
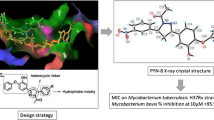
Abstract
In the present work, we have explored triclosan mimic diphenyl ether derivatives as inhibitors of Mycobacterium tuberculosis enoyl acyl carrier protein reductase (InhA) using a structure-based drug design approach. The virtual library of diphenyl ethers was designed and compounds with acceptable absorption, distribution, metabolism, excretion, and toxicity properties were docked. The compounds with higher dock score (5a-g) than triclosan were synthesized, characterized, and evaluated for in vitro antitubercular activity against Mycobacterium tuberculosis H37Rv. Among the synthesized compounds, compounds 5f and 5c appeared to be the most promising with minimum inhibitory concentration of 18 μM and 36 μM respectively. The molecular dynamics simulation study of the most active compound 5f and triclosan was performed, which correlates with its activity in comparison with triclosan. All the compounds were further evaluated for cytotoxicity studies against Vero, and HepG2 were found to be safe. Furthermore, compound 5f was evaluated for in vitro cytotoxicity against mouse macrophage cell lines (RAW 264.7), and the study indicated its safety in eukaryotes at 50-μM concentration. In addition, compounds 5a-g were also screened for their in vitro antibacterial activity against two gram-positive and two gram-negative bacteria by resazurin-based microtiter dilution assay method. Among the synthesized compounds, 5f and 5b appeared to be promising, against various gram-positive and gram-negative microorganisms, indicating its broad-spectrum potential.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) is the second leading cause of death worldwide due to infections [1, 2]. It is a highly infectious airborne disease caused by Mycobacterium tuberculosis. Among the patients infected with HIV, TB is the major cause of mortality [3]. WHO report states that 10 million new cases of TB were reported in the year 2017 [4]. A total of 1.6 million TB patients died in the year 2017, and among them, 300,000 were HIV positive [4]. The emergence of multidrug-resistant TB (MDR-TB), extensively drug-resistant TB (XDR-TB), and totally drug-resistant TB (TDR-TB) has further complicated the situation. The co-infection of TB with HIV has also contributed significantly to the mortality among TB patients. Despite treating tuberculosis by directly observed treatment, short-course (DOTS), the long duration of therapy leads to patient non-compliance, and this is anticipated to be the prime reason for drug resistance [5, 6]. This underlines the urgent need for developing effective and fast-acting new drugs to reduce the global burden of tuberculosis. Despite several endeavors to develop novel molecules to treat TB, isoniazid (INH) still remains to be the first-line drug for the treatment of TB.
Mycobacterium has a unique and complex cell wall structure. The biosynthetic pathway responsible for the synthesis of mycobacterial cell wall presents various drug targets providing an opportunity for the discovery of novel antitubercular agents [7]. Type II pathway fatty acid synthesis (FAS-II) pathway of MTb is involved in the extension of the fatty acid chains that are used for the biosynthesis of mycolic acid. FAS-II system is absent in higher eukaryotes and hence is an attractive target. Inhibitors of FAS-II enzymes are expected to be selectively toxic towards MTb due to the absence of FAS-II system in humans [8]. Fatty acids and mycolic acids form an essential component of the membrane and cell wall of M. tuberculosis, respectively. Mycobacterial enoyl-reductase (InhA), an enzyme belonging to the FAS-II pathway, catalyzes the NADH-dependent reduction of enoyl-ACP in the biosynthesis of fatty and mycolic acids. Isoniazid inhibits mycobacterial enoyl-reductase. Inhibitors of InhA have been shown to have in vivo efficacy, and thus, there is a precedent to support studies aimed at discovering InhA inhibitors [9,10,11,12,13].
INH, which is the first-line antitubercular agent, has been reported to inhibit InhA. Isoniazid is converted to its active form by reacting with the cofactor NAD(H), bound to the active site of the enzyme. This covalent adduct then binds to InhA with high affinity inhibiting its activity. The activation of isoniazid is carried out by the catalase-peroxidase enzyme encoded by katG, and mutation in this gene is the cause of resistance to isoniazid in 64% of clinical isolates. Therefore, molecules that directly inhibit InhA without any bio-activation circumvent this mechanism of resistance and are promising candidates [9,10,11].
Triclosan, which is a diphenyl ether derivative, inhibits InhA directly and has been reported to have activity against Mycobacterium tuberculosis H37Rv (MIC = 43.17 μM) [14]. Unlike INH, triclosan does not require prior activation to bind with ENR [15]. The literature reports on inhibitors of Mtb ENR suggest that restricting the entry of the natural substrate (acyl carrier protein) by inhibitors such as triclosan could be a potential approach to disrupting the essential reduction step in mycobacterial mycolic acid biosynthesis [13, 15,16,17]. Thus, efforts to find diphenyl ether predicated antitubercular agents have been made in recent years.
Diphenyl ether derivatives synthesized previously in our lab have exhibited good antitubercular activity against Mycobacterium tuberculosis H37Rv [18,19,20,21,22]. Encouraged by those results, it was decided to explore further the antitubercular activity of novel diphenyl ethers.
In silico studies like molecular docking of the synthesized compounds were performed to study the nature of interactions of the synthesized compounds in the binding site with the catalytic target residues. The published X-ray crystal structure of M. tuberculosis InhA (pdb 1p45) was downloaded from the protein data bank and used for the docking [23, 24]. This pdb consists of the crystal structure of InhA with triclosan as the co-crystalized ligand. Additionally, the most potent compound 5f complex was subjected to molecular dynamics simulation studies in order to understand the stability of the active compound InhA complex and the non-bonding interaction between the ligand and protein. Further absorption, distribution, metabolism, and excretion (ADME) properties of the synthesized compounds and other drugs like properties were predicted through the QikProp module of the Schrodinger software.
Experimental
Materials and methods
Various chemicals used as initial materials and as catalysts during this study were purchased from Sigma-Aldrich Chemical Pvt. Ltd., India, Spectrochem Pvt. Ltd. India, TCI Chemicals, India, and HiMedia Chemicals, India. All the solvents used were obtained from Merck Chemicals, India. Column chromatography was carried out on silica gel 100–200 mesh. The progress of the chemical reactions was monitored by TLC using aluminum-backed sheets of silica gel 60 F24 (Merck Millipore, India). Melting points were recorded on a laboratory melting point apparatus and are uncorrected. Vero (African Green monkey kidney), HepG2 (human Caucasian hepatocyte carcinoma), and RAW 264.7 (mouse monocyte-macrophage) cell lines used in this study were procured from the National Center for Cell Science, Pune, India, and maintained in high glucose DMEM medium with 10% FBS and 1% antibiotic-antimycotic solution, at 37 °C in a CO2 incubator (CLS-170-B-8, Serial No. 201020569, Esco Micro Pte. Ltd). Dulbecco’s Modified Eagle’s Medium (DMEM), phosphate buffer saline, antibiotic-antimycotic solution, and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5diphenyltetrazolium bromide] were obtained from Sigma-Aldrich Chemical Pvt. Ltd., India. Fetal bovine serum (FBS) was obtained from Invitrogen Bio Services India Pvt. Ltd., Bangalore, India. 1H NMR and 13C NMR spectra were recorded on an NMR spectrometer (AV400—400 MHz High-Resolution Multinuclear FT-NMR spectrometer, Bruker, USA) using DMSO-d6 as the solvent. Mass spectroscopy was performed using LC-MS (Linear ion trap, APCI mode, LC/MS, ThermoFisher Scientific LTQ21532 series, USA). IR spectrum was recorded using FTIR spectrophotometer (IR Affinity-1, Shimadzu, Japan) using KBr pellets.
General procedure of synthesis
Experimental procedure for the synthesis of 3-methoxy-4-phenoxybenzaldehyde, (1)
The 3-methoxy-4-phenoxybenzaldehyde (1) has been synthesized as per the procedures described in the literature [25, 26].
Experimental procedure for the synthesis of 3-hydroxy-4-phenoxybenzaldehyde, (2)
The 3-hydroxy-4-phenoxybenzaldehyde (2) have been synthesized as per the procedures described in the literature [27].
General experimental procedure for the synthesis of compounds, (3a-g)
Synthesis of (E)-3-(3-hydroxy-4-phenoxyphenyl)-1-phenylprop-2-en-1-one (3a) as an example
To a solution of 3-hydroxy-4-phenoxybenzaldehyde (2) (0.5 g, 1.58 mmol), acetophenone (0.187 mL, 1.58 mmol) in absolute alcohol (15.0 mL), ethanolic solution of KOH (0.348 g, 6.13 mmol) was added at 25–27 °C. The reaction mixture was stirred at ambient temperature. After completion of the reaction (14 h), the reaction mixture was poured into ice-cold water (100 mL) with continuous stirring and the residue obtained was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were separated, pooled, washed with water, brine, dried over anhydrous MgSO4, and evaporated under vacuum. The crude compound obtained was purified by column chromatography over silica 100–200 with hexane: ethyl acetate (8:2) as the mobile phase to afford the target compound.
Yield = 0.52 g (88%); mp = 102–104 °C; Rf = 0.64 (hexane: ethyl acetate = 8:2); λmax = 295 nm (MeOH); IR (KBr, cm−1) = 3032, 1658, 1602, 1572, 1481, 1452, 1244, 1149.
General experimental procedure for the synthesis of compounds, (4a-g)
Synthesis of 3-(3-hydroxy-4-phenoxyphenyl)-4-nitro-1-phenylbutan-1-one (4a) as an example
A solution of (E)-3-(3-hydroxy-4-phenoxyphenyl)-1-phenylprop-2-en-1-one (3a) (0.4 g, 1.26 mmol), nitromethane (1.32 mL, 25.30 mmol) and KOH (0.017 g, 0.31 mmol) in ethanol (40 mL) was heated at 60–65 °C. After completion of the reaction (12 h), the reaction mixture was cooled to room temperature. The solvent was removed in vacuum, and the residue was partitioned between ethyl acetate (100 mL) and H2O (100 mL). The organic layer was separated, dried over magnesium sulfate, and evaporated under reduced pressure. The crude compound obtained was purified by column chromatography over silica 100–200 with hexane: ethyl acetate (8:2) as the mobile phase to afford the target compound.
Yield = 0.45 g (95%); mp = 108–110 °C; Rf = 0.34 (hexane: ethyl acetate = 8:2); λmax = 302 nm (MeOH); IR (KBr, cm−1) = 3305, 3142, 1710, 1462, 1430, 1300, 1201.
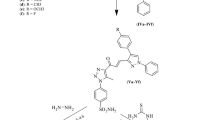
General experimental procedure for the synthesis of compounds, (5a-g)
Synthesis of 2-phenoxy-5-(5-phenyl-3,4-dihydro-2H-pyrrol-3-yl)phenol, (5a)
To a solution of 3-(3-hydroxy-4-phenoxyphenyl)-4-nitro-1-phenylbutan-1-one (4a) (0.3 g, 0.79 mmol) in acetic acid (3 mL), zinc powder (0.52 g, 7.95 mmol) was added in portions at 55 °C. The resultant mixture was stirred at 55 °C. After completion of the reaction (1 h), zinc powder was filtered off, and the filtrate was cooled to 0 °C. The filtrate was diluted with ethyl acetate and neutralized by the addition of sodium hydrogen carbonate. The mixture was extracted with dichloromethane, washed with brine, dried over magnesium sulfate, and evaporated under reduced pressure. The crude compound obtained was purified by column chromatography over silica 100–200 with hexane: ethyl acetate (8:2) as the mobile phase to afford the target compound.
Yield = 0.226 g (60%); mp = 127–129 °C; Rf = 0.34 (hexane:ethyl acetate = 8:2); λmax = 275 nm (MeOH); IR (KBr, cm−1) = 3452, 3057, 1680, 1527, 1483, 1440, 1245, 1132; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 7.89–7.88 (m, 2H), 7.48 (s, 3H), 7.28–7.26 (m, 2H), 6.98 (m, 1H), 6.89–6.88 (m, 1H), 6.82–6.80 (m, 3H), 6.69–6.68 (m, 1H), 4.39 (dd, J = 16.9, 7.3 Hz, 1H), 3.91 (d, J = 14.1 Hz, 1H), 3.58 (s, 1H), 3.50–3.39 (m, 1H), 3.03 (d, J = 15.7 Hz, 1H).13C NMR (100.62 MHz, DMSO-d6): δ 172.08, 158.60, 149.86, 143.14, 141.14, 134.65, 130.94, 129.98, 128.99, 128.04, 122.50, 122.18, 118.39, 116.42, 115.79, 69.44, 43.72, 42.28. LC-MS (APCI) m/z calcd. for [C22H19NO2]: 329.39, found 330.00 (M + H)−.
Synthesis of 2-phenoxy-5-(5-(p-tolyl)-3,4-dihydro-2H-pyrrol-3-yl)phenol, (5b)
Yield = 0.250 g (95%); mp = 141–142 °C; Rf = 0.37 (hexane:ethyl acetate = 8:2); λmax = 278 nm (MeOH); IR (KBr, cm−1) = 3450, 3047, 1680, 1520, 1485, 1445, 1251, 1142; 1H NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 7.79 (s, 2H), 7.28–6.99 (m, 5H), 6.89–6.81 (m, 4H), 6.69 (s, 1H), 4.37 (dd, J = 17.4, 8.8 Hz, 1H), 3.91–3.87 (m, 1H), 3.69–3.56 (m, 1H), 3.01–2.97 (m, 1H), 2.51 (s, 1H), 2.36 (s, 3H). 13C NMR (100.62 MHz, DMSO) δ 171.86, 158.61, 149.85, 143.19, 141.12, 140.65, 132.04, 129.97, 129.55, 128.03, 122.49, 122.18, 118.37, 116.62, 116.42, 115.79, 69.34, 43.69, 42.28, 21.49. LC-MS (APCI) m/z calcd. for [C23H21NO2]: 343.42, found 344.21 (M + H)−.
Synthesis of 5-(5-(4-fluorophenyl)-3,4-dihydro-2H-pyrrol-3-yl)-2-phenoxyphenol, (5c)
Yield = 0.241 g (91%); mp = 147–148 °C; Rf = 0.42 (hexane:ethyl acetate = 8:2); λmax = 279 nm (MeOH); IR (KBr, cm−1) = 3455, 3043, 1681, 1522, 1475, 1402, 1443, 1241, 1143,; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 7.89–7.88 (m, 2H), 7.48 (s, 3H), 7.28–7.26 (m, 2H), 6.98 (m, 1H), 6.89–6.88 (m, 1H), 6.82–6.80 (m, 3H), 4.39 (dd, J = 16.9, 7.3 Hz, 1H), 3.91 (d, J = 14.1 Hz, 1H), 3.58 (s, 1H), 3.50–3.39 (m, 1H), 3.03 (d, J = 15.7 Hz, 1H).13C NMR (100.62 MHz, DMSO-d6): δ 172.08, 158.60, 149.86, 143.14, 141.14, 134.65, 130.94, 129.98, 128.99, 128.04, 122.50, 122.18, 118.39, 116.42, 115.79, 69.44, 43.72, 42.28. LC-MS (APCI) m/z calcd. for [C22H18FNO2]: 347.38, found 348.20 (M + H)−.
Synthesis of 5-(5-(4-methoxyphenyl)-3,4-dihydro-2H-pyrrol-3-yl)-2-phenoxyphenol, (5d)
Yield = 0.220 g (83%); mp = 168–170 °C; Rf = 0.52 (hexane:ethyl acetate = 8:2); λmax = 247 nm (MeOH); IR (KBr, cm−1) = 3452, 3057, 1680, 1527, 1483, 1440, 1245, 1132; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 7.85–7.83 (m, 2H), 7.29 (s, 2H), 7.01–7.00 (m, 3H), 6.90–6.81 (m, 4H), 6.69–6.67 (m, 1H), 4.38–4.32 (m, 1H), 3.89–3.85 (m, 1H), 3.81 (s, 3H), 3.55 (m, 1H), 2.99 (dd, J = 15.7, 0.2 Hz, 1H), 2.01–2.09 (m, 1H). 13C NMR (100.62 MHz, DMSO-d6): δ 171.31, 161.50, 158.61, 149.84, 143.26, 141.10, 129.97, 129.69, 127.42, 122.48, 122.18, 118.38, 116.41, 114.78, 114.31, 69.26, 55.75, 43.68, 42.36, 31.16. LC-MS (APCI) m/z calcd. for [C23H21NO3]: 359.42, found 360.12 (M + H)−.
Synthesis of 5-(5-(3-methoxyphenyl)-3,4-dihydro-2H-pyrrol-3-yl)-2-phenoxyphenol, (5e)
Yield = 0.23 g (88%); mp = 162–165 °C; Rf = 0.56 (hexane:ethyl acetate = 8:2); λmax = 244 nm (MeOH); IR (KBr, cm−1) = 3452, 3057, 1680, 1527, 1483, 1440, 1245, 1132; 1H NMR (400 MHz, DMSO-d6) δ δ 9.47 (s, 1H), 7.48–7.35 (m, 3H), 7.35–7.24 (m, 2H), 7.10–6.86 (m, 3H), 6.85–6.77 (m, 3H), 6.73–6.65 (m, 1H), 4.39 (dd, J = 16.5, 8.3 Hz, 1H), 3.96–3.85 (m, 1H), 3.81 (s, 3H), 3.58 (p, J = 7.7, 7.1 Hz, 1H), 3.44 (dd, J = 17.3, 9.5 Hz, 1H), 3.02 (dd, J = 16.9, 5.8 Hz, 1H). 13C NMR (100.62 MHz, DMSO-d6): δ 171.45, 159.90, 157.89, 149.47, 145.37, 131.77, 130.17, 130.13, 122.88, 121.54, 120.13, 119.39, 117.17, 116.32, 113.09, 111.99, 69.24, 55.43, 43.48. LC-MS (APCI) m/z calcd. for [C23H21NO3]: 359.42, found 360.01 (M + H)−.
Synthesis of 2-phenoxy-5-(5-(m-tolyl)-3,4-dihydro-2H-pyrrol-3-yl)phenol, (5f)
Yield = 0.21 g (80%); mp = 145–146 °C; Rf = 0.60 (hexane:ethyl acetate = 8:2); λmax = 264 nm (MeOH); IR (KBr, cm−1) = 3452, 3043, 1682, 1521, 1480, 1445, 1252, 1142; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 7.79 (s, 2H), 7.28–6.99 (m, 5H), 6.89–6.81 (m, 4H), 6.69 (s, 1H), 4.37 (dd, J = 17.4, 8.8 Hz, 1H), 3.91–3.87 (m, 1H), 3.69–3.56 (m, 1H), 3.01–2.97 (m, 1H), 2.51 (s, 1H), 2.37 (s, 3H). 13C NMR (100.62 MHz, DMSO) δ 171.86, 158.60, 149.85, 143.19, 141.12, 140.65, 132.04, 129.97, 129.55, 128.03, 122.49, 122.18, 118.37, 116.62, 116.42, 115.79, 69.34, 43.69, 42.27, 21.49. LC-MS (APCI) m/z calcd. for [C23H21NO2]: 343.42, found 344.11 (M + H)−.
Synthesis of 5-(5-(2-fluorophenyl)-3,4-dihydro-2H-pyrrol-3-yl)-2-phenoxyphenol, (5g)
Yield = 0.20 g (76%); mp = 149–150 °C; Rf = 0.61 (hexane:ethyl acetate = 8:2); λmax = 270 nm (MeOH); IR (KBr, cm−1) = 3457, 3042, 1680, 1522, 1475, 1401, 1443, 1240, 1143,; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 7.79 (s, 2H), 7.27–6.99 (m, 5H), 6.88–6.81 (m, 4H), 6.69 (s, 1H), 4.37 (dd, J = 17.4, 8.8 Hz, 1H), 3.91–3.87 (m, 1H), 3.69–3.56 (m, 1H), 3.01–2.97 (m, 1H), 2.51 (s, 1H), 2.35 (s, 3H). 13C NMR (100.62 MHz, DMSO) δ 171.85, 158.60, 149.85, 143.19, 141.11, 140.65, 132.04, 129.96, 129.54, 128.03, 122.49, 122.17, 118.36, 116.60, 116.41, 115.79, 69.33, 43.69, 42.27, 21.48. LC-MS (APCI) m/z calcd. for [C22H18FNO2]: 347.38, found 348.45 (M + H)−.
Biological activities
In vitro antitubercular screening
The synthesized compounds were screened against Mycobacterium tuberculosis H37Rv for the determination of minimum inhibitory concentration (MIC) by Microplate Alamar Blue Assay (MABA) protocol using two different media, i.e., regular 7H9-based medium and low-iron GAST with normal pH 6.6 glycerol-alanine-salts (GAS) medium. An inoculum of Mycobacterium tuberculosis H37Rv strain culture suspension was used for this assay. A sterile stock solution of synthesized compounds and standard compounds was prepared in DMSO. The Mycobacterium tuberculosis H37Rv (ATTCC 27294) culture suspension from Lowenstein–Jensen slants in 7H9 broth was mixed and adjusted to a turbidity equivalent to that of a 1 McFarland standard (3 × 108 cfu/mL). The final cell suspension concentration of 2 × 105 cfu/mL was achieved by diluting it with the required medium. The assay was carried out in 96 well plates using serial dilution technique where the medium (50 μL) was added and serial dilutions of compounds was done for attaining the final concentration (50, 25, 12.5, 6.25, 3.125, 1.625, 0.812, 0.406, 0.203, 0.101, 0.050, 0.025 μM). Isoniazid was used as a positive control and DMSO as a negative control. To each well, 50 μL of diluted culture was added. Plates were kept inside a zip lock bag and were incubated at 37 °C for 14 days. At weeks 1 and 2, the plates were read with an inverted enlarging mirror plate reader and graded as either growth, no growth. Furthermore, at week 2, the 1/10th volume of Alamar blue reagent was added to each well of the plate and again incubated for 24 h at 37 °C. The color change from blue to pink represented bacterial growth and the blue color was considered as no bacterial growth. The color developed in compounds was compared with the color present in the control. The MIC was defined as the lowest drug concentration, which inhibited the growth of bacteria [27,28,29].
In vitro antibacterial activity
All the synthesized compounds (5a-g) were investigated for their in vitro antibacterial activity against two gram-positive bacteria Bacillus subtilis (ATCC 6633) and Staphylococcus aureus (ATCC-29213); and two gram-negative bacteria Escherichia coli (ATCC-11229) and Pseudomonas aeruginosa (ATCC-27853) for their inhibitory activity using resazurin-based microtiter dilution assay in 96 well plate in triplicate. One hundred microliters of the test compounds in 10% (v/v) DMSO was added to the first row of the microtiter plate. Fifty microliters of the nutrient broth was added to the remaining wells. Serial dilutions were made using a multichannel pipette. Furthermore, 10 μL of resazurin dye was added to each well, and 30 μL of 3.3× strength broth was added to each well to achieve the volume of single strength. Finally, 10 μL of bacterial suspension (5 × 106 cfu/mL) was added to each well. Each plate was packed loosely with cling film and was placed in the incubator at 37 °C for 18–24 h. Ciprofloxacin and triclosan were used as positive controls. After 24 h, any color change from violet to pink was recorded as positive. The lowest concentration where the color change occurred was taken as the MIC value of the compounds [30].
In vitro cell cytotoxicity screening
The cytotoxicity of the synthesized diphenyl ether derivatives on mammalian Vero cell lines (African Green monkey kidney) and HepG2 (human Caucasian hepatocyte carcinoma) cell lines was investigated using the MTT assay protocol [31,32,33,34]. The most active compounds (5c and 5f) were further examined for its cytotoxicity in RAW 264.7 (mouse monocyte-macrophage) at 50-μM concentration [35].
The test samples were prepared in various concentrations in DMEM and added to the 96 well plates. DMSO concentration was maintained at ≤ 0.1%. One hundred microliters of sample dilutions were added to each well of 96 well plates containing a monolayer of Vero cells (104 cells/well) in a 96-well plate. The plates were incubated at 37 °C with 5% of CO2 inside an incubator. DMSO was used as a blank. Positive control (only inoculum) and negative control (only media) were also maintained on the plate. After 72 h of incubation, the supernatant was removed from the wells and added with 50 μL of MTT (2 mg/mL) in the dark. It was further incubated at 37 °C for 3 h. After the incubation, the supernatant was removed carefully from each well, and 50 μL of sterile DMSO (filtered through a 0.22-μm syringe filter) was added. The plate was transferred to an incubator and kept at 37 °C for 2 h. The optical density (OD) of the wells was measured at 540 nm using an Elisa Reader. In the case of HepG2 cells, 5 × 103 cells/well was added and MTT was added after 24 h of incubation. Optical density (OD) readings from each well were entered into the equations shown below to determine % cell viability and % cell inhibition.
In silico studies
Molecular docking study
The molecular docking studies were performed using the glide dock tool of Schrodinger 2017 for understanding the binding mode of the compounds with the target. The crystal structure of mycobacterial InhA with pdb code 1p45 was downloaded from the protein data bank. The protein was found to be heterodimer consisting of two chains named as chains A and B. Among these chains, after careful observation, it was found that chain B contained one triclosan ligand representing required catalytic interactions at the catalytic site of the protein. By contrast, chain A consisted of two triclosan ligands, and among these two triclosan ligands, one was showing required catalytic interactions, while another ligand was simply lying above demonstrating Van der Waals interactions. Additionally, we thought that the larger cavity of chain A would be detrimental for the residue interactions for the designed ligands. Hence, we retained chain B and deleted chain A from the protein. Furthermore, chain B was optimized using the protein preparation wizard of the Schrodinger 2017 module. Preprocessing of this protein was done by assigning bond orders, adding hydrogen, and treating disulfide. The water molecules within 5 Å of the binding site were removed. Energy minimization of the protein structure to root-mean-square deviation (RMSD) of 0.30 Å was done using the OPLS-2005 force field. Around the co-crystallized triclosan, the ligand-receptor grid was generated using default parameters through the grid construction tool of Schrodinger 2017. The grid was generated so that it can lodge the ligands similar in size of triclosan. The designed molecule structure was drawn using a 2D sketch of Schrodinger 2017. Different conformers were generated through Epik using the Ligprep application of the Maestro 11.04 module of Schrodinger 2017. All the structures were subjected to its energy minimization using the OPLS 2005 force filed in order to get conformer with the least energy. Prepared ligands were subjected to docking using extra precision mode. Van der Wall radii of the ligands were scaled to 0.8, and the partial change cutoff was kept less than 0.15. The ligand position was kept at the center of the 10-Å docking sphere. For all docking, default settings were used in the glide module of Schrodinger 2017. To probe the RMSD and docking parameters, before starting docking of designed ligands, co-crystallized ligand triclosan was extracted from the optimized protein and it was redocked [36, 37]. The docking score and its interactions are analyzed and reported in Table 3.
Molecular dynamics simulation
Molecular dynamics (MD) was run using the Desmond module of the Schrodinger suite. A three-step process comprising of system builder, minimization, and molecular dynamics was used for the study. Using the system builder panel, the orthorhombic simulation box was prepared with the TIP3P explicit water model in such a way that the minimum distance between the protein surface and the solvent surface is 10 Å, and the charge was neutralized by adding counter ions. A 0.15-M NaCl was used to provide an isosmotic salt environment. The system was simulated for 200 ps for equilibration. The simulation was for a total of 30 ns, using NPT ensemble at a temperature of 300 K and atmospheric pressure (1.013 bars) with the default setting of relaxation before simulation. Around 2000 frames were generated during the trajectory of the simulation exercise. To understand the stability of the complex during MD simulation, the protein backbone frames were aligned to the backbone of the initial frame, and then the RMSD was calculated with respect to the initial frame [38].
In silico ADME studies
ADME properties were predicted using the QikProp tool of the Schrodinger suite. Compounds with undesirable features like reactive groups and poor pharmacokinetic properties were removed. Descriptors like lipophilicity (log P), molecular weight, number of nitrogen and oxygen, hydrogen bond donor/acceptor, solubility, % human absorption, number of rotors, and polar surface area (PSA) were taken into consideration for the virtual screening. Lipinski’s parameter was also considered for selecting drug-like compounds for synthesis. Compounds that met absorption, distribution, metabolism, excretion, and toxicity (ADMET) and Lipinski’s drug-like parameters were subjected to a docking study [39].
Determination of logP
HPLC-based method was used for the determination of the logP of the synthesized compound. The chromatographic runs were conducted on HPLC at room temperature using ODS-4 (Intersil ODS-4, 5 μm, 4.6 × 150 mm, GL Science Inc.) column and equipped with UV-Visible detector. Numerical analysis and data processing was done using Lab solution-2013 software. 3-Morpholinopropane-1-sulfonic acid (MOPS, 4.18 g) was added to 900 mL of octanol saturated MilliQ water, and the volume was made up to 1 L. The pH of the buffer was adjusted to 7.4. A mixture of methanol (0.25% v/v octanol) and buffer at the ratio of 60:40, 65:45, and 70:30 were used to elute the test sample. Five microliters of the sample was injected, and the flow rate was kept at 1 mL/min. The logarithm of k’ was extrapolated to a 0% concentration of methanol in the graph. Log k’ at 0% methanol was calculated from the regression equation (R2 = 0.99) generated from the graph to determine logP [40,41,42].
Determination of pKa
HPLC-based method was used for the determination of pKa of the synthesized compound. The chromatographic runs were conducted on HPLC at room temperature using the C18 column (Gemini 5 μ C18 110 Å, 4.6 × 150 mm, Phenomenex) and UV-Visible detector. Numerical analysis and data processing was done using Lab solution-2013 software. Buffers of pH 2, 5, 7.4, and 10 were prepared by mixing universal buffer I [phosphoric acid (1.96 g), glacial acetic acid (1.2 g, 1.14 mL), and boric acid (1.36 g) were added to MilliQ water, and volume was made up to 5 L] and universal buffer II (0.02 M NaOH) at different proportions. The mobile phase contained acetonitrile as an organic modifier. Chromatographic measurements were done at 25–27 °C with an eluent flow rate of 1 mL/min. The compound was eluted using acetonitrile: buffer in the ratio of 50:50 in each different pH point (pH 2, 5, 7.4, and 10). Signal was detected at λmax 270 nm. The sample run time was kept from 15 min to 1 h. Three microliters of samples were injected using an autosampler. Acetonitrile was used as a blank. pH gradient run was applied with a fixed concentration of organic modifier, providing complete suppression of ionization of the test sample at the beginning of the gradient and its full ionization at its end. The retention time values, tR of compound 5f and 5c, was determined from three separate injections [43,44,45].
Evaluation of the extent of protein binding
The extent of binding of compound 5c to serum protein was determined over an HSA (human serum albumin) column using the reverse phase HPLC technique. The chromatographic runs were conducted on HPLC using the Thermo-HSA column (4.6 × 150 mm, 5 μm/025) and UV-Visible detector. Numerical analysis and data processing was done using Lab solution-2012 software. The mobile phase contained isopropyl alcohol as the organic modifier and potassium phosphate buffer (0.067 M, pH 7.4) as the aqueous phase. Compound 5c was dissolved in isopropyl alcohol to prepare a test sample of concentration 10 μg/mL. Chromatographic measurements were done at 25–27 °C with an eluent flow rate of 1–2 mL/min. The HSA column retention characteristics were calibrated using verapamil and metoprolol. Compound 5c was then eluted using buffer and acetonitrile in gradient run at the ratio of 90:10. Signals were detected at λmax 256 nm. Retention time (RT) of isopropanol (IPA) was used as t0 [46].
Result and discussion
Rational design strategy
Diphenyl ether derivatives were the designed possessing phenolic group in ring A as it was found to be essential for antitubercular activity, and functional diversification was performed at the 4th position of the ring A. Pyrroline ring was introduced anticipating that it would provide optimum length between the diphenyl ether moiety and the ring C thus enabling the ligand to fit in the binding pocket.
The presence of the phenolic group in ring A of triclosan is crucial for the antitubercular activity (Fig. 1). Our earlier studies also confirmed that the presence of a phenolic group in ring A of diphenyl ether derivatives is essential for antitubercular activity. A literature survey showed that chlorine atoms present in triclosan are responsible for adverse steric interactions with the enzyme, and their removal from the scaffold increases affinity significantly. Therefore, while designing diphenyl ether derivatives, ring B was not substituted with chlorine atoms. Removal of three chlorine atoms of triclosan also brought down the lipophilicity of the designed molecules substantially. The C-4 position of ring A of diphenyl ether was linked to Ring C through a five-membered heterocyclic ring (pyrroline). Studies carried in our own laboratory previously had suggested that a linker containing two to three atoms is ideal for the antitubercular activity. The introduction of the pyrroline ring between the two phenyl rings also achieves this purpose. Furthermore, the presence of nitrogen also contributes to the lowering of lipophilicity. Functional group diversification was done on ring C to get various derivatives with different lipophilicities. All the synthesized compounds were also subjected to preliminary antibacterial studies to check its inhibitory potential against gram-positive and gram-negative bacteria.

Rational design strategy of compounds 5a-g
Chemistry
The synthetic route to target compounds 5a-g is illustrated in scheme 1 (Fig. 2) 3-Methoxy-4-phenoxybenzaldehyde (1) was synthesized by the condensation of vanillin with phenylboronic acid [23, 28, 29]. Demethylation of the aldehyde 1 was brought about by treating with HBr, resulting in the formation of 3-hydroxy-4-phenoxybenzaldehyde (2) [18, 47]. Compound 2 was then condensed with various substituted benzaldehydes to obtain corresponding chalcones 3a-g [48]. Compounds 4a-g were obtained by the Michael addition of nitromethane to the chalcones using potassium hydroxide as a base [48, 49]. Reductive cyclization of 4a-g was brought about by zinc in glacial acetic acid to obtain the corresponding pyrrolines 5a-g [50]. 1H NMR spectrum of 5e showed a singlet at δ 9.47 (1H) ppm, confirming the presence of the phenolic group. The presence of double doublets for two sets of CH2 protons and one pentet/multiplet for CH protons confirmed the formation of pyrrolines. The disappearance of the band at 1678 cm−1 in the IR spectrum of 5e confirmed the absence of a C=O group of aldehyde.

Synthetic scheme
Antitubercular activity
The synthesized compounds 5a-g were screened for their activity against Mtb H37Rv (ATCC-27294) using the standard micro broth dilution MIC protocol using two different media; regular 7H9-based medium and glycerol-alanine-salts-Tween with iron (GAST-Fe) (pH 6.6) medium (Table 1). INH and triclosan were used as reference drugs. The most potent of all compounds tested was compound 5f having MIC value 18 μM in 7H9-based medium, followed by compound 5c with a MIC value of 36 μM in GAST/Fe medium. The rest of the compounds did not possess promising antitubercular activity. The results indicate that the better activity of compound 5f could be due to the presence of the electron-donating methyl group at the ortho position and a relatively higher value of log P (4.97) compared with other compounds. The effectiveness of the drug in exhibiting an antitubercular activity depends on its accumulation in the cell, leading to cell growth arrest. Higher lipophilic nature of 5f and 5c among the synthesized molecules may correlate with its ability to cross Mtb’s hydrophobic cell wall and phospholipid membrane, leading to higher antitubercular activity.
In vitro cell cytotoxicity assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay technique was used to assess the safety profile of synthesized compounds 5a-g against Vero and HepG2 cell lines. The estimated values of CC50 and the index of selectivity (SI) are shown in (Table 1). The results indicate that all compounds are safe. Most active compounds 5f and 5c had the selectivity index of more than 10. Since Mtb resides within the macrophages, the monocyte-macrophage cell lines (RAW 264.7) were used to check whether the screened compounds were selectively toxic to Mycobacterium and not to macrophages. To prove this, MTT assay was used to evaluate the most active compound 5f for in vitro cytotoxicity in (RAW 264.7) mouse macrophage cell lines. The result showed that the most active compound 5f had 38.60% inhibition at its 50 μM concentration indicated its safety in eukaryotes.
In vitro antibacterial activity
The synthesized (5a-g) compounds were investigated against two gram-positive bacteria Bacillus subtilis (ATCC 6633) and Staphylococcus aureus (ATCC-29213) and two gram-negative bacteria Escherichia coli (ATCC-11229) and Pseudomonas aeruginosa (ATCC-27853) for in vitro antibacterial activity. Triclosan and ciprofloxacin were used as standard drugs for comparison. The MICs of the synthesized compounds are listed in Table 2. Among the compounds screened, 5f was found to be the most active with MICs of 9 μM, 18 μM, and 18 μM against Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa respectively. Compound 5f, however, was less active against Bacillus subtilis (MIC ≥ 73 μM). Compound 5b was also found to be moderately active against with MICs of 18 μM against Staphylococcus aureus and Pseudomonas aeruginosa; however, it was inactive against Bacillus subtilis and Escherichia coli. It was observed that the maximum antibacterial activity was shown by compounds with logP close to 5.0. This can be due to differences in membrane permeability of the synthesized compounds, which also depends on the lipophilicity of the compounds.
In silico molecular modeling and docking study
In order to understand the binding mode of molecules to the active site residues of the target InhA, in silico computational method was used. The docking study was performed on the crystal structure of InhA (pdb: 1P45). Compounds 5a-g and triclosan were docked into the active site of the target enzyme. Molecular docking results (Table 3) of 5a-g revealed that the substituted 2-phenoxy-5-(5-phenyl-3,4-dihydro-2H-pyrrol-3-yl)phenol basic scaffold fits inside the InhA binding pocket [23]. The docking study suggested that the most active compound 5f has established hydrogen bonding with Tyr 158 and NAD 300 with a docking score of − 8.86, which is found to be greater than triclosan (− 7.62). The compound 5c recorded the highest docking score − 10.35 with hydrogen bonding interaction with Gln 100. In all the designed compounds, the hydrogen bonding interaction was observed except compound 5e. The molecules with docking score greater than that of triclosan were taken up for synthesis. The molecules were found to fit well in the active site in the hydrophobic pocket composing of Gly-96, Phe-97, Me-98, Met-161, Lys-165, Leu-216,Glu-219, Ile-215, Pro-193, Ala-157, Gly-104, Met-103, Met-199, and Phe-149 amino acid residues (Figs. 3, 4, 5, and 6).

Triclosan in the binding pocket (PDB:1P45)

Molecular docking interaction of triclosan with Mtb ENR (PDB 1P45) with ligand-interaction diagram

Molecular docking interaction of compound 5c with Mtb ENR (PDB 1P45) with ligand-interaction diagram

Molecular docking interaction of compound 5f with Mtb ENR (PDB 1P45) with ligand-interaction diagram
Molecular dynamics analysis
To compare the binding behavior of 5f and triclosan, their docked complexes with InhA were selected for MD simulations. The RMSD, as a function of the simulation time, was analyzed with respect to the starting structure for each complex, as shown in Fig. 7. The run time was 30.03 nanosecond (ns). In both the simulations, the protein backbone RMSD values attained the plateau phase after about 2.5 ns of simulation time. In the case of 5f, the RMSD recorded during simulation showed slight fluctuation for the initial 5 ns due to the initial protein stabilization for the 5f protein complex. For 5f, the RMSD of the protein backbone was 1.56–2.70 Å, and for the triclosan complex, the values were around 1.8–2.7 Å. Initially, the ligand RMSD for 5f was high (2.70 Å) for the first few nanoseconds as the complex took time to stabilize. After 9 ns, the 5f-InhA complex was completely stabilized, showing ligand RMSD ranging from 2.25 to 1.08 Å, which is evident through lesser fluctuations in the trajectory during the simulation time period. The ligand RMSD should be within 3 Å for the stable complex formation between the protein backbone and the ligand. Though the triclosan RMSD was below 2 Å, the triclosan-InhA complex showed more fluctuations during the simulation time period. In contrast to this, compound 5f, whose ligand RMSD was ranging from 2.25 to 1.08 Å, found to be more stable in comparison to triclosan, which is also evident from the width of the trajectories of both the complexes. The more stable RMSD of compound 5f depicts better stability of the complex, which could be due to the differences in the duration of interactions of 5f (99%) and triclosan (90%) with target residues of the protein. This, in turn, would have contributed to the better activity of 5f when compared with triclosan.

RMSD plot of 5f and triclosan in complex with InhA
Protein-ligand contacts
Non-bonding interactions between the ligand and the protein amino acid residues were studied comparatively for 5f and triclosan (Fig. 8). Triclosan displayed mainly hydrogen bonding and hydrophobic interactions. The hydrophobic interactions were observed with Met 103, Phe 149, Tyr 158, Met 161, Ala 198, Met 199, and Ile 202. Among them, Phe 149 and Met 161 displayed hydrophobic interaction with a maximum occupancy of 29.1% and 15.0%, respectively, and hydrogen bonding interaction was observed with Tyr 158 with a maximum occupancy of 90.5%. The maximum is the occupancy; higher is the contact time between ligand and the amino acid residue. Whereas in the case of 5f, the compound displayed hydrophobic interactions with Met 98, Met 103, Phe 149, Met 155, Met 161, Pro 193, Ala 198, Met 199, Ile 202, Val 203, Ile 215, Leu 218, Trp 222, and Leu 269. Among them, Phe 149, Met 161, Ala 198, and Ile 202 displayed occupancy of 71.3%, 57.4%, 71.5%, and 60.4% respectively. Hydrogen bonding interaction was observed with Tyr 158 with occupancy of 99.4% over the simulation time period. This difference in the types of interaction with amino acid residue and the contact time period between ligand and number of amino acid residues might be important for the activity of the ligand points towards its potency in the in vitro activity of this molecule. Figure 8 shows bar charts of amino acid residues, the nature, and percent of interactions they exhibit during the simulation trajectory time.

Plot (stacked bar charts) of protein interactions with ligand 5f and triclosan as monitored throughout the simulation
The residues which displayed more occupancy, i.e., more than 30% over the simulation time in the trajectory, are shown as ligand-protein contacts (Fig. 9). For compound 5f, the residue B: Tyr158 displayed hydrogen bond interaction with the OH group (A ring of 5f). Triclosan displayed hydrogen bonding interaction between B:Tyr 158 amino acid residue and the OH group of ring A. Similar contact was observed in case of 5f and TCL and as mentioned earlier, the time period for interaction with Tyr 158 was more in the case of ligand 5f-InhA complex when compared with TCL. Therefore, it would have resulted in stronger interaction and better binding between ligand 5f and the target InhA protein as evident through molecular docking results.

The atomic interactions of ligand 5f and triclosan with the key amino acid residues at the active site
Protein RMSF
Root mean square fluctuations (RMSF) display the fluctuations of each protein amino acid residue over the simulation time period (Fig. 10). The most fluctuating amino acid residue was found to be B:Giu 210 and B:Phe 109 with RMSF value of 2.79 Å and 2.86 Å, respectively. The fluctuations also represent the interaction between protein and the ligand. RMSF fluctuation for the remaining protein was found within the range of 0.78 to 2.86 Å. For TCL-InhA complex protein, RMSF fluctuation was observed in the 0.64–2.79 Å. The most fluctuating amino acid residues were B: Glu 209 and B: Ala 206 with RMSF values of 2.79 Å and 2.46 Å respectively.

Protein RMSF plot 5f-InhA complex and triclosan-InhA complex
Ligand RMSF
The fluctuations in the ligand per atom with respect to the protein are depicted by the ligand RMSF plot. It helps in understanding the interactions of the ligand fragments and the protein residues and also the role that these fluctuations play in the binding process. Greater fluctuations in the ligand moieties seem to decrease its interactions with the protein. The ligand RMSF plots of 5f and triclosan are respectively shown in Fig. 11. The X-axis shows the atom index and the Y-axis shows the RMSF values in Å. Carbon atoms at D ring of compound 5f were found to fluctuate the most, and the RMSF value was observed to be in a range from 1.03 to 2.19 Å. Overall, the RMSF value with respect to the protein-ligand complex was observed within the range of 0.80 to 2.19 Å. The 2-methyl group and C-5 at D ring displayed maximum fluctuation with RMSF value of 2.01 and 2.19 Å. The chloro group in ring A at 5th position and at 4th position on ring B of TCL-displayed maximum fluctuation with RMSF value of 1.0 Å and 0.94 Å, respectively. Overall, for TCL-InhA complex, atom fluctuations were observed within the range of 0.76–1.0 Å.

Ligand RMS Plot during MD simulations for compound 5f and triclosan
In silico ADME studies
ADME properties of all the synthesized compounds were predicted using the QikProp tool of the Schrodinger suite. In silico predicted pharmacokinetic descriptors for the synthesized compounds are given in Table 4. The synthesized compounds 5a-g were found to have acceptable values towards the assessed in silico parameters. The predicted log P and polar surface area (5a-g) of the designed compounds indicated that it is comparatively more hydrophilic than triclosan and obeyed the Lipinski’s drug rule. The log p value of triclosan was greater than 5, which was higher than all the synthesized compounds. All the synthesized compounds were found to follow the Lipinski rule of five (Table 1). The predicted oral absorption was found to be 100% for all compounds. QPPCaco predicts Caco-2 cells’ permeability (< 25 is low absorption, > 500 is high absorption). The predicted QPPCaco value for the synthesized compounds were in the range of 2827.367 to 2995.286, thus indicating that all compounds will be easily absorbed through the gut layer. All of the compounds had good predicted pharmacological properties such as HERG K+ (HERG K+ channel). Compounds that met ADMET and Lipinski’s drug-like parameters were subjected to docking study.
Determination of logP
To test the effect of lipophilicity on antitubercular and antibacterial activity, the RP-HPLC method was used to determine experimental logP of the compounds and results are shown in Table 1. Compound 5b, 5c, and 5f showed logP of 4.98, 4.60, and 4.97, which is relatively less than triclosan (log P 5.12) and is within the limit of Lipinski’s five rule.
Determination of pKa
The pKa of the most active 5f compound was determined by RP-HPLC method. Compound 5f was found to be weakly acidic (pKa = 6.24). The weak acidity may be due to the presence of an electron withdrawal group in the molecule. The pKa of compound 5f showed that it remained ionized at pH 7.4 to some extent during the in vitro antitubercular assay. The reason for its antitubercular activity could be due to its significant penetration into the mycobacterial cell wall.
Determination of protein binding
The protein binding capacity of compound 5f was evaluated using the RP-HPLC method. Human serum albumin column was used to assess protein binding. Compound 5f was found to be highly susceptible to protein binding (90.4%).
Conclusion
In summary, we have rationally designed and synthesized novel diphenyl ether derivatives having drug-likeness properties and screened them for in vitro antitubercular and antibacterial activity. Among the tested compounds, 5f appeared to be potent against Mycobacterium tuberculosis strain H37Rv with MIC of 18 μM and satisfactory selectivity index. In addition, compound 5f was also found to be the most active having MICs of 9 μM, 18.22 μM, and 18 μM against Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa respectively. All the synthesized compounds exhibited drug-likeness and ADME properties were found to be within an acceptable range. The molecular dynamics simulation study of the most active compound 5f and triclosan correlates with its activity in comparison to triclosan. The present study indicates that there is further scope to explore the antimicrobial potential of diphenyl ether derivatives.
References
Ghodbane R, Drancourt M (2013). Tuberculosis 93:589–595
Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G (2006). Lancet 4:1575–1580
Isaakidis P, Das M, Kumar AMV, Peskett C, Khetarpal M, Bamne A, Adsul B, Manglani M, Sachdeva KS, Parmar M, Kanchar A, Rewari BB, Deshpande A, Rodrigues C, Shetty A, Rebello L, Saranchuk P (2014) Alarming levels of drug-resistant tuberculosis in HIV-infected patients in metropolitan Mumbai. India PLoS One. https://doi.org/10.1371/journal.pone.0110461
World Health Organization, Global 2018 TB report (2018) https://doi.org/10.1016/j.pharep.2017.02.021
Jarlier V, Aubry A, Robert J, Bernard C, Dournon N, Lafeuille E, Sougakoff W, Le Dû D, Roure F, Veziris N, Caumes E (2016) XDR-tuberculosis in France: community transmission due to non-compliance with isolation precautions. Médecine Mal Infect. https://doi.org/10.1016/j.medmal.2015.12.008
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG (1998) Deciphering the biology of mycobacterium tuberculosis from the complete genome sequence. Nature. https://doi.org/10.1038/31159
Barry CE, Crick DC, McNeil MR (2007). Infect Disord Drug Targets 7:182–202
Bloch K (2006) Control mechanisms for fatty acid synthesis in mycobacterium smegmatis. Adv Enzymol Relat Areas Mol Biol. https://doi.org/10.1002/9780470122907
Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB (1999). Nature 398:383–384
Nguyen M, Claparols C, Bernadou J, Meunier B (2001). ChemBioChem 2:877–883
Zhang Y, Heym B, Allen B, Young D, Cole S (1992). Nature 358:591–593
Zhao X, Yu H, Yu S, Wang F, Sacchettini JC, Magliozzo RS (2006). Biochemistry 45:4131–4140
Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Sacchettini JC (1998). Science 279:98–102
Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ (2006). ACS Chem Biol 1:43–53
Timmins GS, Deretic V (2006). Microbiol 62:1220–1227
Stewart MJ, Parikh S, Xiao G, Tonge PJ, Kisker C (1999). J Mol Biol 290:859–865
Luckner SR, Liu N, Christopher W, Tonge PJ, Kisker C (2010). J Biol Chem 285:14330–14337
Cinu TA, Sidhartha SK, Indira B, Varadaraj BG, Vishnu PS, Shenoy GG (2015) Design, synthesis and evaluation of antitubercular activity of Triclosan analogues. Arab J Chem. https://doi.org/10.1016/j.arabjc.2015.09.003
Ruchi V, Helena IMB, Kriti A, Indira B, Mradul T, Varadaraj GB (2019). J Mol Struct 1197:117–133
Thomas CA, Kar SS, Bair I, Bhat VG, Shenoy VP (2015). Indo Glob J Pharm Sci 5:19–25
Ashutosh PT, Varadaraj BG (2019) Identifying the structural features of diphenyl ether analogues for InhA inhibition: a 2D and 3D QSAR based study. Lett Drug Des Discov. https://doi.org/10.2174/1570180816666190611153933
Kar SS, Bhat GV, Rao PP, Shenoy VP, Bairy I, Shenoy GG (2016). Drug Des Devel Ther 10:2299–2310
Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Sacchettini JC, Fidock DA (2003). J Biol Chem 278:20851–20859
Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth BN, Iwamoto H, Perozzo R, Jacobs JWR, Sacchettini JC, Fidock DA (2003) The protein data bank. https://doi.org/10.2210/pdb4w9n/pdb
Gillmore A, Lauret C, Roberts SM (2003). Tetrahedron 59:4363–4375
Tipparaju SK, Mulhearn DC, Klein GM, Chen Y, Tapadar S, Bishop MH, Yang S, Chen J, Ghassemi M, Santarsiero BD, Cook JL, Johlfs M, Mesecar AD, Johnson ME, Kozikowski AP (2008). ChemMedChem 3:1250–1268
Candice SDM, Feng TS, Van RDW, Gessner RK, Street LJ, Morgans GL, Warner DF, Moosa A, Naran K, Lawrence N, Boshoff HIM, Barry CE, Harris CJ, Gordon R, Chibale K (2015). Bioorg Med Chem 23:7240–7250
Kaniga K, Cirillo DM, Hoffner S, Ismail NA, Kaur D, Lounis N, Metchock B, Pfyffer GE, Venter A (2016). J Clin Microbiol 54:2963–2968
Kuldipsinh MDG, Barot P, Shailesh VJ, Nirzari G, Laurent K, Shubhra S, Vijay BT, Kruti J (2014). Eur J Med Chem 83:245–255
Sarker SD, Nahar L, Kumarasamy Y (2007). Methods 42:321–324
Meerloo JV, Kaspers GJL, Cloos J (2011). Methods Mol Biol 731:237–245
Guo S, Song Y, Huang Q, Yuan H, Wan B, Wang Y, He R, Beconi MG, Franzblau SG, Kozikowski AP (2010). J Med Chem 53:649–659
Ajay A, Singh V, Singh S, Pandey S, Gunjan S, Dubey D, Sinha SK, Singh BN, Chaturvedi V, Tripathi R, Ramchandran R, Tripathi RP (2010). Bioorg Med Chem 18:8289–8301
O’Brien PJ, Irwin W, Diaz D, Howard EC, Krejsa CM, Slaughter MR, Gao B, Kaludercic N, Angeline A, Bernardi P, Brain P, Hougham C (2006). Arch Toxicol 80:580–604
Saxena S, Samala G, Sridevi JP, Devi PB, Yogeeswari P, Sriram D (2015). Eur J Med Chem 92:401–414
Malikanti R, Vadija R, Veeravarapu H, Mustyala KK, Malkhed V, Vuruputuri U (2017). J Mol Struct 1150:227–241
Bari S, Haswani NG (2014). J Saudi Chem Soc 21:S264–S274
Desmond Molecular Dynamics System (2017) D. E. Shaw Research, New York, NY. Maestro-desmond interoperability tools. Schrödinger, New York
Ntie-Kang F (2013). Springerplus 2:1–11
Nasal A, Kaliszan R (2006). Curr Comput Aided-Drug Des 2:327–340
Poole SK, Poole CF (2003). J Chromatogr B Anal Technol Biomed Life Sci 797:3–19
Sophie M, Davy G, Yveline H, Alexandra G, Jean-Luc V, Serge R, Pierre AC (2008) In: Raimund M (ed) Chromatographic approaches for measuring log P. Wiley, New York
Kaliszan R, Haber P, Tomasz B, Siluk D, Valko K (2002). J Chromatogr A 965:117–127
Wiczling P, Markuszewski MJ, Kaliszan R (2004). Anal Chem 76:3069–3077
Demiralay EC, Alsancak G, Ozkan SA (2009). J Sep Sci 32:2928–2936
Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S (2005). J Mol Biol 353:38–52
Kar SS, Bhat VG, Shenoy VP, Bairy I, Shenoy GG (2019). Chem Biol Drug Des 93:60–66
Elamparuthi E, Sarathkumar S, Girija S, Anbazhagan V (2014). Tetrahedron Lett 55:3992–3995
Hua MQ, Cui HF, Wang L, Nie J, Ma JA (2010). Angew Chem 49:2772–2776
Cheruku SR, Padmanilayam MP, Vennerstrom JL (2003). Tetrahedron Lett 44:3701–3703
Acknowledgments
The authors also acknowledge Manipal Schrodinger Centre for Molecular Simulations, Manipal College of Pharmaceutical Sciences, for providing the facilities for in silico studies.
Funding
This work was supported under the Quality Improvement Program (PHR/731/12) and 8–219/RFID/RPS Policy-1/2014-15 of All India Council for Technical Education (AICTE), New Delhi, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 529 kb)
Rights and permissions
About this article

Cite this article
Khade, A.B., Boshoff, H.I.M., Arora, K. et al. Design, synthesis, evaluation, and molecular dynamic simulation of triclosan mimic diphenyl ether derivatives as antitubercular and antibacterial agents. Struct Chem 31, 983–998 (2020). https://doi.org/10.1007/s11224-019-01478-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-019-01478-8