H2O particles with a “rose” type of structure is observed, but here formations of a different type are also present. This is consistent with the XRD data (Table 1), which indicate the presence both of the formed vanadyl hydrophosphate phase and of the original vanadium oxide. Increase in the duration of synthesis (Fig. 1b) leads to the formation of VOHPO4·0.5H2O with a characteristic rose-like structure and the presence of thin lamellar “rose petals.” Similar structures were obtained during traditional syntheses of VPO catalysts [10]. Further increase of the barothermal synthesis time (Fig. 1c) to 10 h led to destruction of the rose-like structure. The “petals” are twisted, and with a longer synthesis time they are enlarged (Fig. 1d) and form a kind of “plait.” The “plaits” have a thickness of 1-1.5 μm(Fig. 1e) and are in the course of time rolled up into clumps measuring 20-40 μm(Fig. 1f). It should be mentioned that the formation of similar structures was observed in [8] after barothermal synthesis in isobutanol for 9 and 72 h. Here both in the present investigation and in [8] only the VOHPO4·0.5H2O phase was present in the samples, and although its specific surface area decreased with increase in the duration of the synthesis the relative content of the basal plane increased. It should be noted that according to the data from AFM the surface of the “plaits” is not flat but represents a set of nanosized peak-like formations (Fig. 2).

The surface morphology of the samples (SEM) prepared by barothermal synthesis with synthesis times of 5 (a), 7 (b), 10 (c), and 18 h (d-f).

