
Abstract
Four new complexes [M(3-tba)2(H2O)4] (1–3) and [Co(4-tba)2(H2O)4] (4) {M = Zn (1), Ni (2), Co (3), 3-Htba = 3-(1H-1,2,4-triazol-1-yl)benzoic acid, 4-Htba = 4-(1H-1,2,4-triazol-1-yl)benzoic acid} have been synthesized under solvothermal conditions and structurally characterized by single crystal X-ray diffraction. Complexes 1–4 are also determined by elemental analysis, X-ray powder diffraction, IR and electronic spectroscopy. Single crystal X-ray diffraction reveals that complexes 1–3 are isostructural and they crystallize in the orthorhombic space group of Pbca, while complex 4 belongs to triclinic system with Pī space group. Based on different intermolecular hydrogen bonding and π···π stacking interactions, complexes 1–4 further assembled into 3D supramolecular frameworks. Hirshfeld surface analysis was used to further study the intermolecular interactions of the complexes. The thermogravimetric analyses (TGA) reveal that these complexes possess good thermal stability, and the differential scanning calorimetry (DSC) analyses show intense exothermic phenomena in the decomposition processes of triazole groups. Besides, the photoluminescence property of complex 1 in the solid state is also determined.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, supramolecular chemistry continues to attract great research interest not only for their appealing structures but also due to their diversified applications in various fields [1,2,3]. Molecular self-assembly is a spontaneous process for setting the disordered molecules into an organized structure or pattern via non-covalent interactions without external direction [4]. Non-covalent interactions, such as hydrogen bonding, electrostatic, π-π stacking, anion-π, and others, are critical for the formation of supramolecular assemblies and crystal packing [5,6,7,8]. So far, a great number of 1D, 2D, and 3D supramolecular architectures have been synthesized by utilizing coordination bonds and non-covalent interactions as the driving forces. Ligands with a combination of donor groups such as carboxylates, imidazoles, and triazoles are of interest in coordination chemistry [9,10,11]. As multidentate ligands, the triazole-carboxylate derivatives are particularly attractive due to their different coordination modes and bridging functions [12]. Recently, many coordination complexes constructed by different triazole-benzoic acids and metal ions have been reported; some of them not only have novel structures, but also promising chemical and physical properties [13,14,15,16,17]. However, the mononuclear coordination complexes based on triazole-benzoic acids ligands with supramolecular architectures are seldom obtained, unless the appropriate reaction conditions are employed [18].
Herein, we report the syntheses and crystal structures of four new transition metal complexes, namely, [Zn(3-tba)2(H2O)4] (1), [Ni(3-tba)2(H2O)4] (2), [Co(3-tba)2(H2O)4] (3), and [Co(4-tba)2(H2O)4] (4) using 3-(1H-1,2,4-triazol-1-yl)benzoic acid (3-Htba) and 4-(1H-1,2,4-triazol-1-yl)benzoic acid (4-Htba) as ligands (Scheme 1). Intermolecular hydrogen bonding and π···π stacking interactions for complexes 1–4 are particularly discussed in the text. Besides, spectroscopic and thermal properties of the prepared complexes are also investigated.

