
Abstract
The role of Ti(II) sandwich complex, [Cp2Ti], generated by the combination of [Cp2TiCl2] and two equivalents of nBuLi, in situ, in toluene has been investigated theoretically for the release of H2 from a series of liquid organic hydrogen carriers (LOHCs), mainly alcohols or diols of low molecular weights. The compounds with hydroxyl functionality considered here are methanol, ethanol, isopropanol, methanediol, and ethylene glycol. Satisfactory results have also been obtained on applying the same mechanism to two other compounds with amino functionality, viz., methanediamine and ethylene diamine. The exploration has been carried out using density functional theory (DFT). Our findings reveal that the reaction is initiated with the formation of an adduct of the Ti center of sandwich complex, [Cp2Ti], with O/N atom of the compound under investigation. From the adduct thus formed, H2-release is possible via two routes. In one of the paths, O/N–H bond breaks first, followed by the cleavage of C–H bond, and in the other path, C–H bond breaking takes place prior to the scission of O/N–H bond. Our results show that the second path is thermodynamically and kinetically more preferable for the release of H2. Thus, [Cp2Ti] can be highly effective in releasing one equivalent of H2 from alcohols and compounds with amino functionalities, and more importantly, removal of two equivalents of H2 from methanediol is possible using [Cp2Ti], which is significant in terms of hydrogen storage purpose.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the rapid depletion of non-renewable fossil fuel reserves, development of alternate renewable sources of power has become a major challenge and research of the utmost importance. Hydrogen, because of its non-polluting, renewable nature and being highly abundant in the forms of H2O and hydrocarbons, is considered to be one of the optimistic solutions to the stumbling block and has been recognized as an ideal energy carrier [1]. However, the actual use of hydrogen as a transportation fuel is limited, and this is mainly because of storage and delivery problems (https://www.energy.gov/eere/fuelcells/hydrogen-storage-challenges). Hydrogen generation from different organic substances like alcohols, formic acid, and amines offers prospective pathways for achieving the target of producing hydrogen from readily available sources [2]. A small organic substance, formic acid, in particular, has an additional advantage as it only gives gaseous co-product CO2, apart from H2-release.
In 1991 and 1993, Corey and co-workers carried out interesting dehydrocoupling reactions of secondary silanes using highly reactive sandwich complexes, generated by the combination of [Cp2MCl2] (M = group 4 metal) and two equivalents of nBuLi [3, 4]. In 2006, Manners and co-workers made use of the Ti(II) sandwich, [Cp2Ti], generated in situ in toluene by combining [Cp2TiCl2] with two equivalents of nBuLi for the dehydrocoupling of amine-borane adducts [5]. They clearly stated the reluctance of Ti(IV) catalysts like Cp2TiMe2 towards dehydrocoupling of phosphine- and amine-borane adducts in their previous work [6] but exhibited, with detailed analysis, the role of less stable intermediate [Cp2Ti] in removing H2 from Me2NH–BH3. Although [Cp2Ti] is highly reactive in the reaction medium, Luo and Ohno carried out a very significant computational investigation in the following year, in which they considered the [Cp2Ti] as the catalyst and suggested that the dehydrogenation of Me2NH–BH3 goes via an intramolecular (and not intermolecular) stepwise mechanism, followed by an off-metal dimerization of Me2N=BH2 [7]. In another account, Chirik and co-workers investigated the dehydrocoupling mechanism using a series of titanium and zirconium sandwiches and concluded that increased substitution of the Cp rings reduces the catalytic activity of the sandwiches [8]. In 2010, Manners and co-workers again carried out in-depth kinetic and mechanistic studies of the dehydrocoupling of a range of amine-borane adducts by a series of metallocenes of group 4 metals, inferring reduced catalytic activity for the metals down the group and also for more substituted Cp ligands [9].
Apart from Amine-Borane and related compounds, liquid organic hydrogen carriers (LOHCs), featuring reasonable energy content, were also proposed as hydrogen storage materials and attracted much attention because of their safe and simple handling of H2 [10,11,12]. Among the LOHCs, cyclic hydrocarbons show evidence of high hydrogen content but, at the same time, are accompanied by high reaction enthalpies [13, 14]. Although hydrogen release from nitrogen heterocycles is accompanied by lower reaction enthalpies, they are not compatible with commonly used acidic proton exchange membranes because of the formation of a non-conductive salt [15]. Another group of LOHCs includes mono and polysubstituted alcohols and diols, devoid of these troubles, and are classified as hydrogen storage materials [16]. Several classes of catalysts were utilized over the years for the dehydrogenation of different alcohols to certain oxidized derivatives [2, 17,18,19,20,21,22,23,24,25,26,27,28,29]. Very recently, Bonitatibus Jr. et al. studied electrochemically the dehydrogenation-hydrogenation of a series of alcohols using homogeneous iron catalysts, establishing the role of the alcohols as aspiring materials for hydrogen storage [15]. Formic acid (FA) has gained much popularity as promising material for hydrogen storage over the years [30,31,32,33,34,35]. Though the hydrogen content of FA is 4.4 wt%, which falls slightly short of the milestones set by the US Department of Energy for 2010 [36], it is compensated by other factors related to simplicity and useable/net capacity, where capacities at ambient temperature are concerned [37]. Dehydrogenation of FA to CO2 and H2 is thermodynamically favorable by ∆G° = − 32.9 kJ mol−1, as mentioned by Mellmann et al. [30]. Another advantage of using FA as a hydrogen storage material is that 100% of the stored hydrogen is available for catalytic storage [30]. However, a side reaction resulting in the formation of CO and H2O from FA is also thermodynamically plausible [38], demanding selective catalysts for the dehydrogenation pathway. Formic acid was also considered as a fuel in direct formic acid fuel cells (DFAFCs) [39].