The surface morphology of a VPO sample after barothermal synthesis for 18 h (AFM).
The results of investigation of the catalytic activity of the synthesized samples in the oxidation of n-butane using two reaction mixtures with different concentrations of the hydrocarbon are presented in Table 2. Published data on the activity of other VPO samples synthesized by the barothermal method are also included. Unfortunately, only one reaction mixture with the traditional content of n-butane was used in the work presented in Table 2. As seen from the presented data, in the given reaction mixture the best results (activity, selectivity, and yield of maleic anhydride respectively) are obtained on the samples represented by VOHPO4·0.5H2O while the maximum results are obtained in the presence of the “rose”-type morphology. This result is fully consistent with the data obtained on similar structures synthesized by various traditional methods. Breakdown of the structure (the formation of the “plaits”) reduces the yield of the product.
A different picture is observed when a reaction mixture rich in the hydrocarbon is used (Table 2). In this case the samples with the “plait” morphology give better results than the rose-like structure (selectivity for MA and yield for MA). When the catalyst obtained by the barothermal method and having the rose-like structure is used (Table 2, the VPO-7 structure) the yield of maleic anhydride is at the level attained on the VPO catalysts produced by traditional methods [11,12,13,14,15]. At the same time the catalysts with the “plait” morphology (VPO-10, VPO-18) exhibit higher selectivity and product yield. This may be due to the fact that a larger relative amount of the basal plane is found in the samples with the “plait” form (Table 1) and also, possibly, to the high stability of the samples against adiabatic heating of the catalyst grains.
Comparison of the results obtained on the VPO-10 and VPO-18 samples of reaction mixtures with various n-butane concentrations shows that although the yield of maleic anhydride is decreased in an excess of the hydrocarbon this is not however as substantial as on the samples synthesized by the traditional methods [11,12,13,14,15] or on their analog VPO-7. Here, with increased n-butane content in the reaction mixture for the given samples a substantially higher productivity with respect to maleic anhydride (VPO-10 1.29 mol of MA/h·kg VPO; VPO-18 1.10 mol of MA/h·kg VPO) is obtained than with the traditional mixture (VPO-10 0.73 mol of MA/h·kg VPO; VPO-18 0.66 mol of MA/h·kg VPO). It should be noted that the values of the productivity for MA are higher than the values obtained on VPO-7 in the case where the traditional mixture was used (1.05 mol of MA/h·kg VPO). In view of the fact that the conversion of the hydrocarbon (Table 2) with increased n-C4H10 content is not greater than 50% it is expedient to use n-butane recycling, proposed in [16, 17], after isolation of the maleic anhydride. Another version of the oxidation of n-C4H10 can also be proposed. After passing through the first reactor, working on a mixture with an excess of the hydrocarbon, and isolation of the maleic anhydride the gas mixture (the concentration of unreacted n-butane amounts to about 1.7 vol.%) passes on to a second reactor with a catalyst of the VPO-7 type (or a similar catalyst synthesized by the traditional method), at which the n-butane is oxidized with productivity of 1.05 mol of MA/h·kg VPO (see above). Here it is not necessary to add the further amount of hydrocarbon required to support recycling. The overall productivity can reach 2.34 mol of MA/h·kg VPO.
The main problem in the oxidative dehydrogenation of ethane is not only the fairly low yield of ethylene but also the high temperature of the process (>500 °C) [19,20,21]. Optimum selectivity here is obtained with 10%-12% conversion of ethane and amounts to about 90%, while the selectivity decreases sharply with increase of the degree of conversion. Table 3 shows the results for oxidative dehydrogenation of ethane obtained on VPO samples synthesized by the barothermal method. As seen from the presented data, first, the best results with respect to selectivity and yield of the product are obtained on the VPO-10 and VPO-18 catalysts that have surface morphology in the form of “plaits.” Second, the samples synthesized by the barothermal method make it possible to lower the reaction temperature to less than 500 °C. Third, as on other known catalysts of this process maximum selectivity for the olefin is obtained at small degrees of conversion of the paraffin and decreases as the conversion increases. Fourth, maximum product yield is obtained at 475 °C and a hydrocarbon conversion about 25%, which makes it possible to realize a recycling process.
The obtained results as a whole demonstrate the effect of the surface morphology of the catalysts on their properties (selectivity and product yield) and the potential of barothermal synthesis of VPO catalysts for use in the oxidation of C2 and C4 hydrocarbons conducted with increased concentration of the hydrocarbon in the reaction mixture. With the VPO catalysts synthesized by this method it is possible to increase the product yield and the productivity compared with the catalysts prepared by traditional methods.
References
V. A. Zazhigalov, V. É. Yaremenko, and V. G. Il’in, Kinet. Katal., 40, No. 1, 141-144 (1999).
V. A. Zazhigalov, V. É. Yaremenko, I. V. Bacherikova, and J. Stoch, Teor. Éksp. Khim., 35, No. 2, 103-106 (1999). [Theor. Exp. Chem., 35, No. 2, 99-102 (1999) (English translation).]
V. V. Sidorchuk, E. A. Diyuk, and V. A. Zazhigalov, Neorgan. Materialy, 43, No. 4, 471-476 (2007).
V. V. Sidorchuk, V. A. Zazhigalov, and S. V. Khalameida, Neorgan. Materialy, 43, No. 10. 1239-1245 (2007).
J. K. Bartley, C. J. Kiely, R. P. K. Wells, and G. J. Hutchings, Catal. Lett., 72, Nos. 1/2, 99-105 (2001).
J. K. Bartley, J. A. Lopez-Sanchez, and G. J. Hutchings, Catal. Today, 81, No. 2, 197-203 (2003).
J. A. Lopez-Sanchez, L. Griesel, J. K. Bartley, et al., Phys. Chem. Chem. Phys., 5, No. 16, 3525-3533 (2003).
L. Griesel, J. K. Bartley, R. P. K. Wells, et al., Catal. Today, 99, No. 1, 131-136 (2005).
E. Kleimenov, H. Blihm, M. Havecker, et al., Surface Sci., 575, 181-188 (2005).
G. Centi, F. Cavani, and F. Trifiro, Selective Oxidation by Heterogeneous Catalysis, Kluwer Acad. Publ., New York (2001).
F. Trifiro and R. K. Grasseli, Top. Catal., 57, Nos. 14-16, 1188-1195 (2014).
K. Kourtakis and P. Gai, J. Mol. Catal. A, 220, Nos. 1/2, 93-102 (2004).
Y. Kamiya, E. Nishikawa, T. Okuhara, and T. Hattori, Appl. Catal. A, 206, Nos. 1/2, 103-112 (2001).
J.-A. T. Schwartz, “Process for manufacture of an attrition resistant catalyst,” USA Pat. 6362128 B1, IC1 B 01 J 27/128, Publ. March 26, 2002.
M. E. Davis, C. J. Dillon, J. H. Holles, et al., “Polyoxometallate catalysts and catalytic processes,” USA Pat. 7019165 B2, IC1 C 07 C 63/14, C 07 C 63/307, C 07 C 68/313, Publ. March 28, 2006.
A. Bertola and S. Cassarino, “High productivity process to produce maleic anhydride from n-butane,” USA Pat. 6040460 A, IC1 C 07 C 51/216, B 01 J 27/198, C07 D 307/60, B 01 J 37/08, Publ. March 21, 2000.
G. S. Patience and R. E. Bockrath, Appl. Catal. A, 376, Nos. 1/2, 4-12 (2010).
P. R. Blum and E. C. Milberger, “Process for the oxydehydrogenation of ethane to ethylene,” USA Pat. 4410752, IC1 C 07 C 5/38, C 07 C 5/48, Publ. Oct. 18, 1983.
P. M. Michalakos, M. C. Kung, I. Jahan, and H. H. Kung, J. Catal., 140, No. 2, 226-242 (1993).
J. M. Lopez Nieto, V. A. Zazhigalov, B. Solsona, and I. V. Bacherikova, Stud. Surface Sci. Catal., 130, 1853-1858 (2000).
F. Cavani, N. Ballarini, and A. Cericola, Catal. Today, 127, No. 1, 113-131 (2007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Teoreticheskaya i Éksperimental’naya Khimiya, Vol. 54, No. 1, pp. 61-66, January-February, 2018.
Rights and permissions
About this article

Cite this article
Zazhigalov, V.A., Diyuk, E.A. Barothermal Synthesis and Catalytic Properties of Vanadium–Phosphorus Oxide Systems in Oxidative Transformations of Butane and Ethane. Theor Exp Chem 54, 66–72 (2018). https://doi.org/10.1007/s11237-018-9547-9
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11237-018-9547-9