
Abstract
Encapsulation of chlorambucil, cyclophosphamide, and melphalan with (9,9) carbon nanotube was investigated in aqueous solution. The structures were modeled in gas phase applying density functional calculations. Then, solvation free energies and association free energies of considered structures in water were studied by Monte Carlo simulation and perturbation method. Outcomes of gas phase study revealed that all three drugs can encapsulate into carbon nanotube. Monte Carlo simulation results showed that electrostatic interactions play a crucial role in the intermolecular energies after encapsulation in aqueous media. It is found that among three drugs, solvation free energy of chlorambucil was increased after encapsulation. Moreover, solvation of carbon nanotube was enhanced after encapsulation of drugs which improve the pharmaceutical applications. Calculated association free energies indicated that chlorambucil–carbon nanotube was the only stable encapsulated drug in aqueous solution.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alkylating agents have been applied for the medication of different cancers, such as leukemias and lymphomas. While efficiency of alkylating agents is different, some of them have analogous mechanisms. Their actions are affected on DNA directly, resulting in its cross-linking and initiating DNA strand breaks, causing abnormal base pairing, following to inhibit cell division and finally resulting in cell death. Alkylating agents have capability to produce strong electrophiles by the formation of carbonium ion intermediates or to yield transition complexes with the objective molecules that covalently bind an alkyl group or a substituted alkyl group to any nucleophilic structure, containing the N-7 position on guanine, which is a main site of action [1]. When both alkyl groups bind with pairs of guanine residues, a cross-linking of DNA is formed. Finally, the DNA sequence is broken, and cell death.
The side effects of alkylating agent anticancer drugs are permanent infertility due to decreasing sperm creation in males and producing menstruation cessation in females, bone marrow suppression, associated improved risk for infections followed by decreasing host resistance nausea and vomiting and reversible hair loss [2].
To reduce the toxic side effects of alkylating agents, various attempts have been made. Limiting the amount of the drug, its side effects can be reduced but all of the tumor tissue may not be exposed to an appropriate dose of drug. To reduce side effects and enhance efficiency of treatment, the use of nanomaterials as drug delivery system in anticancer therapies has been investigated. Pharmacological properties of customary chemotherapeutics can be developed by the use of nanocarriers for instance liposomes (lipid-based nanoparticles) [3,4,5], micelles [6, 7], protein [8], and polymers [9, 10]. They can interact to or encapsulate therapeutic drugs (e.g., encapsulation of doxorubicin [11, 12] and Paclitaxel [13] by liposomal). Nanocarriers can be freely extravasated from circulation through vascular defects naturally existing at tumor sites due to their small size (~ 100 nm or less) [14] and then they can deliver encapsulated cytotoxic agents to tumor tissue. Carbon nanotubes (CNTs) are other nanomaterials that have been applied as drug delivery systems and enhancement of the treatment efficiency while reducing unfavorable side effects to normal tissues. The properties of nanotubes can be improved by adding of impurities. Mixed nanotube can be considered another vehicle for applications on nanomedicine. First, principle studies of some compositions of hybrid BN/C [15] nanotubes have revealed that surface distribution charge modify in these nanotubes. Determination of structural, electronic, and energetic properties of some compositions of hybrid boron nitride/carbon nanotubes has indicated that these nanostructures have low reactivity and high polarity that can be applied as drug delivery system.
CNTs have many attractive properties, such as great mechanical strength, admirable electronic and chemical properties, high surface area, hollow cavities, and thermal stability [1,2,3,4,5,6,7]. CNTs can be applied in several biological applications for example drug delivery [16,17,18,19], tumor therapy [20], and biosensors [21]. They can enter cells and deliver molecules into the cytoplasm [22]. CNT labeled with a fluorescent agent could be tracked into the cytoplasm or the nucleus of fibroblasts by epifluorescence and confocal microscopy [23]. CNTs have a high surface area for attachment of targeting ligands, and also have an interior cavity which encapsulates either therapeutic or diagnostic agents. Various substances like anticancer drugs can be bind with CNT. To construct CNT-drug nanocarriers, computational molecular modeling has also been applied [24, 25]. Molecular dynamics simulations have been indicated that a DNA structure could be loaded into CNT in aqueous solution [26]. It was indicated that van der Waals interactions have important role in binding of DNA and CNT. Molecular simulation studies on the structural properties of the interaction of CNTs with β-alanine and histidine in water have been indicated that adsorption of these amino acids can rise solubility of CNT [27]. Encapsulation behaviors of cisplatin into CNT and BNNT in aqueous solutions have been studied [28] by density functional calculation and Monte Carlo simulation. Computed complexation free energies have confirmed the stability of encapsulated drug [28].
Learning adsorption properties of carnosine loaded on CNT was the aim of another research [29]. The possible bindings of carnosine and CNT were concluded via quantum mechanical calculations of binding energies in gas phase. Monte Carlo simulations have been applied for estimating solvation free energies. Interactions with carnosine raise solubility of CNT and therefore decrease its toxicity. Complexation free energies in water approve stability of CNT-carnosine systems and eventually confirm the great potential of CNT in the field of nanomedicine.
Anticancer gemcitabine drug has been loaded into CNTs [30] in an alternative research. The results indicated great activity toward lymph node metastasis in mice [30]. In another study, camptothecin drug has been interacted with polyvinyl alcohol-functionalized CNTs. In treatment of breast and skin cancers, efficiency of the functionalized CNT has been approved [31].
In all mentioned researches, large inner volume of CNT, capability of CNTs to enter into cells, and deep potential of CNT interior [32,33,34,35,36] are the advantages of CNTs to encapsulate of drugs. On the other hand, side effects of anticancer drugs can be reduced by encapsulating into CNTs. Therefore, encapsulation of alkylating agent anticancer drugs into CNT was the main purpose of this research.
In this study, chlorambucil, cyclophosphamide, and melphalan were chosen as anticancer drugs which could be encapsulated into CNT. Density functional calculations were applied to evaluate bindings in gas phase and then Monte Carlo simulation method was used to determine solvation free energies. Association free energies were also calculated in physiological media to investigate the possibility of encapsulation of these drugs into CNT.
Computational method
To deactivate the drugs until receiving cancer cells, CNT has been used as a transporter. Therefore, capability of the CNT to carriage of drug molecules and also solubility of its compound in physiological media has been considered. The vital concern of this investigation was the study of bindings of drugs (chlorambucil, cyclophosphamide, melphalan) and CNT in aqueous solution and evaluation of interaction energies and solvation of their compounds. The study contained two sections: (a) quantum mechanical calculation (QM) and (b) Monte Carlo (MC) simulation. In the first section, QM, after optimization of separated structures, binding energies of molecules were determined in gas phase. In the MC simulation part, we focused on determination of solvation free energies and association free energies in water.
QM
Optimization of CNT, drugs (chlorambucil, cyclophosphamide, melphalan), and drug encapsulated into CNT was organized by DFT/B3LYP [37,38,39] method and 6–31 G* [40, 41] basis set.
To avoid dealing with asymmetry effects in the case of non-chiral zigzag structures, the armchair (9,9) CNT with 378 atoms and the length of 24.916 Å was considered a drug carrier in our research.
The electronic properties and chemical reactivity of CNTs mainly depend on their chirality and curvature [42]. Zigzag nanotubes act as semiconductors, whereas armchair nanotubes indicate metallic behavior [43]. The electronic properties of CNTs affect the adsorption of drug molecules. It has been found that for CNTs with almost similar diameters, binding affinity of armchair nanotubes toward molecules is weaker than zigzag counterparts [44, 45]. DFT calculations [44] have been indicated that adsorption of pyrazinamide antitubercular drug molecules onto (9,0) CNT represents higher binding energy than (5,5) CNT, whereas the release of drug inside the biological environments from (5,5) CNT has been more preferred than (9,0) CNT due to its low adsorption energy [44].
By comparing of (5,5) and(9,0) CNT, it has indicated that the frontier orbital of (5,5) CNT is delocalized along nanotube side walls, whereas HOMO and LUMO of (9,0) CNT localize along the edge states [44]. It has been proposed that the active sites lie along nanotube edges of (9,0) CNT [46]. It has been shown that the higher binding affinity of zigzag nanotubes with respect to armchairs is due to the larger polarizability values of zigzag nanotubes [45]. Furthermore, the outcomes of comparing the various armchair and zigzag CNTs have shown that the zigzag CNTs display higher HOMO energies which leads to larger binding affinity with respect to those of armchair CNTs [45].
Hence, we used armchair CNTs due to electronic properties that induce lower interaction energies compared with zigzag CNTs to facile drug releasing. Although it has been indicated that energy difference between the zigzag and armchair CNT complexes is lower than the flat systems [45], the chirality has no significant effect on the interactions with drugs in comparing with curvature of CNT.
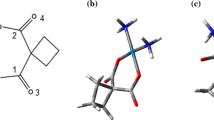
Molecular structures of drugs are presented in Fig. 1. In QM calculations, two ends of CNT were coated with hydrogens to accomplish a precise calculation to unlimited nanotube. Neutral charge was considered for CNT, drugs, and CNT-drug complexes. All structures were stabilized with multiplicity 1 and neutral charge. Moreover, to confirm the correct ground state of CNT and CNT-drug structures, multiplicity 3 was also examined. GAMESS-US quantum chemistry package [47] was implemented for all QM calculations.

Structure of drugs. a Chlorambucil. b Cyclophosphamide. c Melphalan
MC
In this part, possibility of considered alkylating agent encapsulation into CNT in water media was studied. Metropolis sampling [48] with NVT ensemble was applied in MC simulation calculations. A rectangular box of dimensions 40 × 40 × 56 Å3 and about 3000 water molecules with density of 0.993 g/cm3 at 298 K [49, 50] was used in simulations. Dilute solutions of each solute were used. Each species was immersed in the box and some water molecules were snuff out from box according to solute size.
On each MC cycle, a random selected molecule was translated (in the range of ± 0.13 Å) and rotated (in the range of ± 10°) arbitrarily using periodic boundary conditions with total acceptance of 50% for produced configurations. Each separated run involved107 configurations which were extended to reduce statistical error.
Total potential energy comprised intermolecular and intramolecular interaction energies. The intermolecular energies in aqueous solution were containing of water–water and solute–water interactions that both of them involve van der Waals and electrostatic energies.
To model water–water interactions, transferable intermolecular potential function (TIP3) [51, 52] was applied with parameters q, A, and C for electrostatic, repulsive, and attractive van der Waals interactions respectively. Interactions between solute (CNT, drugs, and drug encapsulated CNT) and water were distinct by Lennard-Jones potential for van der Waals interactions (with parameters ε and σ) and Coulomb potential for electrostatic interactions (with parameter q). Lennard-Jones parameters for C (in CNT) [53] and drug atoms [54] were implemented. The site-site LJ parameters were attained by Lorenz-Berthelot combining rule [55]. Parameter q was determined through QM calculations in the previous section.
Free energy perturbation (FEP) theory using Zwanzig equation [56] was applied to estimate free energy variation:
The averaging (< >) is done over the ensemble corresponding to the initial state, A. Actually, a parameter λ is introduced to the potential function and then free energy is varied from initial to final configurations via a set of related states.
Solvation free energy (ΔGsolv(A)) is free energy differences of transporting a species (A) from gas phase to solution. To compute solvation free energy by perturbation method, free energy is calculated when a species is disappeared in two phases (gas and solution phases):
In fact, the simulations were divided into a series of steps where λ running from 0 to 1 with step size, Δλ, is equal to 0.1.
Association free energies of encapsulation of drugs into CNT were calculated using thermodynamic cycle (Fig. 2). By considering Eq. 2 and thermodynamic cycle, association free energies (∆Gass(D - CNT)) were calculated by:

Thermodynamic cycle for CNT-drug complex
∆Gsol(D → 0), ∆Gsol(CNT → 0), and ∆Gsol(D − CNT → 0) denote free energy differences of disappearing drug ( ), CNT, and drug encapsulated CNT in solution.
Results and discussion
QM
The aim of the QM part was the evaluation of binding affinity of CNT and drugs in gas phase.
Earlier researches have indicated that interaction of CNT and encapsulated drugs has been stronger than adsorbed drug on the outer nanotube surface [57, 58]. Leonor et al. revealed that encapsulated doxorubicin inside the nanotube exhibits stronger intermolecular interactions than the outside [57]. In fact π-π stacking was one of the important components of the attraction of doxorubicin-CNT forces and nanotube exhibited a dual strong interaction with the encapsulated drug. In another research adsorption of oleuropein, a phenolic compound with many biological activities, inside and outside the CNT, has been studied by molecular dynamic simulation method [58]. It has been revealed that interaction of CNT and encapsulated oleuropein was stronger than outer surface. We also considered encapsulation of drugs into CNT due to higher binding energies of encapsulated drugs than surface adsorption reported in earlier researches. However, we examined one of the drugs (chlorambucil) through surface functionalization to confirm predominance of encapsulation rather than outer surface adsorption. A systematic analysis has been done on several orientation modes of the adsorption of drug on the surface of CNT (Figure S1). According to our calculations on the studied models, the binding energies for the adsorption of chlorambucil on the outer surface of CNT are positive (see Table S1) which indicate these complexes are energetically unstable and therefore, encapsulation of drug into (9,9) CNT is preferred.
We considered encapsulation of three nitrogen mustard anticancer agents, chlorambucil, cyclophosphamide, and melphalan, into (9,9) armchair CNT. As it is mentioned before, neutral charge with multiplicity 1 and 3 was considered for all studied structures. Resulting relative energies are found in Table S2. The results approve the ground state of CNT and CNT-drug structures with multiplicity 1.
Vibrational analysis was performed on the optimized structures and the character of stationary points was checked within the harmonic approximation for CNT, drugs, and CNT-drug complexes. No imaginary vibration frequencies were found that confirm the stability of the obtained structures.
Optimized structure of chlorambucil-CNT compound is displayed in Fig. 3.

Optimized structure of chlorambucil encapsulated into (9,9) CNT
Binding energy (Eb) was calculated according to the expression:
where E(CNT−D), E(D), and E(CNT) specified energy of optimized individual structures of drug encapsulated CNT, drug, and CNT respectively. The calculations were performed with counterpoise corrections for basis set superposition error (BSSE) [59]. The results are presented in Table 1.
As it could be seen, BSSE corrected binding energies of chlorambucil, cyclophosphamide, and melphalan complexes with CNT were − 60.247, − 55.875, and − 64.157 kcal/mol respectively, so melphalan was preferred to encapsulate into CNT, although these QM results indicate that all three drugs can encapsulate into CNT in the gas phase.
Susceptibility of encapsulation depends on reactivity of drug and also steric factors [60]. Actually, different sizes of CNTs have been tried before using (9,9) one. The steric hindrance of smaller CNTs induced smaller and positive binding energy.
Apart from all mentioned researches that confirm the importance of encapsulation, the significant advantage is the total protection of the encapsulated drugs during transportation. By comparing of dipole moments in Table 1, it is concluded that dipole moments of the drug are reduced after encapsulations. Therefore, the passage of the drugs through nonpolar cell membrane is facilitated.
In the QM part, probability of nitrogen encapsulation mustard drugs into CNTs was studied. To evaluate the possible encapsulation of the three anticancer drugs and CNT in aqueous solution and its applicability as drug delivery system, MC simulation method was applied. The optimized QM structures of drugs and their complexes with CNT were used as the initial structures for MC simulations and also QM charges have been used in subsequent simulation step.
MC
To evaluate encapsulation of considered drugs into CNT in aqueous solution, total interaction energies were determined initially. Accordingly, one molecule of solute (CNT, chlorambucil, cyclophosphamide, melphalan, and their complexes) was immersed in water and total energies were determined from MC simulations. Snapshot of configuration that delivered from MC simulation of pure CNT and chlorambucil encapsulated CNT in solution is presented in Fig. 4. The figure provides qualitative arrangement of hydration shell around CNT.

Snapshot of the simulation of a CNT and b chlorambucil–CNT in aqueous solution
Total energy (Etot) in solution is elucidated by the sum of the contributions of solute–H2O, H2O–H2O, and intramolecular interaction energies.
MC simulation calculated energies are gathered in Table 2. This table contains number of water molecules (NH2O) in the box, energy contributions of solute–H2O interaction energy (Esoln), electrostatic contribution, and van der Waals contribution in solute–H2O interaction energy.
The outcomes of MC simulations specified that total energy of pure CNT was less than CNT encapsulated with drugs. Total energies of CNT drugs in solution were in the following order: CNT-cyclophosphamide > CNT-melphalan > chlorambucil.
Encapsulations of drugs into CNT enhance total interaction energy by increasing electrostatic contribution in solute–H2O interaction energy. In fact, interaction encapsulated drug with tube wall increases atomic charge on CNT and therefore enhances electrostatic contribution in CNT–H2O interaction energy. van der Waals contributions in solute–H2O interaction energies of CNT and drug encapsulated CNTs are the same (Table 2). These results indicated that van der Waals interactions do not affect intermolecular interactions after encapsulation in water. These consequences are compatible with our earlier research [28]. Simulation study of the encapsulation of cisplatin into carbon nanotubes has also indicated that after interaction of drug and CNT, electrostatic contributions were increased, whereas van der Waals interactions did not alter.
Solvation free energies of CNTs, drugs, and their complexes were computed using thermodynamic perturbation and Eq. 5. Computed solvation free energies were offered in Table 3. As it is presented, the order of solvation free energies of drugs is as follows:
cyclophosphamide > melphalan > chlorambucil.
In charged systems, contributions of electrostatic interactions in solute–H2O interaction energy are larger than van der Waals. Therefore, it is predicted that solvation free energies of these systems are considerable. Electrostatic contributions of drug–water interaction energy of cyclophosphamide, melphalan, and chlorambucil are − 117.905, − 27.158, and − 2.375 kcal/mol respectively (Table 2), so it is predictable that solvation free energies must be in the same order. According to these outcomes, cyclophosphamide is the most stable drug in water. When it is interacted with nanotube, resultant complex should be more stable than individual reactant in water to support the possibility of reaction in aqueous solution.
Solvation free energies of cyclophosphamide, melphalan, and chlorambucil encapsulated into CNT are − 191.728, − 155.883, and − 154.600 kcal/mol. Melphalan and chlorambucil complexes have the same solvation free energies approximately and cyclophosphamide CNTs have a little more solvation in water. In fact, encapsulation of these drugs in CNT reduces their interactions with bulk water. Only outer tube wall contacts with bulk water molecules directly. Therefore, all drug CNTs exhibit the same solvation approximately. Encapsulation of drugs into CNT is induced by atomic charges on the tube wall and so enhanced solvation free energy compared with pure CNT.
Association free energies were also calculated by using Eq. 4 and the results were also presented in Table 3. As it is seen, association free energy of interaction of chlorambucil and CNT is negative. Thus, only this alkylating agent can be encapsulated into CNT in aqueous solution. Actually, the encapsulation of cyclophosphamide and melphalan into CNT is unspontaneous in solution. This evidence is due to high stability of these two drugs in water. Actually, cyclophosphamide and melphalan do not incline to encapsulate into nanotube that will be produced by lower stable compounds. It should be a reminder that electrostatic energy has the main contribution in drug and H2O interactions. By considering the data in Table 2, higher electrostatic interactions of cyclophosphamide and melphalan with water (50 and 11 as much than chlorambucil respectively) induced higher stability of these drugs in water, whereas electrostatic interaction energies of all three drug-CNT compounds and water are the same. Therefore, chlorambucil can encapsulate into CNT due to suitable solvation free energy and association free energy. Consequently, CNT can act as a substantial applicant for encapsulation of this drug in aqueous solution.
In order to evaluate distribution of water molecules inside and outside the nanotube, radial distribution functions were also computed.
Radial distribution function (RDF, g(r)) is determined as the relation of local density at the distance r from the central axis of nanotube (ρ(r)) to the bulk density. It is a recognizable property which illustrates the configuration of fluid phase.
RDFs of pure CNT and chlorambucil capsulated CNT are presented in Fig. 5. These graphs compare g(r) of H2O molecules versus r (distance from the nanotube central axis). The CNTs inside and outside the diagrams are separated by a dashed line. RDF graphs of CNT inside include two peaks, which is located at the center of the nanotube and about 3 Å from the central axis. As it is seen, central peak of pure CNT is higher than capsulated CNT. Since drug is located at the center of the nanotube, density of water molecules in capsulated CNT is lower than pure CNT.

RDFs of water molecules from the center of nanotube. a CNT (9,9). b CNT-chlorambucil
RDF diagrams outside the pure CNT include two peaks which indicate the first and second shell-like of solvent molecules around the exterior of the nanotube. Outside diagrams of capsulated CNT have three peaks. The first peak occurs closer to the surface than that of the pure CNT. Actually, atomic charges due to encapsulation caused more solubility of capsulated CNT which enhance RDF around the CNT. Therefore, solvent molecules within the shells point to each other and produce wider peak than pure CNT. Moreover, encapsulated CNT indicates another peak at about 8 Å from CNT surface which corresponds to a third layer of solvent molecules. As it is seen, RDF of pure CNT shows limit of bulk water at distance larger than 7 Å from surface, whereas in encapsulated CNT, water molecules adopt the bulk structure at about 9 Å due to more hydrophilicity.
Conclusions
Potential usage of CNTs as drug transporter of alkylating agent anticancer drugs was studied in this research. Encapsulation behavior of chlorambucil, cyclophosphamide, and melphalan into inner volume of (9,9) CNT was considered in solution. In the CNT, three alkylating agent drugs and their complexes were first optimized by QM calculations and then association free energies of the structures in water were calculated using MC simulation.
DFT results revealed that all three drugs could encapsulate into CNT in the gas phase. To evaluate the actual application of CNT to transport these drugs in biological systems, encapsulation in aqueous media was the main purpose of this research.
Monte Carlo simulation consequences indicated that encapsulation of drugs into CNT enhance total interaction energy of nanotube by increasing electrostatic interactions. Computed solvation free energies also revealed enhancement of solvation of CNT after interacting with drugs. However, solvation free energy of cyclophosphamide and melphalan decreased after complexation with CNT. Encapsulation of chlorambucil into CNT enhanced the solvation free energy and also stability of this drug in physiological media. Moreover, computed association free energies indicated that among the three drugs, chlorambucil could only encapsulate into CNT. Hence, it is predicted that CNT is the ideal drug transporter system for chlorambucil to carry it to target cells.
References
Ralhan R, Kaur J (2007) Alkylating agents and cancer therapy. Expert Opin Ther Pat 17:1061–1075
Skeel R (2007) Antineoplastic drugs and biologic response modifiers: classification, use and toxicity of clinically useful agents (Chapter 4). Handbook of cancer chemotherapy 7th edition New York: Lippincott Williams & Wilkins
Rivera E (2003) Liposomal anthracyclines in metastatic breast cancer: clinical update. Oncologist 8:3–9
Wang W-S, Chiou T-J, Liu J-H, Fan FS, Yen C-C, Chen P-M (2000) Vincristine-induced dysphagia suggesting esophageal motor dysfunction: a case report. Jpn J Clin Oncol 30:515–518
Batist G, Gelmon KA, Chi KN, Miller WH, Chia SK, Mayer LD et al (2009) Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin Cancer Res 15:692–700
Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H et al (2007) A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br J Cancer 97:170–176
Wilson R, Plummer R, Adam J, Eatock M, Boddy A, Griffin M et al (2008) Phase I and pharmacokinetic study of NC-6004, a new platinum entity of cisplatin-conjugated polymer forming micelles. J Clin Oncol 26:2573
Miele E, Spinelli GP, Miele E, Tomao F, Tomao S (2009) Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. Int J Nanomedicine 4:99
Agarwal N, Fletcher D, Ward J (2007) Obesity and treatment of prostate cancer: what is the right dose of Lupron Depot? Clin Cancer Res 13:4027
Duncan R (2006) Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer 6:688–701
Chonn A, Cullis PR (1995) Recent advances in liposomal drug-delivery systems. Curr Opin Biotechnol 6:698–708
Torchilin VP (2010) Antinuclear antibodies with nucleosome-restricted specificity for targeted delivery of chemotherapeutic agents. Ther Deliv 1:257–272
Yang T, Choi M-K, Cui F-D, Lee S-J, Chung S-J, Shim C-K et al (2007) Antitumor effect of paclitaxel-loaded PEGylated immunoliposomes against human breast cancer cells. Pharm Res 24:2402–2411
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K (2000) Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 65:271–284
Juárez AR, Anota EC, Cocoletzi HH, Ramírez JS, Castro M (2017) Stability and electronic properties of armchair boron nitride/carbon nanotubes. Fullerenes, Nanotubes, Carbon Nanostruct 25:716–725
Bhirde AA, Patel V, Gavard J, Zhang G, Sousa AA, Masedunskas A et al (2009) Targeted killing of cancer cells in vivo and in vitro with EGF-directed carbon nanotube-based drug delivery. ACS Nano 3:307–316
Chen D, Wu X, Wang J, Han B, Zhu P, Peng C. Morphological observation of interaction between PAMAM dendrimer modified SWCNT and pancreatic cancer cells
Cui D, Zhang H, Sheng J, Wang Z, Toru A, He R et al (2010) Effects of CdSe/ZnS quantum dots covered multi-walled carbon nanotubes on murine embryonic stem cells. Nano Biomed Eng 2:236–244
Tian Z, Shi Y, Yin M, Shen H, Jia N (2011) Functionalized multiwalled carbon nanotubes-anticancer drug carriers: synthesis, targeting ability and antitumor activity. Nano Biomed Eng;3
Thakare VS, Das M, Jain AK, Patil S, Jain S (2010) Carbon nanotubes in cancer theragnosis. Nanomedicine. 5:1277–1301
Chen RJ, Bangsaruntip S, Drouvalakis KA, Kam NWS, Shim M, Li Y et al (2003) Noncovalent functionalization of carbon nanotubes for highly specific electronic biosensors. Proc Natl Acad Sci 100:4984–4989
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY (2013) Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev 65:1866–1879
Pantarotto D, Briand J-P, Prato M, Bianco A (2004) Translocation of bioactive peptides across cell membranes by carbon nanotubes. Chem Commun:16–17
Liu Z, Robinson JT, Tabakman SM, Yang K, Dai H (2011) Carbon materials for drug delivery & cancer therapy. Mater Today 14:316–323
Hilder TA, Hill JM (2008) Carbon nanotubes as drug delivery nanocapsules. Curr Appl Phys 8:258–261
Gao H, Kong Y, Cui D, Ozkan CS (2003) Spontaneous insertion of DNA oligonucleotides into carbon nanotubes. Nano Lett 3:471–473
Rahmani L, Ketabi S (2015) Solvation of alanine and histidine functionalized carbon nanotubes in aqueous media: a Monte Carlo simulation study. J Mol Liq 208:191–195
Roosta S, Hashemianzadeh SM, Ketabi S (2016) Encapsulation of cisplatin as an anti-cancer drug into boron-nitride and carbon nanotubes: molecular simulation and free energy calculation. Mater Sci Eng C 67:98–103
Ketabi S, Rahmani L (2017) Carbon nanotube as a carrier in drug delivery system for carnosine dipeptide: a computer simulation study. Mater Sci Eng C 73:173–181
Yang F, Jin C, Yang D, Jiang Y, Li J, Di Y et al (2011) Magnetic functionalised carbon nanotubes as drug vehicles for cancer lymph node metastasis treatment. Eur J Cancer 47:1873–1882
Sahoo NG, Bao H, Pan Y, Pal M, Kakran M, Cheng HKF et al (2011) Functionalized carbon nanomaterials as nanocarriers for loading and delivery of a poorly water-soluble anticancer drug: a comparative study. Chem Commun 47:5235–5237
Avti PK, Sitharaman B (2012) Luminescent single-walled carbon nanotube-sensitized europium nanoprobes for cellular imaging. Int J Nanomedicine 7:1953
Fabbro C, Ali-Boucetta H, Da Ros T, Kostarelos K, Bianco A, Prato M (2012) Targeting carbon nanotubes against cancer. Chem Commun 48:3911–3926
Zhang W, Zhang Z, Zhang Y (2011) The application of carbon nanotubes in target drug delivery systems for cancer therapies. Nanoscale Res Lett 6:555
Elhissi A, Ahmed W, Hassan IU, Dhanak V, D’Emanuele A (2012) Carbon nanotubes in cancer therapy and drug delivery. J Drug Deliv, 2012
Wang X, Liu Z (2012) Carbon nanotubes in biology and medicine: an overview. Chin Sci Bull 57:167–180
Becke AD (1993) Becke’s three parameter hybrid method using the LYP correlation functional. J Chem Phys 98:5648–5652
Becke AD (1998) A new inhomogeneity parameter in density-functional theory. J Chem Phys 109:2092–2098
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785
Petersson G, Al-Laham MA (1991) A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J Chem Phys 94:6081–6090
Petersson A, Bennett A, Tensfeldt TG, Al-Laham MA, Shirley WA, Mantzaris J (1988) A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J Chem Phys 89:2193–2218
Niyogi S, Hamon M, Hu H, Zhao BB, Bhowmik P, Sen R, Itkis ME, Haddon RC (2002). Acc Chem Res 35:1105
Umadevi D, Panigrahi S, Sastry GN (2014) Noncovalent interaction of carbon nanostructures. Acc Chem Res 47:2574–2581
Saikia N, Deka RC (2013) A comparison of the effect of nanotube chirality and electronic properties on the π–π interaction of single-wall carbon nanotubes with pyrazinamide antitubercular drug. Int J Quantum Chem 113:1272–1284
Umadevi D, Sastry GN (2013) Impact of the chirality and curvature of carbon nanostructures on their interaction with aromatics and amino acids. ChemPhysChem. 14:2570–2578
Bilic A, Gale JD (2008) Chemisorption of molecular hydrogen on carbon nanotubes: a route to effective hydrogen storage? J Phys Chem C 112:12568–12575
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH et al (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) Equation of state calculations by fast computing machines. J Chem Phys 21:1087–1092
Haar L, Gallagher J, Kell G (1984) National Bureau of Standards/National Research Council Steam Tables. Hemisphere Publishing Corp.: Bristol, PA
Marsh K (1987) International Union of Pure and Applied Chemistry: recommended reference materials for the realization of physicochemical properties. Section Optical Refraction, Blackwell Scientific Publications, Oxford, UK ;500
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Jorgensen WL (1981) Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J Am Chem Soc;(United States), 103
Jia Y, Wang M, Wu L, Gao C (2007) Separation of CO2/N2 gas mixture through carbon membranes: Monte Carlo simulation. Sep Sci Technol 42:3681–3695
Rappé AK, Casewit CJ, Colwell K, Goddard III WA, Skiff WM (1992) UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc 114:10024–10035
Hansen J-P, McDonald IR (2013) Theory of simple liquids: with applications to soft matter: academic press
Zwanzig RW (1954) High-temperature equation of state by a perturbation method. I. Nonpolar gases. J Chem Phys 22:1420–1426
Contreras ML, Torres C, Villarroel I, Rozas R (2019) Molecular dynamics assessment of doxorubicin–carbon nanotubes molecular interactions for the design of drug delivery systems. Struct Chem 30:369–384
Ghasemi-Kooch M, Dehestani M, Housaindokht MR, Bozorgmehr MR (2017) Oleuropein interactions with inner and outer surface of different types of carbon nanotubes: insights from molecular dynamic simulation. J Mol Liq 241:367–373
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Menon M, Richter E, Mavrandonakis A, Froudakis G, Andriotis AN (2004) Structure and stability of SiC nanotubes. Phys Rev B 69(11):115322–115334
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 131 kb)
Rights and permissions
About this article

Cite this article
Moradi, V., Ketabi, S., Samadizadeh, M. et al. Potentiality of carbon nanotube to encapsulate some alkylating agent anticancer drugs: a molecular simulation study. Struct Chem 32, 869–877 (2021). https://doi.org/10.1007/s11224-020-01658-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01658-x