The homogeneous dehydrogenation of alcohols of low molecular weight, viz., methanol, ethanol, and isopropanol, and decomposition of formic acid can be deployed as important methods for the production of H2 as a sustainable fuel source [2]. Recently, Alberico et al. efficiently carried out H2 generation from methanol along with CO2 using iron pincer complex as catalyst under mild conditions, which is highly significant in the context of hydrogen economy [40]. Heim et al. reported methanediol as a hydrogen storage material, with weight efficiency of 8.4 wt% [28]. Based on exactly how Luo and Ohno modeled the catalyst and reaction conditions in their theoretical investigation [7], in our present work, we have investigated the scope of Ti(II) sandwich complex, [Cp2Ti], as an effective assisting reagent in a broader spectrum in the release of H2 from a series of compounds with hydroxyl functionality which undergo dehydrogenation in nearly thermoneutral or slightly endothermic pathways and, thus, may be effective in hydrogen storage purpose. To the best of our knowledge, there is no previous experimental or theoretical report on the production of H2 by using [Cp2Ti] from the hydroxyl-containing compounds. We have further applied the same mechanism to carry out dehydrogenation from compounds with amino functionality, viz., methanediamine and ethylene diamine. Smaller amino compounds like methanamine and ethanamine although have high gravimetric weight % of H2, but overall Gibbs energy values involved in H2-release is highly positive and, thus, are not acceptable as potential hydrogen storage systems. The potential energy surface (PES) associated with the hydrogen release route in all the systems has also been scanned at the same level of theory to get a graphical idea of the pathway. The whole assisting phenomena in solvent medium have been investigated through the implementation of conductor-like screening solvation model (COSMO), considering toluene as the bulk solvent medium.
Computational details
All electronic structure calculations have been carried out using the Gaussian 09 suite of quantum chemistry program, Revision D.01 [41] suite of quantum chemistry program. The geometries of all the adducts, intermediates, and transition states and products involved have been optimized by employing density functional theory (DFT). We have employed Head−Gordon’s long-range-corrected hybrid functional ωB97X-D [42, 43] in conjunction with all electron 6-31++G** basis set for main group elements (C,H,N,O) and effective core potential LANL2DZ [44, 45] basis set for Ti. Altogether, the basis set is designated as the genECP basis set. In order to further validate the results, single point calculations of all the geometries involved have been carried out at two other levels of theory: (a) M06-2X [46]/cc-pVTZ with Grimme’s dispersion effect (D3) and (b) B3LYP [47, 48]/cc-pVTZ with Grimme’s dispersion effect (D3). The connecting first order saddle points that are the transition states between the equilibrium geometries have been obtained by the synchronous transit-guided quasi-Newton (STQN) method. Normal-mode analysis has been carried out at the same level of theories to confirm whether the optimized structures are local minima (no imaginary frequency) or transition states (one imaginary frequency) on the potential energy surfaces (PESs). The potential energy surfaces (PESs) have been constructed using relative energies. Parallel intrinsic reaction coordinate (IRC) calculations [49, 50] have been carried out to confirm the connections between the transition states and local minima. Gibbs energy (G) has been calculated at 298 K temperature and 1-atm pressure using the in-built algorithm provided in Gaussian 09, following the ideal gas equation (http://gaussian.com/thermo/).The hydration sphere calculations have also been carried out at the same level of theory as in gas phase study. The self-consistent reaction field (SCRF) method has been implemented using COSMO [51, 52] to take into account the effect of bulk solvent medium, considering toluene as the solvent. SCRF requests that a calculation be performed in the presence of a solvent by placing the solute in a cavity within the solvent reaction field.
Results and discussion
The mechanism suggested by Luo and Ohno involves activation of the N–H bond of Me2NH.BH3 upon interaction with Cp2Ti(II), thereby forming a Ti(IV) intermediate with a Ti–H bond (the hydrogen coming from N–H bond cleavage) [7]. In the following step, the formation of another Ti–H bond via the B–H bond scission generates Cp2TiH2, from which molecular hydrogen can be removed easily. In case of the compounds studied here, we have encountered two modes of coordination to the Ti center of [Cp2Ti]: one through the O/N atom and the other through a hydrogen atom (H(C)) bonded to the carbon atom next to O/N. Between these two adducts, the O/N-coordinated one is found to be highly stabilized thermodynamically in comparison to the latter. So, we have considered the O/N-coordinated pathway only. The structures of the highly unstable H(C)-coordinated adducts have been provided in the supporting information, mentioning the extent by which they are unstable compared to the O/N-coordinated adducts.
Departure of H2 from compounds with hydroxyl functionality
Being inspired by the work of Bonitatibus Jr. et al. [15], we have considered a series of simple alcohols, viz., methanol (M), ethanol (E), isopropanol (I), and two other polyhydroxy compounds, viz., methanediol (MO) and ethylene glycol (EG), and studied the effect of [Cp2Ti] for releasing H2.
Elimination of H2 from simple alcohols: methanol, ethanol and isopropanol
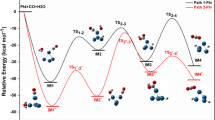
In this section, at first, we have considered the simplest compound with hydroxyl functionality, methanol (M) and then analyzed the effects of addition of extra methyl group(s) on the structures of the stationary points along the PES and also the reaction energies. M is found to coordinate to the Ti center of Cp2Ti through O-center, generating adduct M1, which is 0.3 kcal mol−1 more stable than the unreacted starting materials. Once M1 is formed, the reaction may proceed either through the cleavage of O–H bond or C–H bond. First, we have explored the path in which the reaction goes on with O–H bond scission, which is shown in Scheme 1. The cleavage of O–H bond followed by the formation of Ti–O and Ti–H bonds occurs through the transition state (TS), M12ts[o], of Gibbs energy barrier of 21.7 kcal mol−1. As a result, the intermediate, M2[o], is produced, which is found to be stabilized by 26.1 kcal mol−1 than M1[o]. From M2[o], the reaction proceeds with complete C–H bond scission, supported by the bond lengths collected in Table S1, which brings H(C) closer to Ti–H(O), thus forming a dihydride complex, M3[o]. However, the formation of M3[o] is highly endothermic and is achieved through a TS, M23ts[o] of barrier height of 24.0 kcal mol−1. From M3[o], the H2 molecule is released via a simple TS, M34ts[o] (depicted in Fig. 1) of barrier height of 0.8 kcal mol−1, producing M4. Removal of HCHO from M4 may give back [Cp2Ti].

Pathway of [Cp2Ti]-promoted H2-release from compounds with hydroxyl functionality initiated by O–H bond cleavage

Gibbs energy of [Cp2Ti]-promoted H2-release from methanol in kcal mol-1. Relative Gibbs energies of adducts, intermediates, and transition states are given from top to bottom at three levels of theory: ωB97X-D/genECP, M06-2X/cc-pVTZ with Grimme’s dispersion effect (D3), and B3LYP/cc-pVTZ considering Grimme’s dispersion effect (D3)
In the second path, the reaction proceeds with the breaking of C–H bond from the initial adduct, M1, as illustrated in Scheme 2. The C–H bond cleavage takes it to an intermediate, M2[c], with the newly developed Ti–H(C) and Ti–C bonds along with a Ti...O interaction, as perceived by the Ti–H(C) (1.65 Å), Ti–C (2.10 Å), and Ti...O (2.22 Å) contacts in Table S2. The transformation takes place via a TS, M12ts[c], of Gibbs energy barrier of 13.8 kcal mol−1. From M2[c], a bond rotation brings the O–H and Ti–H(C) bonds closer, resulting in the formation of another intermediate, M3[c], as shown in Fig. S1. Once H(C) and H(O) attain close proximity, the cleavages of Ti–H(C) and O–H bonds occur and the intermediate directly leads to release of a molecular H2 via TS, M34ts[c], of free energy activation barrier of 10.0 kcal mol−1, resulting in the formation of M4. Since the C–H bond activation from the initial adduct M1 takes place via a TS of lower activation energy, methanol is more likely to adopt the second path.

Pathway of [Cp2Ti]-promoted H2-release from compounds with hydroxyl functionality initiated by C–H bond cleavage
In case of ethanol (E), the initial adduct E1 (shown in Fig. S2) is stabilized by 0.2 kcal mol−1, which is found to be 1.6 kcal mol−1 less stable in case of the isopropanol analogue I1(exhibited in Fig. S4). In the first path, the breaking of O–H bond of ethanol takes place via transition state E12ts[O] (the same is I12ts[O] for isopropanol) of Gibbs energy barriers of 22.0 kcal mol−1 (23.9 kcal mol−1 for isopropanol) giving intermediate E2[O] (I2[O] for isopropanol). From E2[O] and I2[O], the reactions proceed to the subsequent intermediates, E3[O] and I3[O], in which C–H bond cleaves and the H(C) comes closer to Ti–H(O) forming a dihydride complex (perceived from the distances furnished in Tables S3 and S5). Similar to the previous reaction with methanol, the transformation in case of both ethanol and isopropanol are highly endothermic and the conversions go via transition states E23ts[O] and I23ts[O] of activation barriers 25.5 and 29.0 kcal mol−1, respectively. The removal of H2 from the intermediate E3[O] follows the same trend as seen in case of methanol and goes via barrierless TS, E34ts[O], producing E4. However, in case of isopropanol, this step goes via the TS I34ts[O] of barrier height 6.7 kcal mol−1, giving the aldehyde complex I4.
In the second path, the reaction proceeds with the C–H bond scissions from the initial adducts E1 and I1. The resulting intermediate thus formed, E2[c] is 5.0 kcal mol−1 less stable than E1, while the formation of I2[c] is endothermic by 9.3 kcal mol−1. The transformations are achieved via transition states, E12ts[c] (exhibited in Fig. S3) and I12ts[c] (Fig. S5) of activation barriers of 14.7 and 18.6 kcal mol−1 respectively, with an elongated C–H bond of 2.3 Å, as evident from Tables S4 and S6. O–H bond involved in the E2[c] intermediate of ethanol rotates to come in proximity of the Ti–H(C) bond and another intermediate, E3[c], is formed, which is higher in energy compared to the previous one, E2[c], by 2.0 kcal mol−1. The isopropanol analogue I3[c] is also 2.1 kcal mol−1 more energetic than I2[c]. From E3[c] and I3[c], molecular H2 is released via the transition states E34ts[c] and I34ts[c] of Gibbs energy barriers of 9.9 and 10.3 kcal mol−1 leaving E4 and I4, respectively.