The molecular structures of 3-Htba and 4-Htba
Experimental
Materials and methods
All chemicals included ligands that were purchased from commercial sources and used as received. FTIR spectra were recorded from KBr pellets in the range of 4000–400 cm−1 on a Varian 640-IR spectrometer. The elemental analysis for C, H, and N was carried out with a Perkin-Elmer 2400 Series II element analyzer. The crystalline phase of the prepared metal complex was identified by a Bruker D8 Advance X-ray diffractometer equipped with Cu Kα radiation (λ = 1.5406 Å). UV–Vis absorption spectra were measured on a Persee TU-1901 (Beijing) spectrometer. The solid-state luminescent spectroscopy was performed on a Hitachi F-4500 fluorescence spectrometer equipped with a xenon lamp. Thermogravimetric analyses (TGA) and differential scanning calorimetry (DSC) analyses were carried out with a Mettler Toledo TGA/DSC 1/1600 (Switzerland) thermal analyzer from room temperature to 800 or 900 °C under N2 atmosphere at a heating rate of 10 °C min−1. Molecular Hirshfeld surface calculations were performed by using the CrystalExplorer 3.1 program, and all the Hirshfeld surfaces were generated using a high surface resolution.
Synthesis of [Zn(3-tba)2(H2O)4] (1)
A mixture of ZnCl2 (27 mg, 0.2 mmol) and 3-Htba (19 mg, 0.1 mmol) in 6 mL solvent (DMF 2 mL, H2O 4 mL) was firstly stirred at room temperature for 15 min; then, the mixed solution was sealed into a pressure glass bottle (15 mL) equipped with a Teflon lid and heated at 120 °C for 48 h. Colorless needle crystals of complex 1 were obtained (yield 70%, based on ligand). Anal. Calcd. (%) for C18H20N6O8Zn: C, 42.08; H, 3.92; N, 16.36. Found (%): C, 42.12; H, 3.95; N, 16.31. IR (KBr, cm−1): 3273 (br, m), 3125(m), 1558(s), 1366(s), 1234(s), 991(m), 754(m), 669(m).
Synthesis of [Ni(3-tba)2(H2O)4] (2)
A mixture of NiCl2·6H2O (24 mg, 0.1 mmol), 3-Htba (19 mg, 0.1 mmol), and 5 mL water was sealed into a 25-mL Teflon-lined autoclave and then heated at 150 °C for 48 h. After cooling to room temperature, green block crystals of complex 2 were obtained (yield 37%, based on ligand). Anal. Calcd. (%) for C18H20N6O8Ni: C, 42.63; H, 3.98; N, 16.57. Found (%): C, 42.58; H, 3.93; N, 16.46. IR (KBr, cm−1): 3265 (br, m), 3125(m), 1556(s), 1367(s), 1234(m), 991(m), 770(m), 654(w).
Synthesis of [Co(3-tba)2(H2O)4] (3)
Synthesis of 3 was similar to that of 1, but with CoCl2·6H2O (24 mg, 0.1 mmol) instead of ZnCl2. Red needle crystals of complex 3 were obtained in 68% yield based on ligand. Anal. Calcd. (%) for C18H20N6O8Co: C, 42.61; H, 3.97; N, 16.57. Found (%): C, 42.57; H, 3.94; N, 16.49. IR (KBr, cm−1): 3263 (br, m), 3125(m), 1556(s), 1367(s), 1232(m), 991(m), 771(m), 652(w).
Synthesis of [Co(4-tba)2(H2O)4] (4)
A mixture of Co(NO3)2·6H2O (29 mg, 0.1 mmol) and 4-Htba (19 mg, 0.1 mmol) in 8 mL mixed solvent (DMF 2 mL, CH3OH 4 mL, H2O 2 mL) was firstly stirred at room temperature for 15 min; then, the mixed solution was sealed into a pressure glass bottle (15 mL) equipped with a Teflon lid and heated at 100 °C for 48 h. Pink block crystals of complex 4 were obtained (yield 73%, based on ligand). Anal. Calcd. (%) for C18H20N6O8Co: C, 42.61; H, 3.97; N, 16.57. Found (%): C, 42.58; H, 3.92; N, 16.50. IR (KBr, cm−1): 3400(br, m), 3117(m), 1531(s), 1387(s), 1150(m), 978(m), 785(m), 673(w), 509(w).
X-ray structure determinations
Crystals of complexes 1–4 having good morphology were chosen for three-dimensional intensity data collection. The X-ray diffraction measurement of complexes was performed on a Bruker SMART APEX II CCD area detector using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Structures were solved using the direct methods procedure in SHELXS-97 [19] and refined by full-matrix least squares on F2 using SHELXL-97 [20] and SHELXL-2014 [21]. The non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were added theoretically. CCDC numbers 1537386, 1537387, 1537388, and 1537389 for 1–4, respectively, contain the supplementary crystallographic data for this paper. Details of the crystal parameters, data collection procedure, and refinement results for complexes 1–4 are summarized in Table 1. Selected bond lengths and angles are listed in Table 2.
Results and discussion
Structural description of complexes 1–3
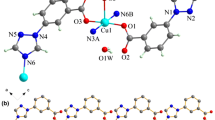
Single crystal X-ray diffraction studies reveal that complexes 1–3 are isostructural with different central metal ions, and they all belong to the orthorhombic system with Pbca space group. Here, the structure of complex 1 will be discussed in detail as an example. The asymmetric unit of 1 consists of one half occupied Zn(II) atom, one 3-tba− ligand, and two coordinated water. As shown in Fig. 1, the central Zn(II) ion is six-coordinated and adopts a distorted octahedral coordination geometry, defined by two N atoms from triazole rings of different ligands and four O atoms from coordinated water molecules. The bond length of Zn–N is 2.1062(14) Å, while the Zn–O(w) distances are ranged from 2.0944(11) to 2.1572(11) Å. The carboxylate group of ligand 3-tba is deprotoned but not coordinate with Zn(II) ion.

Coordination environment of Zn(II) atom in complex 1, symmetry transformations used to generate equivalent atoms, A: 1 − x, 1 − y, 2 − z
Classical O−H···O hydrogen bonds are found in the packing structure of complex 1. As shown in Fig. 2a, all of the four water molecules coordinated with central Zn(II) ion participate in the formation of hydrogen bonds with non-coordinated carboxylate oxygen, and the hydrogen bonding networks are formed. Every coordinated water molecule acts as H donor to form two hydrogen bonds with carboxylate oxygen atoms from different ligands. Furthermore, as the H acceptor, O1 from carboxylate group interacts with three water molecules to form three hydrogen bonds, while O2 only forms one hydrogen bond with water molecule. In complex 1, the d(H···A) of four different hydrogen bonds are 1.875, 1.996, 2.015, and 2.020 Å, respectively. The similar hydrogen bonds are also found in the packing structure of complexes 2 and 3. The structural parameters of hydrogen bonds for complexes 1–3 are shown in Table 3. It is noteworthy that the H···A distances in complexes 1–3 are ranged from 1.848 to 2.042 Å, and the angles of hydrogen bonds are ranged from 159.3 to 177.2°, which indicate the strong intermolecular hydrogen bonding interactions in complexes 1–3.

a The packing structure of complex 1 showing intermolecular hydrogen bonding interactions (The hydrogen bonds are represented by dashed lines). b Supramolecular framework of complex 1 (view along a axis)
As shown in Fig. 3, strong face-to-face π···π stacking interactions between the adjacent benzene rings and triazole rings can be obviously observed. For complex 1, the centroid to centroid distances between triazole ring (Cg5) and the two adjacent benzene rings (Cg6) on both sides are 3.546 and 3.772 Å (3.597 and 3.799 Å for 2; 3.539 and 3.771 Å for 3). Interestingly, for the same 3-Htba ligand, the centroid to centroid distances between benzene ring (Cg6) to two adjacent triazole rings (Cg5) on both sides are 3.772 and 3.546 Å, respectively, which indicate that the two adjacent aromatic rings are not exactly parallel and the dihedral angle between the two adjacent planes is 1.599°. The π···π stacking mode between adjacent aromatic rings in complexes 1–3 can be simplified as (···Cg5···Cg6···Cg5···Cg6···)n. Complexes 1–3 are further connected by water–carboxylate hydrogen bonds and π···π interactions to form a stable 3D supramolecular structure (Fig. 2b).

π···π stacking interactions of 1–3 between the adjacent benzene rings and triazole rings
Structural description of complex 4
X-ray crystallographic analysis demonstrates that complex 4 belongs to triclinic crystal system with P-1 space group. Compounds 4 and 3 are isomeric structures with different ligands [22]. The asymmetric unit of 4 consists of one half occupied Co(II) atom, one 4-tba− ligand, and two coordinated water. As shown in Fig. 4, the central Co(II) ion is six-coordinated and defined by two N atoms from triazole rings of 4-tba− ligands and four O atoms from coordinated water molecules. The bond length of Co–N is 2.140(2) Å, while the Co–O(w) distances range from 2.081(2) to 2.146(2) Å. Similarly, the carboxyl group of ligand 4-tba is deprotoned and not coordinates with Co(II) ion.

Coordination environment of Co(II) atom in complex 4, symmetry transformations used to generate equivalent atoms, A: 1 − x, 1 − y, − z
Figure 5a shows the rich hydrogen bonds in the packing structure of complex 4 (dashed lines), in which the four coordinated water molecules act as H donors, while the two oxygen atoms from carboxylate groups act as H acceptors. As shown in Fig. 5a, every coordinated water molecule forms two hydrogen bonds with carboxylate oxygen atoms from different ligands. Moreover, O1 from carboxylate group interacts with three coordinated water molecules to form three hydrogen bonds, while O2 interacts with one coordinated water molecule to form one hydrogen bond. For complex 4, the d(H···A) of four different hydrogen bonds are 1.858, 1.907, 2.050, and 2.090 Å, respectively (Table 3).

a The intermolecular hydrogen bonding interactions of complex 4 (the hydrogen bonds are represented by dashed lines). b Packing diagram of the complex 4 showing π···π stacking interactions between adjacent aromatic fragments; coordinated water molecules are omitted for clarity. c Supramolecular framework of complex 4 (view along b axis)
As shown in Fig. 5b, the motif of π···π stacking interactions in complex 4 is different to that of 1–3. The centroid to centroid distance between triazole (Cg5) ring and adjacent benzene ring (Cg6) is 3.768 Å, while the centroid to centroid distance between the two adjacent benzene rings (Cg6) is 3.852 Å. The π···π stacking mode between adjacent aromatic rings in complex 4 can be simplified as Cg5···Cg6···Cg6···Cg5. Complex 4 further packed into a 3D supramolecular framework through different water–carboxylate O−H···O hydrogen bonds and strong face-to-face π···π interactions (Fig. 5c). Different coordination modes are found in other transition metal(II) complexes of 4-Htba ligand. For example, in the crystal structure of complex [Zn(4-tba)2]n, the Zn(II) center has a tetrahedral coordination geometry, and the 4-tba− ligand acts as μ2-linker to connect the Zn(II) atoms via one triazole N atom and one carboxylate O atom, forming a fivefold interpenetrating diamondoid network [14]. However, in complexes [M(4-tba)(CH3COO)·solvent]n (M = Mn or Co), the central metal atoms adopt distorted octahedral coordination geometry, and the 4-tba− ligand acts as μ3-linker to connect the metal atoms via one triazole N atom and two carboxylate O atoms, giving a 3D framework [23].
Hirshfeld surface analysis
Hirshfeld surface analysis was used to further study the intermolecular interactions of the two different crystal structures. Hirshfeld surface gives a detailed explanation on the immediate environment of a molecule in a crystal [24, 25]. The 3D Hirshfeld surface and 2D fingerprint plots of complex 3 (as an example) are illustrated in Fig. 6. The 3D Hirshfeld surfaces have been mapped over dnorm, shape index, and curvedness. The surfaces are shown as transparent to allow visualization of the molecular moiety around which they were calculated. As shown in Fig. 6a, the deep red spots on the dnorm Hirshfeld surface indicate the close-contact interactions, which are mainly responsible for the significant intermolecular hydrogen bonding interactions. The deep red spots labeled as 1a and 1b in Fig. 6a can be assigned to the hydrogen bonding interactions formed by carboxylate group and coordinated H2O, respectively. The shape index is most sensitive to very subtle changes in surface shape; the red triangles on them represent concave regions indicating atoms of the π···π stacked molecule above them [25]. In the case of complex 3, the red triangles are π···π intermolecular interactions. The curvedness is the measurement of “how much shape”; the flat areas of the surface correspond to low values of curvedness, while sharp curvature areas correspond to high values of curvedness, indicating interactions between neighboring molecules [25]. The large flat region indicated by a blue outline on the curvedness surface refers to the π···π stacking interactions of the molecule (Fig. 6c). The π···π stacking information conveyed by the shape index and curvedness plots are consistent with the crystal structure analyses. Hirshfeld 2D fingerprint plots allow a quick and easy identification of the significant intermolecular interactions map on the molecular surface [26, 27]. As shown in Fig. 6e, the strong O···H/H···O hydrogen bonding interactions cover 33.5% of the total Hirshfeld surface with two distinct spikes in the 2D fingerprint plots, indicating hydrogen bonding interactions are the most significant interaction in the crystal. As shown in Fig. 6f, in the middle of scattered points in the 2D fingerprint plots, H···H interactions cover 32.5% of the total Hirshfeld surface. However, according to the de and di value, the H···H interactions are not very strong in the crystal. Besides the above interactions, the presence of relatively weak N···H (7.2%) and C···H (7.8%) interactions is also observed, as shown in Fig. S1.

Hirshfeld surface mapped with dnorm (a), shape index (b), curvedness (c), and fingerprint plots (d–f) for complex 3
The Hirshfeld surfaces of 4 are illustrated in Fig. 7; the deep red spots on the dnorm Hirshfeld surface (Fig. 7a) indicate the significant intermolecular hydrogen bonding interactions formed by carboxylate groups (labeled as 2a) and coordinated H2O molecules (labeled as 2b). The red triangles on the shape index surface (Fig. 7b) correspond to π···π intermolecular interactions. The flat region indicated by a blue outline on the curvedness surface (Fig. 7c) also refers to the π···π stacking interactions of the molecule. The decomposition of the fingerprint plot shows that the O···H/H···O hydrogen bonding interactions appear as two distinct spikes in the 2D fingerprint plot which cover 33.0% of the total Hirshfeld surface (Fig. 7e). The H···H intermolecular interactions appear as a small spike, which have the most significant contribution to the total Hirshfeld surfaces, comprising 34.9% (Fig. 7f). The proportions of N···H and C···H interactions comprise 6.6 and 6.5% of the total Hirshfeld surfaces (Fig. S2), respectively.

Hirshfeld surface mapped with dnorm (a), shape index (b), curvedness (c), and fingerprint plots (d–f) for complex 4
XRD patterns
X-ray powder diffraction data for complexes 1–4 were collected (2θ range, 5–50°). As shown in Fig. 8, the experimental XRD patterns agree well with the simulated patterns generated on the basis of the single-crystal analyses for complexes 1–4, which suggest the good phase purity and homogeneity of the synthesized samples.

PXRD patterns for complexes 1–3 (a) and 4 (b) in solid state
IR spectra
As shown in Fig. 9, the isostructural complexes 1, 2, and 3 have the similar IR spectra. In the IR spectra of complexes 1–3, no strong band is found in the region 1690–1730 cm−1 which indicates deprotonation of the carboxyl groups [28, 29]. The asymmetric and symmetric stretching vibrations of carboxyl groups are observed at about 1556 and 1367 cm−1, respectively [30]. The peaks at about 3125 cm−1 are attributed to sp2 C–H stretching vibrations of the aromatic rings [31], and the broad bands appeared between 3200 and 3400 cm−1 can be assigned to O–H vibrations of coordinated water molecules. The IR spectrum of complex 4 shows two strong bands at 1531 and 1387 cm−1, which can be assigned to the asymmetric and symmetric stretching vibrations of carboxyl groups, respectively. In addition, the peak at 3117 cm−1 is attributed to sp2 C–H stretching vibrations.

IR spectra of complexes 1–4
Electronic spectra and luminescent property
Electronic spectra of complexes 1–4 and their organic ligands were measured in methanol solution at ambient temperature (concentrations are ca. 10−4 M). As shown in Fig. 10a, complexes 1–3 and free 3-Htba display similar absorption bands in the UV region. The strong bands centered at 232, 216, and 231 nm for 1, 2, and 3 are attributed to the ligand-centered π → π* transitions, which show blue shift of 7, 23, and 8 nm when compared with free ligand 3-Htba (239 nm), respectively. The weak bands at about 282 nm for complexes 1–3 may be assigned to n → π* transitions of the triazole group of ligand. As shown in Fig. 10b, complex 4 exhibits two strong absorption bands at 207 and 255 nm, which can be assigned to π → π* electron transitions [28]. The two bands of complex 4 show blue shift of 6 and 3 nm when compared with the bands of free ligand 4-Htba.

The electronic spectra for complexes 1–4 and the free ligands in the UV region at room temperature
The solid-state emission properties of Zn(II) complex (1) and free ligand (3-Htba) were investigated at room temperature using powder samples. As shown in Fig. 11, ligand 3-Htba exhibits a fluorescent emission band at 349 nm (λex = 302 nm), which can be assigned to the π* → π transitions [32]. Complex 1 displays a strong blue fluorescent emission band at 356 nm, when excited at 290 nm. The emission of 1 is probably attributed to the intraligand π* → π transitions. Complex 1 exhibits red shift of 7 nm when compared to free ligand 3-Htba, which mainly due to a metal-to-ligand or ligand-to-metal charge transfer [33]. The high intensity of luminescence in d10 complexes can be attributed to the ligand chelation to the metal ion causing the increased rigidity of the ligand and the reduction of energy loss by radiation-less decay [34, 35]. Complexes 2–4 do not exhibit detectable emission.

Solid-state emission spectrum of complex 1 (excited at 290 nm) and 3-Htba (excited at 302 nm) at room temperature
Thermal analyses
Based on the isostructures of complexes 1–3, the thermal behaviors of [Co(3-tba)2(H2O)4] (3) and [Co(4-tba)2(H2O)4] (4) will be discussed in detail as an example. The thermal gravimetric analyses (TGA) have been performed under a flow of nitrogen. As shown in Fig. 12, in the TGA curve of complex 3, the first step takes place in the range of 130–200 °C with a weight loss of 14.4%, corresponding to the release of four coordinated water (calcd. 14.2%). Then, a rapid and significant weight loss of 26.3% is observed in the range of 340–440 °C, which could be assigned to the decomposition of triazole group of 3-Htba− ligands (calcd. 26.8%). Further heating leads to more weight loss, which corresponds to the continue decomposition of the remaining skeleton. In the case of complex 4, the TGA curve shows a 14.2% weight loss at about 114–227 °C, corresponding to the loss of four water molecules (calcd. 14.2%). Similarly, a rapid weight loss of 26.6% can be discerned in the range of 284–402 °C, corresponding to the collapse of triazole groups of 4-Htba− ligands (calcd. 26.8%). Following this, a continuous mass loss is seen until 900 °C with a residual mass rate of 12.4%, and the remaining part may decompose completely to CoO (calcd. 14.7%).

TGA curves of complexes 3 and 4 measured in nitrogen atmosphere at a heating rate of 10 °C min−1
Complexes 3 and 4 are also characterized by differential scanning calorimetry (DSC) analyses. In the DSC curve of complex 3 (Fig. S3), the endothermic dehydration process occurs in the temperature range of 110–200 °C, with a peak temperature of 168 °C. In addition, a strong exothermic process from 240 to 275 °C with a peak temperature of 258 °C can be assigned to collapse of the triazole groups. In the case of complex 4 (Fig. S4), the endothermic dehydration process occurs in the temperature range of 110–230 °C, with a peak temperature of 193 °C. Moreover, a moderate exothermic process from 297 to 360 °C with a peak temperature of 308 °C may be ascribed to collapse of the triazole groups.
Conclusion
In summary, four new transition metal complexes based on 3-(1H-1,2,4-triazol-1-yl)benzoic acid and 4-(1H-1,2,4-triazol-1-yl)benzoic acid have been synthesized under solvothermal conditions. The structures of the complexes were determined by single-crystal X-ray diffractions. The four complexes are further constructed into 3D supramolecular structures via different intermolecular hydrogen bonds and interesting π–π stacking interactions. Moreover, Hirshfeld surface analysis was used to further study the intermolecular interactions of the synthesized complexes; the results agree well with their single-crystal structure analyses. The packing structure of complex 4 is different to the other three complexes, which is just effected by the position of carboxylate group of the ligand. Thermal behavior investigations indicate that the structures of complexes 3 and 4 possess good thermal stabilities, with obvious exothermic phenomena in the decomposition processes. Besides, complex 1 shows a strong emission band at 356 nm when excited at 290 nm. Further studies based on triazole-carboxylate ligands are under way in our laboratory.
References
Zhou P, Shi RF, Yao JF, Sheng CF, Li H (2015). Coord Chem Rev 292:107
Santo AD, Echeverría GA, Piro OE, Perez H, Altabef AB, Gil DM (2017). J Mol Struct 1134:492
Zhang X, Liu LW, Hao ZC, Cui GH (2016). Transit Met Chem 41:459
Mirzaei M, Eshtiagh-Hosseini H, Bazargan M (2015). Res Chem Intermed 41:9785
Sadhu MH, Mathoniere C, Patil YP, Kumar SB (2017). Polyhedron 122:210
Yajima T, Takamido R, Shimazaki Y, Odani A, Nakabayashi Y, Yamanchi O (2007). Dalton Trans 36:299
Mitra A, Clark RJ, Hubley CT, Saha S (2014). Supramol Chem 26:296
Jin F, Zhang Y, Wang HZ, Zhu HZ, Yan Y, Zhang J, Wu JY, Tian YP, Zhou HP (2013). Cryst Growth Des 13:1978
Song Y, Fan RQ, Gao S, Wang XM, Wang P, Yang YL, Wang YL (2015). Inorg Chem Commun 53:34
Zhao H, Chen JM, Lin JR, Wang WX (2011). J Coord Chem 64:2735
Hong JL, Sun LN, Zhai ZR, Zhao H (2014). Chinese J Inorg Chem 30:1678
Li J, Xiong PP, Bu HY, Chen SP (2014). Acta Phys -Chim Sin 30:1354
Du M, Li CP, Chen M, Ge ZW, Wang X, Wang L, Liu CS (2014). J Am Chem Soc 136:10906
Wang YL, Fu JH, Wei JJ, Xu X, Li XF, Liu QY (2012). Cryst Growth Des 12:4663
Li T, Liu X, Huang ZP, Lin Q (2014). Inorg Chem Commun 39:70
Li T, Yang J, Hong XJ, Ou YJ, Gu ZG, Cai YP (2014). CrystEngComm 16:3848
Mu YH, Ge ZW, Li CP (2014). Inorg Chem Commun 48:94
Du PY, Su J, Lv R, Gu W, Liu X (2016). Polyhedron 115:86
Sheldrick GM (1997) SHELXS-97. Program for crystal structure resolution. University of Göttingen, Germany
Sheldrick GM (1997) SHELXL-97. Program for crystal structure refinement. University of Göttingen, Germany
Sheldrick GM (2015). Acta Crystallogr Sect A A71:3
Ying SM (2012). Inorg Chim Acta 387:366
Nandi S, Haldar S, Chakraborty D, Vaidhyanathan R (2017). J Mater Chem A 5:535
Dhamodharan P, Sathya K, Dhandapani M (2017). Physica B 508:33
Li Y, Zhang CG, Cai LY, Wang ZX (2013). J Coord Chem 66:3100
Luo YH, Liu QL, Yang LJ, Wang W, Ling Y, Sun BW (2015). Res Chem Intermed 41:7059
Zhang JL, Zhang CL, Xiao Y, Qin Y, Zhang SH (2016). Supramol Chem 28:231
Zhang X, Zhao YQ, Hao ZC, Cui GH (2016). J Inorg Organomet Polym. https://doi.org/10.1007/s10904-016-0397-4
Feng X, Ling XL, Liu L, Song HL, Wang LY, Ng SW, Su BY (2013). Dalton Trans 42:10292
Boghaei DM, Gharagozlou M (2007). