
Abstract
We designed 12 novel cage-shaped high energy density compounds by incorporating –O– moiety and replacing up to two –NO2 groups at the different positions by –NH2 groups into the basic 2, 4, 6, 8, 10, 12, 13, 14, 15-nonanitro-2, 4, 6, 8, 10, 12, 13, 14, 15 nonaazaheptacyclo [5.5.1.13, 11. 15, 9] pentadecane (NNNAHP) skeleton. Their geometrical structures, electronic structures, heats of formation, detonation properties, thermodynamic properties, thermal stability, impact sensitivity, and free spaces were studied by using density functional theory. The favorable substitution position is a very important factor to tune the detonation properties, thermal stability, and impact sensitivity of the designed compounds. The thermal stability of the designed compounds was analyzed based on the bond dissociation energy of the weakest bond among all the bonds in a compound. The impact sensitivity and free spaces of the designed compounds were compared with well-known cage high energy density compound CL-20. Due to large densities, excellent detonation performance, suitable thermal stability, and low sensitivity, 11 compounds were chosen as potential high energy density compounds having huge potential for their synthesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the recent past, a lot of work has been focused on nitrogen-rich energetic compounds for developing new energetic compounds having both low sensitivity and high energetic properties together in the same compound [1,2,3,4,5,6,7,8,9,10]. But all the energetic compounds cannot fulfill this requirement. One among many reasons is that the energetic properties of parent nitrogenous heterocycles are not good enough; hence, they need energetic fragment substitution to enhance their energetic properties, which causes an increase in their sensitivity simultaneously. On the other hand, to decrease the number of energetic fragments will hurt their energetic properties. Therefore, keeping a good balance between sensitivity and energetic properties is still a major challenge in the field of high energy density compounds (HEDCs).
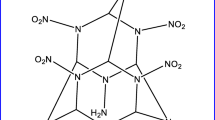
Energetic cage-shaped compounds emerged as one of the important classes of HEDCs due to their high heats of formation (HOFs) and high strain coupled with large crystal densities and compact structure [11,12,13,14,15,16,17,18,19]. For example, hexanitrohexaazaprismane [16] (Fig. 1) has high HOFs (1052.8 kJ/mol) and excellent detonation properties (P = 49.58 GPa, D = 10.13 km s−1, and ρ = 2.10 g cm−3). Because of the superior detonation performance, the synthesis and design of new cage HEDCs have become a hot field. One new energetic cage compound 2, 4, 6, 8, 10, 12, 13, 14, 15-nonanitro-2, 4, 6, 8, 10, 12, 13, 14, 15 nonaazaheptacyclo [5.5.1.13, 11. 15, 9] pentadecane (NNNAHP) [20] was designed recently, as shown in Fig. 1. NNNAHP has high positive HOFs (772.45 kJ/mol) and excellent detonation properties (P = 43.14 GPa, D = 9.55 km s−1, and ρ = 2.01 g cm−3). However, to the best of our knowledge, this compound has not been reported experimentally. Probably the highly nitrated cage structure gives it high sensitivity and makes its synthesis difficult. A large number of nitro (–NO2) groups are also associated with high sensitivity [21, 22]. Therefore, to decrease the sensitivity and make the experimental synthesis possible, it is essential to replace the nitro groups in NNNAHP by other structural fragments.

Molecular structures of HNHAH, CL-20, and NNAHP
Prior studies [23,24,25] show that the incorporation of oxygen (–O–) into the structure of energetic compounds is a very effective way to get thermally stable and low-sensitive energetic materials. Similarly, combining both nitro (–NO2) and amino (–NH2) groups in the same compound greatly decreases the sensitivity [26,27,28]. This sparks the idea of designing new cage-shaped compounds based on the skeleton of NNNAHP by incorporating –O– and replacing some of the –NO2 groups with –NH2 to combine both low sensitivity and high energy together in the same compounds.
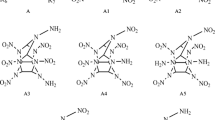
In this work, we studied the HOFs, densities, detonation velocity, detonation pressure, thermal stability, free space, impact sensitivity, and electronic structures of energetic NNNAHP derivatives (Fig. 2). The prime purpose of our study is to investigate the roles of –O– and –NH2 in designing insensitive cage-shaped compounds with superb detonation performance.

Molecular numbering of the designed compounds
Computational methodology
All the calculations were performed using the Gaussian 09 program package [29]. The DFT-B3LYP method with the 6-311G (d, p) basis set was employed to optimize the structures of the title compounds. Many studies show that the 6-311G (d, p) basis set is suitable to investigate the molecular structure and energy of energetic compounds [2, 30, 31].
Based on the total energies (E0), thermal corrections (ET), and zero-point energies (ZPE), the gas-phase phase HOFs (HOFsf, gas) were calculated via isodesmic reactions. As the number of all sorts of bonds on both reactants and products is the same in isodesmic reaction, therefore, the errors in the HOFs can be effectively decreased. The plotted isodesmic reactions used to evaluate the HOFsf, gas are displayed in Scheme 1.

Isodesmic reactions for the designed compounds (isodesmic reaction a for the compound P, b for the compound A, c for the compounds B–E, and d for the compounds F–L)
The heat of reactions ΔH298 K at room temperature can be calculated by the following expression:
where ΔHf, P and ΔHf, R are the HOFs of the product and reactants, respectively at 298 K. The value of ΔH298 K can be calculated using the following expression:
where ΔE0 and ΔZPE are the changes in total energies and zero-point energies between the products and reactants at 0 K, respectively, ΔET is the thermal correction from 0 to 298 K, and Δ(PV) is the pressure-volume work term and is equal to ΔnRT for the reactions of ideal gas.
It is well-known that most of the energetic compounds exist are solid-state; hence, the calculation of detonation properties (heat of detonation, detonation velocity, and detonation pressure) needs to calculate the solid-phase HOFs (ΔHf, solid) first. According to Hess’s law of constant heat summation, ΔHf, solid can be calculated from ΔHf, gas and heats of sublimation (ΔHsub).
The ΔHsub can be calculated by the following equation proposed by Politzer et al. [32]:
where A is the surface area of the 0.001 electrons/bohr3 isosurface of the electronic density of a molecule; \( \upsilon {\sigma}_{\mathrm{tot}}^2 \) is the electrostatic interaction index; and a, b, and c are 2.670× 10−4 kcal mol−1 Å−4, 1.650 kcal mol−1, and c = 2.9660 kcal mol−1 [33].
The crystal densities for the designed compounds were calculated using the method suggested by Politzer et al. [34]
where M is the molecular mass of the molecule (g/mol), V(0.001) is the volume enclosed by 0.001 electrons/bohr3 (cm3/molecule), and \( \upsilon {\sigma}_{\mathrm{tot}}^2 \) is the electrostatic interaction index factor. The constants α, β, and ϒ are equal to 0.9183, 0.0028, and 0.0443, respectively.
The detonation properties (detonation velocities and detonation pressures) were calculated based on the Kamlet-Jacobs equation [35].
where D and P are detonation velocity (GPa) and detonation pressure (km s−1), respectively, N is the number of detonation gases produced per gram of energetic materials, \( \overline{M} \) is the average of the produced gases, Q is the heat of detonation (cal/g) which can be calculated the HOF differences between products and reactants, and ρ is the density (g/cm3) of the energetic materials.
Bond dissociation energy (BDE) is one of the important parameters to understand the thermal decomposition and bonding strength of energetic materials. The homolytic BDE at 0 K can be calculated by the following expression:
Finally, the BDE with zero-point energy corrections was evaluated with the help of the following equation:
where ΔZPE denotes the differences between the zero-point energies of products and reactants.
The impact sensitivities for the designed compound can be calculated by using the positive variance (\( {\sigma}_{+}^2 \)) and the balance between the charges (υ) as follows [36]:
where h50 represents the height in centimeters from which 2.5 kg is dropped upon the energetic compound, and the chance of detonation is 50%; \( {\sigma}_{+}^2 \) shows the strength of the positive surface; υ is the balance of the charges on the surface; and α, β, and ϒ are the coefficients of − 0.0064, 241.42, and − 3.43, respectively.
Free space represented as ΔV is another important parameter used to know the impact sensitivity of energetic materials [37]. The higher the ΔV value is, the more sensitive the compound is. The ΔV can be calculated using the following empirical equation:
where ΔV is the free space, Veff is the effective volume which is needed to exactly fill the crystal, and the V0.003 is the volume enclosed by 0.003 electrons/bohr3. The effective volume can be calculated by dividing molecular mass over crystal density as follows:
Results and discussion
Molecular geometry and electronic structure
It is important to study the geometrical structures first before studying other properties. Figure 3 displays the optimized structures of the designed compounds. Table 1 enlists the bond lengths of some selective bonds for the title compounds. In every NNNAHP (P) derivative, one of the C–N bonds in the cage was replaced by the C–O bond. As clear in Table 1, in every compound, the average bond length of the C–O bond connecting two rings is smaller than that of the C–N bond connecting two rings. This indicates that connecting two rings in the cage structure via a C–O bond instead of a C–N bond could make it more compact crystal packing. The majority of the designed compounds have smaller average C–N (in the six-member ring) bond length than the parent one. Similarly, the average value of the N–N (linked to the cage) bond length is smaller than that of normal N–N bond (1.45 Å) for every designed compound.

Optimized structures of the title compounds
Figure 4 shows the 3D plots of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) as well as the HOMO and LUMO energies and their energy gaps for the title compounds. The energy gap is an important parameter which gives valuable knowledge about the reactivity of compounds [30, 38]. All the derivatives have suitable energy gaps ranging from 4.61 to 5.83 eV. Only the compound A has higher energy gap than the parent compound P, which means that all the compounds except for A are chemically more active than P. In the 3D plots of HOMO and LUMO, the negative and positive phases are shown in green and red, respectively. It is clear from Fig. 4 that all of the C–N orbits take part in the HOMO level, while there are only few C–N orbits to participate in the LUMO level. This indicates that the removal of an electron from the HOMO level could make the cage structure more unstable than the acceptance of an electron by the LUMO level. Additionally, in comparison with –NH2, the LUMO is more localized over the –NO2 group.

HOMO and LUMO energy levels and energy gaps of the title compounds
Molecular electrostatic potentials (MEPs) are critical to know the charge distribution and intermolecular forces in a molecule [39]. Prior research [40] shows that those compounds become more sensitive due to electron deficiency (positive potential) in the cage structure. Figure 5 shows the molecular electrostatic potential of the title compounds. The MEPs are for 0.001 electrons/bohr3 isosurface of electron density. The color varies from − 0.02 to 0.02 hartree. The blue and red indicate the electron-deficient (positive potential) and electron-rich (negative potential) regions, respectively. It can be seen in Fig. 5 that the negative potential is spread mainly around the N–NO2 portion, while the positive potential locates around the center of the cage. The compounds B, C, and D have the same number of –NO2, –NH2, and –O–. However, the positive potential in the compound C is more located at the center of the cage which might give it high sensitivity. Similarly, among E, F, G, H, I, J, K, and L, the positive potential in the case of J focuses more deep at the cage center than that of the other compounds. This might make it more sensitive.

Molecular electrostatic potentials of the title compounds
The regions with the most positive potential and most negative potential are most likely to be attacked by nucleophiles and electrophiles, respectively. The ratio of positive and negative surface area of the compounds P, A, B, C, D, E, F, G, H, I, J, K, and L are 1.12, 1.16, 1.0, 1.02, 1.08, 0.96, 0.96, 0.99, 0.95, 0.99, 0.99, 1.02, and 1.1, respectively. These values indicate that the introduction of the –O– moiety and increasing the number of –NH2 groups tend to keep the ratio around 1 (a kind of balance between the positive and negative surface areas) and so decrease the chances of nucleophilic and electrophilic attacks. This makes the molecules chemically more stable. The molecular surface areas in each electrostatic potential region were calculated and displayed in Fig. 6. Furthermore, the global molecular electrostatic potential maxima of the compounds A, B, C, D, E, F, G, H, I, J, K, and L are 52.8, 52.8, 55.0, 45.6, 47.6, 52.1, 50.5, 52.7, 50.7, 52.2, 49.9, and 39.4 kcal mol−1, respectively. Similarly, the global molecular electrostatic potential minima of the compounds A, B, C, D, E, F, G, H, I, J, K, and L are − 14.3, − 18.1, − 17.2, − 18.2, − 19.1, − 21.4, − 20.1, − 23.0, − 21.5, − 18.8, − 18.7 and − 19.8 kcal mol−1, respectively.

Percentage area in each electrostatic potential region for the title compounds
The contour line maps of the title compounds are displayed in Fig. 7. The high peaks are associated with the heavy nucleus’s nuclear charges (such as O and N) which intensify the electron accumulation. Due to the high electronegativity, it is found that the electron density around the oxygen is the highest, while that around hydrogen is the lowest. The decrease in the electronic density is noted in the regions A-1, A-2, B-1, B-2, C-1, C-2, D-1, D-2, E-1, E-2, F-1, F-2, G-1, G-2, H-1, I-1, I-2, I-3, J-1, J-2, K-1, K-2, K-3, L-1, L-2, and L-3 due to the repulsive forces of the heavy nucleuses with O and N.

Contour line map of electronic densities on the title compounds
Gas- and solid-phase HOFs
Table 2 enlists the total energies (E0), zero-point energies (ZPE), thermal corrections (ET), and HOFs for the reference compounds in the plotted isodesmic reactions. The experimental HOFs of CH4, NH3, CH3CH3, CH3OCH3, H2NNH2, and CH3NHCH3 were available in the literature [42], while the HOFs of C3H9N3 and H2NNO2 were unavailable. Additional calculations were performed at the G2 level [43] for atomization reaction CaHbOcNd → aC + bH + cO + dN to obtain their theoretical HOFs. The G2 theory is able to predict the HOFs of small molecules [44].
Table 3 shows the total energies, ZPEs, ET, heats of sublimation (ΔHsub), ΔHf, gas, and solid-phase HOFs (ΔHf, solid) of NNNAHP (P) and its derivatives. As clear from Table 2, all the title compounds have large positive gas-phase HOFs (478.72–547.27 kJ/mol) and solid-phase HOFs (287.92–369.62 kJ/mol). Among all the title compounds, the compound A has the lowest ΔHf,gas, while G has the highest one. When –O– is incorporated to the parent (P) to construct the compound A, the HOF decreases dramatically. But after introducing one amino (–NH2) (B, C, and D) and two amino groups (E, F, G, H, I, J, K, and L) to P, the HOFs gradually increase. This may be because of the strong interactions between the –NO2 and –NH2 groups. The compounds B, C, and D have the same chemical composition, but their HOFs are different from one another. The same is the case of E, F, G, H, I, J, K, and L. These results show that the substituents’ position plays a very important effect on the HOFs of the title compounds.
Figure 8 presents a comparison of solid- and gas-phase HOFs for the title compounds. Qualitatively, the solid-phase HOFs follow the same trend of gas-phase HOFs. This result indicates the variation trend of the gas- and solid-phase HOFs of the NNNAP-based compounds in the presence of –O– and –NH2 groups are consistent with one another.

Comparison of solid- and gas-phase HOFs of the title compounds
Detonation performance
Table 4 shows the oxygen balance (OB), ρ, the heat of detonation (Q), D, and P of the title compounds. In order to compare our results, the calculated detonation properties of NNNAHP and experimental ones of two famous energetic compounds CL-20 and HMX are also tabulated in Table 4. It is noted that the ρ values from 1.95 to 2.03 g cm−3 are all higher than that of HMX but lower than that of NNNAHP and CL-20. This is because the replacement of –NO2 by –O– and –NH2 brings down the density values in the compounds A–L. The D and P values of the title compounds range from 9.42 to 9.71 km s−1 and 41.2 to 44.48 GPa, respectively. All the title compounds have larger D values than CL-20 and HMX. Similarly, their P values are either greater or very close to that of CL-20 (Fig. 9).

Fig. 9
OB is a measure of excess or deficiency of the oxygen content in the HEDC needed to oxidize carbon and hydrogen to carbon mono oxide or carbon dioxide and water, respectively. Generally the high negative and high positive values of OB are not favorable to get high Q values. It is because the high negative value does not let the complete oxidation of carbon and hydrogen. Similarly, the high positive OB values cause the production of additional oxygen gases (O2) which withdraw a lot of energy during the detonation process. Therefore, it is good to keep the OB value around zero while designing HEDCs. All the designed compounds have high Q values than the parent molecule ranging from 1483.98 to 1595.4 cal g−1. It is important to note that the compounds B, C, and D have the OB values very close to zero (−2.94), and their Q values are the highest one among all derivatives. This result depicts that the oxygen balance values play a very important effect on the heat of detonation.
In the energetic compounds CaHbOcNd, CO or CO2, H2O, and N2 are the major gaseous species produced during the detonation. Kistiakowsky and Wilson [46, 47] proposed the rules which can be used to know the number of moles and types of gases produced during the detonation process. Table 5 shows the types of gases, their number of moles, and the total number of gases produced during the detonation for the title compounds.
The volume of gaseous products provides valuable knowledge about work done by the HEDC. Standard conditions (273 K temperature and 1 atm pressure) were applied to calculate the total volume of the gases, because the changing condition causes a change in the volume. The amount of gas produced by 1 g of energetic compound (V) can be calculated by dividing total volume over the molecular volume.
The explosive power of energetic compounds can be obtained by the multiplication of Q and volume of gases produced by 1 g of energetic compound (V) as follow:
Then, the explosive power value of an energetic compound can be compared with a standard explosive picric acid (PA) to calculate the explosive power index of the title compounds.
Table 6 presents the explosive power index values of the PA, parent compound (P), and title compounds. In terms of the explosive power index, all the title compounds except for A are better than PA and P. The explosive power index values of the designed compounds range from 97.68 to 120.10.
Thermodynamic properties
Based on the principle of statistical thermodynamics, some important thermodynamic properties such as standard molar entropies\( \left({S}_{\mathrm{m}}^{\theta}\right) \), standard molar heat capacities\( \left({C}_{\mathrm{P},\mathrm{m}}^{\theta}\right) \), and standard molar enthalpies\( \left({H}_{\mathrm{m}}^{\theta}\right) \) were calculated at different temperatures ranging from 100 to 600 K and are tabulated in Table 7. The corresponding equations for these three parameters at various temperatures (100–600 K) can be expressed as follows:
where a, b, and c are constants and are displayed in Table 7. It is clear in Table 7 that all the thermodynamic properties obviously increase with the increasing temperature. It is because the vibrational movement becomes intensive at higher temperature. However, their growth rates are different from each other. The growth rate of \( {H}_m^{\theta } \) gradually increases with the increasing temperature, while that of \( {S}_{\mathrm{m}}^{\theta }\ \mathrm{and}\ {C}_{\mathrm{P},\mathrm{m}}^{\theta } \) gradually decreases. It is also observed that at a specific temperature, the \( {S}_{\mathrm{m}}^{\theta }\ \mathrm{and}\ {C}_{\mathrm{P},\mathrm{m}}^{\theta } \) successively decrease after replacing one and two –NH2 groups by one and two –NO2 groups. However, for \( {H}_{\mathrm{m}}^{\theta } \), no such trend is noted.
Thermal stability
BDC gives important information about the stability of HEDCs. The larger the value of BDE is, the stronger the bond is, and the more difficult the bond breaks. To study the thermal stability of the title compounds, first, the bond with the smallest Mulliken bond order (BO) among all the bonds was determined, and then this bond was broken to calculate its BDE. It is found that all the bonds having the lowest BO values are the N–NO2 bonds. This means that the N–NO2 bonds are the weakest bonds and may rupture in the thermal decomposition process.
Table 8 lists the BDEs for the parent and title compounds. Among the designed compounds, E has the smallest BDE value (127.16 kJ/mol), while G has the largest one (144.37 kJ/mol). All the designed compounds have higher BDE values than the parent one except for the derivative E. When –O– was incorporated into the parent molecule to get the molecule A, its BDE increases, which shows that the incorporation of –O– into the cage helps to thermally stabilize its derivative. Replacing one –NO2 by one –NH2 further increases in its BDE. When the second –NO2 was replaced by –NH2, the situation was not clear as some of the double-substituted –NH2 compounds that have higher BDEs than single NH2-substituted compounds, while the others have lower BDEs.
It is understood that HEDCs should not only have high detonation properties but should also have suitable thermal stability. The energetic compounds having BDE values greater than 83.0 kJ/mol are considered to be thermally stable [49]. In this regard, all of our designed compounds are thermally stable.
Sensitivity
The impact sensitivity (h50) is the height from which impacting 2.5 kg mass on sample energetic compound leads 50% chance of initiating explosion [50, 51]. The larger the value of h50 is, the more insensitive the compound is. Table 9 lists the h50 values of the title compounds. In order to compare our results, the experimental h50 value of CL-20 is also enlisted in Table 9.
As shown in Table 9, all the designed compounds have larger h50 values than the parent one. In comparison with CL-20, it is found that only 4 compounds (A, B, C, and D) have smaller h50 values than CL-20, while all others (E, F, G, H, I, J, K, and L) have larger ones. The incorporation of –O– into the parent cage increases the h50 value of its derivatives. When one –NO2 group is replaced by –NH2, a further increase in the h50 value is observed. Similarly, when two –NO2 are replaced by two –NH2 groups, its h50 values continue to increase. These results indicate that incorporation of –O– moiety and –NH2 groups is very helpful to decrease the sensitivity of the energetic derivatives. The h50 values of the designed compounds range from 6.26 to 21.40 cm. It is worthy to note that compounds B, C, and D have the same chemical composition, but their h50 values are different from one another. The same situation is true for E, F, G, H, I, J, K, and L. This shows that the different substitution positions of the substituents can be used to tune the impact sensitivity of energetic materials. The predicted sensitivities of the designed molecules seem to be overall fairly high (low h50) because they still have a lot of the –NO2 groups that are in fact electron-withdrawing groups even after introducing –O– and –NH2 groups into the cage. Due to the electron-withdrawing effect of the –NO2 groups, the cage structure becomes more positive (electron-deficient), leading to the increase in the sensitivity of energetic materials. This is supported by some studies [39, 40]. However, it should be noted that all of our designed compounds are less sensitive as compared with the parent compound.
Free space denoted as (ΔV) is another parameter used to analyze the impact sensitivity of energetic materials [36, 52]. When external force such as shock or impact is applied on energetic compound, it gets compressed [53,54,55,56]. Due to compression, the temperature in the compound increases [56, 57]; the larger is the compression (decrease in volume), the larger is the increase in the temperature. The smaller or larger decrease in the volume is dependent on the free space available in the compound. The smaller the ΔV value is, the more insensitive the compound is. All the designed compounds have smaller ΔV values than that of the parent one but larger than CL-20. This means that all the compounds are less sensitive than the parent molecule but more sensitive than CL-20. However, it is noted that their ΔV values are very close to that of CL-20. The compound A has the highest ΔV value (105.46 Å3), while the compound I has the smallest one (92.07 Å3) among all the title compounds. The incorporation of –O– moiety is found to decrease their ΔV values. Similarly, the introduction of an increasing number of –NH2 groups into the cage can gradually decrease their ΔV values. This shows that our designing strategy is very useful to decrease the impact sensitivity of the designed compounds.
Figure 10 presents the ΔV and h50 profile of the designed compounds. It is clear that the compounds with large h50 values (more insensitive) have small ΔV values and vice versa, which is according to previous studies [26, 37, 52].

ΔV and h50 profile of the title compounds
Conclusions
In the present study, molecular structures, HOFs, detonation properties, thermodynamic properties, thermal stability, and impact sensitivity of 12 novel cage skeleton compounds based on the skeleton of NNNAHP were examined by employing the DFT-B3LYP method with 6-311G (d, p) basis set. The results show that favorable substitution position of –NH2 is very helpful to increase HOFs, detonation properties, thermal stability, and insensitivity of the designed compounds. All the 12 compounds have densities, detonation velocities, and detonation pressures over than 1.95 g cm−3, 9.42 km s−1, and 41.2 GPa, respectively. The incorporation of the –O– moiety and –NH2 groups is found to increase thermal stability (and to decrease impact sensitivity) for the designed compounds as compared with the parent molecule (NNNAHP). The majority of the compounds (8) have h50 values greater (less sensitive) than CL-20.
In addition, the findings of this work show that our designing strategy of incorporating –O– into the cage and replacing up to two –NO2 groups attached to the cage by the –NH2 groups is very useful to combine large density, excellent detonation property, suitable thermal stability, and low sensitivity all together in the same compound. Our strategy provides new ways to design novel cage skeleton compounds which can hold both low sensitivity and excellent detonation performance in one compound.
References
Talawar M, Sivabalan R, Senthilkumar N, Prabhu G, Asthana S (2004) Synthesis, characterization and thermal studies on furazan- and tetrazine-based high energy materials. J Hazard Mater 113(1–3):11–25
Wei T, Zhu W, Zhang X, Li Y-F, Xiao H (2009) Molecular design of 1, 2, 4, 5-tetrazine-based high-energy density materials. J Phys Chem A 113(33):9404–9412
Wei T, Zhu W, Zhang J, Xiao H (2010) DFT study on energetic tetrazolo-[1, 5-b]-1, 2, 4, 5-tetrazine and 1, 2, 4-triazolo-[4, 3-b]-1, 2, 4, 5-tetrazine derivatives. J Hazard Mater 179(1–3):581–590
Zhu W, Zhang C, Wei T, Xiao H (2011) Computational study of energetic nitrogen-rich derivatives of 1, 1′-and 5, 5′-bridged ditetrazoles. J Comput Chem 32(10):2298–2312
Chi W, Li L, Li B, Wu H (2012) Density functional calculation on a high energy density compound having the formula C2OH4−n (NO2)n. Struct Chem 23(6):1837–1841
Klapötke TM, Piercey DG, Stierstorfer J, Weyrauther M (2012) The synthesis and energetic properties of 5, 7-dinitrobenzo-1, 2, 3, 4-tetrazine-1, 3-dioxide (DNBTDO). Propellants Explos Pyrotech 37(5):527–535
Liu H, Du H, Wang G, Liu Y, Gong X (2012) Molecular design of new nitramine explosives: 1, 3, 5, 7-tetranitro-8-(nitromethyl)-4-imidazolino [4, 5-b] 4-imidazolino-[4, 5-e] pyridine and its N-oxide. J Mol Model 18(4):1325–1331
Klapötke TM, Leroux M, Schmid PC, Stierstorfer J (2016) Energetic materials based on 5, 5′-Diamino-4, 4′-dinitramino-3, 3′-bi-1, 2, 4-triazole. Chem Asian J 11(6):844–851
Jin X, Xiao M, Zhou G, Zhou J, Hu B (2019) Molecule design and properties of bridged 2, 2-bi (1, 3, 4-oxadiazole) energetic derivatives. RSC Adv 9(10):5417–5430
Khan RU, Zhu S, Zhu W (2019) DFT studies on nitrogen-rich pyrazino [2, 3-e] [1, 2, 3, 4] tetrazine–based high–energy density compounds. J Mol Model 25(9):283. https://doi.org/10.1007/s00894-019-4167-4
Paquette LA, Fischer JW, Engel P (1985) Synthesis and X-ray crystal structure analysis of a vicinally dinitro-substituted bishomopentaprismane. J Org Chem 50(14):2524–2527
Ghule V, Jadhav P, Patil R, Radhakrishnan S, Soman T (2009) Quantum-chemical studies on hexaazaisowurtzitanes. J Phys Chem A 114(1):498–503
Tan B, Long X, Li J (2012) The cage strain energies of high-energy compounds. Comput Theor Chem 993:66–72
Lin H, Zhu S-g, Zhang L, Peng X-h, Chen P-y, Li H-z (2013) Theoretical investigation of a novel high density cage compound 4, 8, 11, 14, 15–pentanitro-2, 6, 9, 13–tetraoxa-4, 8, 11, 14, 15-pentaazaheptacyclo [5.5. 1.1 3, 11. 1 5, 9] pentadecane. J Mol Model 19(3):1019–1026
Wang Y, Qi C, Song J-W, Zhao X-Q, Sun C-H, Pang S-P (2013) Trinitromethyl/trinitroethyl substituted CL-20 derivatives: structurally interesting and remarkably high energy. J Mol Model 19(3):1079–1087
Wu Q, Zhu W, Xiao H (2014) Computer-aided design of two novel and super-high energy cage explosives: dodecanitrohexaprismane and hexanitrohexaazaprismane. RSC Adv 4(8):3789–3797
Wu Q, Tan L, Hang Z, Wang J, Zhang Z, Zhu W (2015) A new design strategy on cage insensitive high explosives: symmetrically replacing carbon atoms by nitrogen atoms followed by the introduction of N-oxides. RSC Adv 5(113):93607–93614
Nielsen AT, Chafin AP, Christian SL, Moore DW, Nadler MP, Nissan RA, Vanderah DJ, Gilardi RD, George CF, Flippen-Anderson JL (1998) Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron 54(39):11793–11812
Zhang MX, Eaton PE, Gilardi R (2000) Hepta-and octanitrocubanes. Angew Chem Int Ed 39(2):401–404
Zhang J-y, Du H-c, Wang F, Gong X-d, Ying S-j (2012) Crystal structure, detonation performance, and thermal stability of a new polynitro cage compound: 2, 4, 6, 8, 10, 12, 13, 14, 15-nonanitro-2, 4, 6, 8, 10, 12, 13, 14, 15-nonaazaheptacyclo [5.5. 1.1 3, 11. 1 5, 9] pentadecane. J Mol Model 18(6):2369–2376
Zhang C, Shu Y, Huang Y, Zhao X, Dong H (2005) Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds. J Phys Chem B 109(18):8978–8982
Cao C, Gao S (2007) Two dominant factors influencing the impact sensitivities of nitrobenzenes and saturated nitro compounds. J Phys Chem B 111(43):12399–12402
Liang L, Huang H, Wang K, Bian C, Song J, Ling L, Zhao F, Zhou Z (2012) Oxy-bridged bis (1H-tetrazol-5-yl) furazan and its energetic salts paired with nitrogen-rich cations: highly thermally stable energetic materials with low sensitivity. J Mater Chem 22(41):21954–21964
Pan Y, Zhu W (2018) Designing and looking for novel cage compounds based on bicyclo-HMX as high energy density compounds. RSC Adv 8(1):44–52
Pan Y, Zhu W, Xiao H (2018) Molecular design on a new family of azaoxaadamantane cage compounds as potential high-energy density compounds. Can J Chem 97(2):86–93
Wu Q, Zhu W, Xiao H (2014) A new design strategy for high-energy low-sensitivity explosives: combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule of 1-amino-5-nitrotetrazole-3 N-oxide. J Mater Chem A 2(32):13006–13015
Jiao Y, Liu Z, Zhu W (2018) Searching for a new family of modified CL-20 cage derivatives with high energy and low sensitivity. Struct Chem 29(3):837–845
Politzer P, Murray JS (2016) High performance, low sensitivity: conflicting or compatible? Propellants Explosives, Pyrotechnics 41(3):414–425
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Mennucci B, Petersson G (2009) Gaussian 09 package. Gaussian Inc, Pittsburgh
Wang F, Du H, Zhang J, Gong X (2011) Computational studies on the crystal structure, thermodynamic properties, detonation performance, and pyrolysis mechanism of 2, 4, 6, 8-tetranitro-1, 3, 5, 7-tetraazacubane as a novel high energy density material. J Phys Chem A 115(42):11788–11795
Pan Y, Li J, Cheng B, Zhu W, Xiao H (2012) Computational studies on the heats of formation, energetic properties, and thermal stability of energetic nitrogen-rich furazano [3, 4-b] pyrazine-based derivatives. Comput Theor Chem 992:110–119
Politzer P, Murray JS, Edward Grice M, Desalvo M, Miller E (1997) Calculation of heats of sublimation and solid phase heats of formation. Mol Phys 91(5):923–928
Byrd EF, Rice BM (2006) Improved prediction of heats of formation of energetic materials using quantum mechanical calculations. J Phys Chem A 110(3):1005–1013
Politzer P, Martinez J, Murray JS, Concha MC, Toro-Labbe A (2009) An electrostatic interaction correction for improved crystal density prediction. Mol Phys 107(19):2095–2101
Kamlet MJ, Jacobs S (1968) Chemistry of detonations. I A simple method for calculating detonation properties of C–H–N–O explosives. J Chem Phys 48(1):23–35
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2010) A possible crystal volume factor in the impact sensitivities of some energetic compounds. J Mol Model 16(5):895–901
Politzer P, Murray JS (2014) Impact sensitivity and crystal lattice compressibility/free space. J Mol Model 20(5):2223
Ravi P, Gore GM, Sikder AK, Tewari SP (2012) A DFT study on the structure-property relationship of aminonitropyrazole-2-oxides. Int J Quantum Chem 112(6):1667–1677
Murray JS, Politzer P (2011) The electrostatic potential: an overview. Wiley Interdiscip Rev Comput Mol Sci 1(2):153–163
Rice BM, Hare JJ (2002) A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A 106(9):1770–1783
Scott AP, Radom L (1996) Harmonic vibrational frequencies: an evaluation of Hartree− Fock, Møller− Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J Phys Chem 100(41):16502–16513
Lide D (2004) The 84th edition of the CRC handbook of chemistry and physics. CRC Press, Boca Raton
Curtiss LA, Raghavachari K, Trucks GW, Pople JA (1991) Gaussian-2 theory for molecular energies of first-and second-row compounds. J Chem Phys 94(11):7221–7230
Curtiss LA, Raghavachari K, Redfern PC, Pople JA (1997) Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J Chem Phys 106(3):1063–1079
Talawar M, Sivabalan R, Mukundan T, Muthurajan H, Sikder A, Gandhe B, Rao AS (2009) Environmentally compatible next generation green energetic materials (GEMs). J Hazard Mater 161(2–3):589–607
Mader CL (1961) Detonation performance calculations using the Kistiakowsky-Wilson equation of state. Los Alamos Scientific Lab, N. Mex
Türker L, Varisļ S (2017) Defence technology. Interaction
Keshavarz MH, Oftadeh M (2004) New method for estimating the heat of formation of CHNO explosives in crystalline state. High Temp High Pressures 36(4):499
Chung G, Schmidt MW, Gordon MS (2000) An ab initio study of potential energy surfaces for N8 isomers. J Phys Chem A 104(23):5647–5650
Anders G, Borges Jr I (2011) Topological analysis of the molecular charge density and impact sensitivy models of energetic molecules. J Phys Chem A 115(32):9055–9068
Oliveira MA, Borges Jr I (2019) On the molecular origin of the sensitivity to impact of cyclic nitramines. Int J Quantum Chem 119(8):e25868
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2011) Sensitivity and the available free space per molecule in the unit cell. J Mol Model 17(10):2569–2574
Tsai D, Armstrong R (1994) Defect-enhanced structural relaxation mechanism for the evolution of hot spots in rapidly compressed crystals. J Phys Chem 98(43):10997–11000
Whitea C, Barretta J, Mintmirea J, Elert M, Robertson D (1995) Effects of Nanoscale Voids on the Sensitivity of Model Energetic Materials MRS Online Proceedings Library Archive 418
Rice BM, Mattson W, Trevino SF (1998) Molecular-dynamics investigation of the desensitization of detonable material. Phys Rev E 57(5):5106
Tarver CM, Urtiew PA, Tran TD (2005) Sensitivity of 2, 6-diamino-3, 5-dinitropyrazine-1-oxide. J Energ Mater 23(3):183–203
Lemons DS, Lund CM (1999) Thermodynamics of high temperature, Mie–Gruneisen solids. Am J Phys 67(12):1105–1108
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 21773119) and Science Challenging Program (no. TZ2016001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khan, R.U., Zhu, W. Designing and looking for novel low-sensitivity and high-energy cage derivatives based on the skeleton of nonanitro nonaaza pentadecane framework. Struct Chem 31, 1387–1402 (2020). https://doi.org/10.1007/s11224-020-01506-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01506-y