Our detailed study suggests that for all three simple alcohols, viz., methanol, ethanol, and isopropanol, the pathway in which C–H bond breaks first followed by the cleavage of O–H bond is found to be kinetically more favorable. As evident from Fig. 2, the barrier height of the rate-determining step of this path increases with the addition of extra methyl group(s) from methanol to isopropanol. However, it should be mentioned here that the dissociation of M4 to HCHO and [Cp2Ti], as displayed in Fig. 1, requires about 31 kcal mol−1, which makes the dissociation of M4 very difficult. This can be achieved only by trapping HCHO in some exergonic and kinetically favorable reaction, releasing [Cp2Ti], which can then be treated as a catalyst. However, potential energy surfaces depicted in Fig. S3 and S5 suggest that the dissociation of the corresponding aldehyde complexes (E4, I4) derived from larger alcohols, viz., ethanol and isopropanol is more favorable, allowing to use [Cp2Ti] as a catalyst for H2-removal from these alcohols without using a trapping reagent.

Activation barriers associated with the rate-determining steps of the two pathways involved in [Cp2Ti]-promoted H2-release from all chosen compounds (in kcal mol-1) at ωB97X-D/genECP level
Elimination of two equivalents of H2 from methanediol
After establishing the mechanism for small alcohols, we paid our attention towards methanediol (MO). The study of H2-release from MO is central to this work, as it is the smallest geminal diol, and removal of two equivalents of H2 is possible. The catalytic effect of [Cp2Ti] in the removal of two equivalents of H2 has been observed stepwise.
Release of first equivalent of H2
MO coordinates to the Ti center via one of the oxygen atoms [O(1)] and the resultant adduct is named as MO1. As observed in previous cases, the route which is initiated with the O–H bond splitting from the adduct, MO1, generates an intermediate, MO2[o], via a TS, MO12ts[o], of Gibbs energy barrier 25.3 kcal mol−1. The intermediate, MO2[o], with newly developed Ti–H(O) and Ti–O bonds (established by the bond distances in Table S7) is stabilized by 25.4 kcal mol−1. Similar to previous cases, here also from the intermediate, MO2[o], C–H bond cleavage brings H(C) closer to H(O), resulting in the formation of a dihydride complex, MO3[o]. This transformation goes via a TS, MO23ts[o], of barrier height of 27.2 kcal mol−1. The removal of H2 from MO3[o] is accomplished via the TS, MO34ts[o], of Gibbs energy activation barrier of 1.7 kcal mol−1 with respect to MO3[o] and, thereby, giving MO4 (shown in Fig. S6).
In the other path, the adduct MO1 undergoes C–H bond splitting, which takes it to a new intermediate, MO2[c], with newly developed Ti–H and Ti–C bonds and a mild Ti...O(1) interaction, as perceived from Table S8. The transformation takes place via a transition state, MO12ts[c], of Gibbs energy barrier of 18.4 kcal mol−1. This step is also the rate-determining step of this pathway. Once MO2[c] is formed, there are two possibilities—(a) the H atom bonded to O(2), which is already in proximity of the H(C) bonded to Ti, may undergo concerted bond scission along with H(C), releasing one molecule of H2; (b) a bond rotation may take place, bringing the H bonded to O(1) closer to the Ti–H(C), followed by a C–H bond breaking prior to eliminating a H2 molecule. In the first pathway, the H2 molecule is released via a transition state, MO25ts[c], that involves lengthening of O(2)–H and Ti–H(C) bonds and bringing the two hydrogen atoms closer. However, the four-membered TS, MO25ts[c], (shown in Scheme 3 in Supporting Information) involves Gibbs energy barrier of 37.7 kcal mol−1, exhibited in Fig. S7, which is significantly high, and the pathway is not likely to be followed. H2 release in this pathway gives MO5[c]. In the other pathway, the bond rotation generates another intermediate, MO3[c] (2.0 kcal mol−1 higher in energy), bringing O(1)–H in proximity of the Ti–H(C) bond. The concerted cleavages of Ti–H(C) and O(1)–H bonds liberate a molecule of H2, generating the complex, MO4. This step goes via a four-membered transition state, MO34ts[c], of Gibbs energy barrier of 16.8 kcal mol−1 with respect to MO3[c]. As evident from Fig. S7, the second route which goes via the TS, MO12ts[c], followed by TS, MO34ts[c], is kinetically the preferred one.
Release of second equivalent of H2
Our investigation unveils the fact that the new complex, MO4, thus generated, may further undergo H2-release. The O–H bond cleavage takes it to an intermediate, FA1[o], via a TS, FA41ts[o], of Gibbs energy barrier of 12.8 kcal mol−1, as shown in Fig. S8. The route has been schematically demonstrated in Scheme 4 in supporting information. The intermediate thus formed, with newly developed Ti–H(O) and Ti–O(2) bonds, is confirmed from the bond distances in Table S9 and is stabilized by 36.9 kcal mol−1 compared to MO4. From FA1[o], H(C) comes closer to H(O) (bonded to Ti), and by the cleavage of C–H bond, forms a dihydride complex, FA2[o]. This transformation goes via a TS, FA12ts[o], of Gibbs energy activation barrier of 23.6 kcal mol−1 with respect to FA1[o]. The release of H2 takes place via a TS, FA23ts[o], as depicted in Fig. S9, of Gibbs energy barrier of 0.9 kcal mol−1 with respect to FA2[o] and FA3 is formed. Removal of CO2 from FA3 regenerates the catalyst [Cp2Ti].
A second pathway is possible from the complex, MO4. In this path, as illustrated in Scheme 5 in supporting information, the reaction proceeds through C–H bond scission via transition state, FA41ts[c], of Gibbs energy activation barrier of 11.7 kcal mol−1, leading to an intermediate, FA1[c], made up of new Ti–C and Ti–H(C) bonds, as apparent from the distances 1.66 Å between Ti and H(C) and 2.05 Å between Ti and C provided in Table S10. The intermediate, FA1[c], is more stable than MO4 by 10.7 kcal mol−1. It is evident from Fig. S8 that the intermediate, FA1[c], attains an orientation where Ti–H(C) and O–H are in proximity. The reaction proceeds with breaking of C–H and Ti–H(C) bonds, thereby releasing an H2 molecule and FA3 is formed. The conversion of FA1[c] to FA3 goes via a four-membered planar TS, FA13ts[c], of barrier height of 16.6 kcal mol−1 with respect to FA1[c].
Thus, our study endorses the fact that removal of two H2 molecules from methanediol and regeneration of the catalyst is achievable using [Cp2Ti] as catalyst, and the overall process is exothermic.
Elimination of one equivalent of H2 from ethylene glycol
The mechanism established so far for the release of molecular hydrogen from the previous compounds holds good for another molecule with hydroxyl functionality, ethylene glycol (EG), which embraces two hydroxyl groups. But unlike methanediol, removal of the second equivalent of H2 is accompanied with high endothermic value (40 kcal mol−1), for which we have restricted our investigation up to removal of one equivalent of H2. The formation of ethylene glycol-Cp2Ti adduct, EG1, by the coordination of O(1) of EG to Ti center of [Cp2Ti] is absolutely thermoneutral, as evident from Fig. S11. In the first path, the adduct proceeds to the intermediate, EG2[o], with newly developed Ti–H(O) and Ti–O(1) bonds, which is confirmed from the bond distances summarized in Table S11, via three-membered TS, EG12ts[o], for which it needs to overcome a Gibbs energy barrier of 23.2 kcal mol−1. EG2[o] is stabilized by 27.0 kcal mol−1 compared to EG1[o]. From EG2[o], C–H bond cleavage takes H(C) closer to Ti–H(O) in the dihydride complex, EG3[o]. This transformation proceeds via TS, EG23ts[o], of Gibbs energy activation barrier of 25.0 kcal mol−1. The coordinated H2 molecule is detached from the second intermediate, EG3[o], via a TS, EG34ts[o], of barrier height of 0.4 kcal mol−1 with respect to EG3[o], displayed in Fig. S10, giving EG4. Removal of the dehydrogenated OHCH2CH=O molecule from EG4 may give back [Cp2Ti].
In the second pathway, from EG1, the cleavage of C(1)–H bond takes it to the intermediate, EG2[c], via TS, EG12ts[c], of Gibbs energy barrier of 14.1 kcal mol−1. As apparent from Fig. S11 and Table S12, EG2[c] involves formation of Ti–H(C) and Ti–C(1) bonds and is stabilized by 5.0 kcal mol−1 in comparison to EG1[c]. From EG2[c], a bond rotation brings O(1)–H bond nearer to the Ti–H(C) in the newly formed intermediate, EG3[c]. The concerted cleavage of H(C)–Ti and H(O)–O bonds from EG3[c] removes molecular H2 via a four-membered TS, EG34ts[c], of activation barrier of 17.6 kcal mol−1 with respect to EG3[c], giving the adduct, EG4. As observed in the previous compounds, here also, the second path is kinetically more preferable.
Departure of H2 from compounds containing amino group
Unlike alcohols with very small hydrocarbon chains, the small and simple primary amines, viz., methyl amine, ethyl amine, are not usually considered to be effective for hydrogen storage purpose because of very high endothermic values of the overall dehydrogenation process. However, there are certain compounds containing amino group, for which the Gibbs energy change of the whole dehydrogenation process is comparable to the compounds which have already been tagged effective for hydrogen storage. We have considered two such compounds, viz., methanediamine, usually available commercially as methylenediamine dihydrochloride (with H2 content of 13.04%) and ethylene diamine (with 13.33% of H2 by mass).
Elimination of H2 from methanediamine
The dehydrogenation pathway of methanediamine is studied computationally, for which the adduct, MA1, formed by the coordination of Ti center to N(1), is stabilized by 8.5 kcal mol−1. Two dehydrogenation paths have been detected, one of which is initiated by the N(1)–H and the other by the C–H bond cleavages, as shown schematically in Scheme S6 and Scheme 7 in supporting information. In the first path, MA1 is converted to MA2[n] with newly developed Ti–H(N) and Ti–N bonds, which is evident from Ti–H(N) (1.67 Å) and Ti–N (1.93 Å) contacts provided in Table S13. The transformation from MA1 to MA2[n] is achieved on gaining an activation energy of 27.8 kcal mol−1 via a TS, MA12ts[n]. The scission of C–H bond from MA2[n] brings the H(C) closer to Ti–H(N) resulting in the formation of a dihydride complex, MA3[n], as shown in Fig. S12. This transformation is achieved via a cyclic four-membered TS, MA23ts[n], of Gibbs energy activation barrier of 28.4 kcal mol−1, as illustrated in Fig. S13. The coordinated H2 molecule is released via a TS, MA34ts[n], of barrier height of 1.6 kcal mol−1 with respect to MA3[n], giving MA4. From the complex, MA4, detachment of NH2CH=NH gives back [Cp2Ti].
The second dehydrogenation path starts with the breaking of C–H bond from MA1. The reaction goes to an intermediate,MA2[c], developing new Ti–H(C) and Ti–C bonds and Ti...N interaction, which is evident from Ti–H(C) (1.67 Å), Ti–C (2.13 Å), and Ti...N (2.20 Å) contacts summarized in Table S14. The transformation of MA1[c] to MA2[c] is accomplished via a TS, MA12ts[c], of activation barrier of 13.1 kcal mol−1. Once MA2[c] is reached, there are two routes possible, as observed earlier in case of MO also. However, the route via MA25ts[c] (shown in Scheme 8 in supporting information) needs to attain a very high energy activation barrier of 49.7 kcal mol−1 with respect to MA2[c], as demonstrated in Fig. S13, and is not likely to be followed. Instead, the reaction is likely to follow the other route, in which a bond rotation brings H(N) in proximity of the hydrogen bonded to Ti and a new intermediate, MA3[c], is formed, which is 3.0 kcal mol−1 higher in energy than MA2[c]. The concerted cleavages of Ti–H(C) and N(1)–H bonds liberate a molecule of H2, generating the complex, MA4, via a cyclic four-membered TS, MA34ts[c], of activation barrier of 20.3 kcal mol−1 with respect to MA3[c], which is also the rate-determining step of this path.
Elimination of H2 from ethylene diamine
EA coordinates to the Ti center through N (N(1) labeled in Fig. S14), generating an adduct EA1. In the first path, EA1 proceeds to the intermediate, EA2[n], via a three-membered TS, EA12ts[n], for which it has to overcome a Gibbs energy barrier of 27.9 kcal mol−1. N(1)–H(N) bond breaking results in the formation of Ti–H(N) and Ti–N(1) bonds in EA2[n], as apparent from the bond distances in Table S15, and the intermediate is further stabilized by 8.8 kcal mol−1. From EA2[n], another intermediate, EA3[n], is formed via a four-membered TS, EA23ts[n], of activation energy of 25.4 kcal mol−1, where C(1)–H(C) bond completely breaks and H(C) comes closer to H(C) forming a dihydride complex. H2 molecule is then released and EA4 is formed via a TS, EA34ts[n], of Gibbs energy of 0.9 kcal mol−1 with respect to EA3[n]. The removal of NH2CH2CH=NH from EA4 gives back Cp2Ti.
In the second path, C(1)–H bond scission from the adduct, EA1, via a TS, EA12ts[c], of activation barrier of 13.7 kcal mol−1 takes it to an intermediate, EA2[c]. New Ti–H(C) and Ti–C(1) bonds and a mild Ti...O(1) interaction are developed in EA2[c] by the cleavage of C(1)–H bond, as exhibited by the bond distances in Table S16. A bond rotation brings (H(N)) in proximity of the H(C), bonded to Ti, producing another intermediate, EA3[c], as depicted in Fig. S15, which is 2.5 kcal mol−1 higher than EA2[c] in terms of energy. Concerted N–H(N) and Ti–H(C) bond cleavages from EA3[c] generate a H2 molecule, which is removed directly from the complex, giving EA4, as demonstrated in Fig. S8. The removal of H2 from EA3[c] occurs via a TS, EA34ts[c], of Gibbs energy activation barrier of 20.7 kcal mol−1 with respect to EA3[c] and the step is endothermic by 1.4 kcal mol−1.
In case of the compounds with amino functionality, the path which proceeds with the cleavage of C–H bond followed by N–H bond breaking, is found to be the kinetically preferred one. Rate-determining steps of both the pathways have been depicted in Fig. 2. Comparable barrier heights of the rate-determining steps have been observed for methanediamine and ethylene diamine.
Since there is no experimental data available for comparison, we have carried out calculations at different levels of theory. The single point calculations of all the adducts, intermediates, and transition states were carried out at two other levels of theory: (a) M06-2X [46]/cc-pVTZ (D3) and (b) B3LYP [47, 48]/cc-pVTZ with Grimme’s dispersion effect (D3) in both cases, having been depicted in all the potential energy surfaces. We have obtained similar trends in all cases, which further strengthen the predictions.
Conclusions
In the present article, we have theoretically investigated the potentiality of Ti(II) sandwich complex, [Cp2Ti], in releasing hydrogen from a series of liquid organic hydrogen carriers (LOHCs), mainly alcohols or diols of low molecular weights, which have already been cited as hydrogen storage materials because of their ability to release molecular hydrogen effectively. Usually for amines, the H2-removal is highly endothermic. However, we have tried to apply the same mechanism of hydrogen release to two compounds with amino functionality, viz., methane diamine and ethylene diamine, for which this dehydrogenation is moderately endothermic. We have found pathways with considerable values of activation energies of the rate-determining steps, ensuring catalytic activity of [Cp2Ti] for such compounds as well. The chosen alcohols and amines are found to coordinate to the Ti center of [Cp2Ti], via O/N. Another possibility is the coordination through H bonded to the carbon atom adjacent to O/N. However, such adducts are found to be thermodynamically highly unstable compared to the O/N-coordinated adducts, for which we have discarded any investigation with such thermodynamically unstable complexes. Once the O/N-coordinated adduct is formed, stepwise cleavage of O/N–H and C–H bonds releases molecular H2. For the compounds we have considered here, two pathways have been espied once the O/N-coordinated adduct is formed. In one of the pathways, the O/N–H bond breaks first prior to the cleavage of C–H bond. In the other path, C–H bond breaking takes place earlier, followed by the O/N–H bond scission. Our detailed study endorses the fact that both the pathways are accessible; however, the one involving C–H bond breaking followed by O–H bond cleavage is kinetically more preferable. After establishing the assisting role of [Cp2Ti] in releasing H2 from small alcohols, we have applied the same mechanism to study methanediol, for which the overall reaction is exothermic. Our investigation gives strong evidence in favor of the removal of two equivalents of H2 from methanediol, with the regeneration of catalyst. Based on our exploration, we conclude that the [Cp2Ti] sandwich may play significant role for dehydrogenation of alcohols, methanediol, and compounds with amino functionalities.
References
Zuttel A, Borgschulte A, Schlapbach L (eds) (2008) Hydrogen as a Future Energy Carrier. Wiley VCH, Weinheim
Johnson TC, Morris DJ, Wills M (2010) Hydrogen generation from formic acid and alcohols using homogeneous catalysts. Chem Soc Rev 39:81–88
Corey JY, Zhu XH, Bedard TC, Lange LD (1991) Catalytic dehydrogenative coupling of secondary silanes with Cp2MCl2-BuLi. Organometallics 10:924–930
Corey JY, Huhmann JL, Zhu XH (1993) Coupling of deuteriosilanes in the presence of Cp2MCl2/nBuLi. Organometallics 12:1121–1130
Clark TJ, Russell CA, Manners I (2006) Homogeneous, titanocene-catalyzed dehydrocoupling of amine−borane adducts. J Am Chem Soc 128:9582–9583
Jaska CA, Temple K, Lough AJ, Manners I (2003) Transition metal-catalyzed formation of boron−nitrogen bonds: catalytic dehydrocoupling of amine-borane adducts to form aminoboranes and borazines. J Am Chem Soc 125:9424–9434
Luo Y, Ohno K (2007) Computational study of titanocene-catalyzed dehydrocoupling of the adduct Me2NH·BH3: an intramolecular, stepwise mechanism. Organometallics 26:3597–3600
Pun D, Lobkovsky E, Chirik PJ (2007) Amineborane dehydrogenation promoted by isolable zirconium sandwich, titanium sandwich and N2 complexes. Chem Commun 3297–3299
Sloan ME, Staubitz A, Clark TJ, Russell CA, Lloyd-Jones GC, Manners I (2010). J Am Chem Soc 132:3831
Yadav M, Xu Q (2012) Liquid-phase chemical hydrogen storage materials. Energy Environ Sci 5:9698
Teichmann D, Arlt W, Wasserscheid P, Freymann R (2011) A future energy supply based on liquid organic hydrogen carriers (LOHC). Energy Environ Sci 4:2767–2773
Jiang HL, Singh SK, Yan JM, Zhang XB, Xu Q (2010) Liquid-phase chemical hydrogen storage: catalytic hydrogen generation under ambient conditions. ChemSusChem 3:541–549
Bond GC (2006) Metal-catalysed reactions of hydrocarbons. Springer, Berlin
Soloveichik GL, Lemmon JP, Zhao J-C (2008) US Patent Appl 2008/0248339
Bonitatibus PJ, Chakraborty S, Doherty MD, Siclovan O, Jones WD, Soloveichik GL (2015) Reversible catalytic dehydrogenation of alcohols for energy storage. Proc Natl Acad Sci U S A 112:1687–1692
Cheung K-C, Wong W-L, Ma D-L, Lai T-S, Wong K-Y (2007) Transition metal complexes as electrocatalysts—development and applications in electro-oxidation reactions. Coord Chem Rev 251:2367–2385
Prechtl MHG, Wobser K, Theyssen N, Ben-David Y, Milstein D, Leitner W (2012) Direct coupling of alcohols to form esters and amides with evolution of H2 using in situ formed ruthenium catalysts. Catal Sci Technol 2:2039–2042
Royer AM, Rauchfuss TB, Gray DL (2010) Organoiridium pyridonates and their role in the dehydrogenation of alcohols. Organometallics 29:6763–6768
Fujita K-i, Furukawa S, Yamaguchi R (2002) Oxidation of primary and secondary alcohols catalyzed by a pentamethylcyclopentadienyliridium complex. J Organomet Chem 649:289–292
Yuan J, Sun Y, Yu G-A, Zhao C, She N-F, Mao S-L, Huang P-S, Han Z-J, Yin J, Liu S-H (2012) Phenyl substituted indenylphosphine ruthenium complexes as catalysts for dehydrogenation of alcohols. Dalton Trans 41:10309–10316
Zhang D-D, Chen X-K, Liu H-L, Huang X-R (2014) A computational mechanistic study of pH-dependent alcohol dehydrogenation catalyzed by a novel [C,N] or [C,C] cyclometalated Cp*Ir complex in aqueous solution. New J Chem 38:3862–3873
Winter M, Brodd RJ (2004) What are batteries, fuel cells, and Supercapacitors? Chem Rev 104:4245–4270
Chakraborty S, Lagaditis PO, Förster M, Bielinski EA, Hazari N, Holthausen MC, Jones WD, Schneider S (2014) Well-defined Iron catalysts for the acceptorless reversible dehydrogenation-hydrogenation of alcohols and ketones. ACS Catal 4:3994–4003
Lyalin A, Shimizu K-i, Taketsugu T (2017) Interface effects in hydrogen elimination reaction from isopropanol by Ni13 cluster on θ-Al2O3(010) surface. J Phys Chem C 121:3488–3495
Hou C, Jiang J, Li Y, Zhao C, Ke Z (2017) When bifunctional catalyst encounters dual MLC modes: DFT study on the mechanistic preference in Ru-PNNH pincer complex catalyzed dehydrogenative coupling reaction. ACS Catal 7:786–795
Dutta I, Sarbajna A, Pandey P, Rahaman SMW, Singh K, Bera JK (2016) Acceptorless dehydrogenation of alcohols on a diruthenium(II,II) platform. Organometallics 35:1505–1513
Hasanayn F, Harb H (2014) A metathesis model for the dehydrogenative coupling of amines with alcohols and esters into carboxamides by Milstein’s [Ru(PNN)(CO)(H)] catalysts. Inorg Chem 53:8334–8349
Heim LE, Schlörer NE, Choi J-H, Prechtl MHG (2014) Selective and mild hydrogen production using water and formaldehyde. Nat Commun 5:3621
Gunanathan C, Milstein D (2013) Applications of acceptorless dehydrogenation and related transformations in chemical synthesis. Science 341:1229712
Mellmann D, Sponholz P, Junge H, Beller M (2016) Formic acid as a hydrogen storage material - development of homogeneous catalysts for selective hydrogen release. Chem Soc Rev 45:3954–3988
Grasemann M, Laurenczy G (2012) Formic acid as a hydrogen source - recent developments and future trends. Energy Environ Sci 5:8171–8181
Singh AK, Singh S, Kumar (2016) A hydrogen energy future with formic acid: a renewable chemical hydrogen storage system. Catal Sci Technol 6:12–40
Barnard JH, Wang C, Berry NG, Xiao J (2013) Long-range metal-ligand bifunctional catalysis: cyclometallated iridium catalysts for the mild and rapid dehydrogenation of formic acid. J Chem Sci 4:1234–1244
Onishi N, Ertem MZ, Xu S, Tsurusaki A, Manaka Y, Muckerman JT, Fujita E, Himeda Y (2016) Direction to practical production of hydrogen by formic acid dehydrogenation with Cp*Ir complexes bearing imidazoline ligands. Catal Sci Technol 6:988–992
Mellone I, Gorgas N, Bertini F, Peruzzini M, Kirchner K, Gonsalvi L (2016) Selective formic acid dehydrogenation catalyzed by Fe-PNP pincer complexes based on the 2,6-diaminopyridine scaffold. Organometallics 35:3344–3349
Office of Energy Efficiency and Renewable Energy; The FreedomCAR and Fuel Partnership, Targets for onboard hydrogen storage systems for light-duty vehicles, September (2009), http://www1.eere.energy.gov.
Demirci UB, Miele P (2011) Chemical hydrogen storage: ‘material’ gravimetric capacity versus ‘system’ gravimetric capacity. Energy Environ Sci 4:3334–3341
Loges B, Boddien A, Gärtner F, Junge H, Beller M (2010) Catalytic generation of hydrogen from formic acid and its derivatives: useful hydrogen storage materials. Top Catal 53:902–914
Yu X, Pickup PG (2008) Recent advances in direct formic acid fuel cells (DFAFC). J Power Sources 182:124–132
Alberico E, Sponholz P, Cordes C, Nielsen M, Drexler H-J, Baumann W, Junge H, Beller M (2013) Selective hydrogen production from methanol with a defined Iron pincer catalyst under mild conditions. Angew Chem 52:14162–14166
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M., Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K., Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox, DJ (2013) Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA
Chai J-D, Head-Gordon M (2008) Systematic optimization of long-range corrected hybrid density functionals. J Chem Phys 128:084106
Chai J-D, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J Chem Phys 82:270
Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J Chem Phys 82:299
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215–241
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B: Condens Matter Mater Phys 37:785–789
Becke ADJ (1993) Density-functional thermochemistry. III. The role of exact exchange. Chem Phys 98:5648–5652
Gonzalez C, Schlegel HB (1989) An improved algorithm for reaction path following. J Chem Phys 90:2154
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
Klamt A, Schüürmann G (1993) COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc, Perkin Trans 2:799–805
Klamt A, Moya C, Palomar J (2015) A comprehensive comparison of the IEFPCM and SS(V)PE continuum solvation methods with the COSMO approach. J Chem Theory Comput 11:4220–4225
Funding
AG is grateful to the University Grant Commission (UGC); TD and TA are grateful to the Council of Scientific and Industrial Research (CSIR), Government of India, for providing them research fellowships. AKD is grateful to the Council of Scientific & Industrial Research (CSIR), Govt. of India, for a research grant under scheme number: 01(2846)/16/EMR-II.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 5856 kb)
Rights and permissions
About this article

Cite this article
Ghosh, A., Ash, T., Debnath, T. et al. H2-release from alcohols, diols, and compounds with amino functionality promoted by titanium(II) sandwich complex, [Cp2Ti]: a theoretical approach. Struct Chem 30, 681–690 (2019). https://doi.org/10.1007/s11224-018-1207-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1207-0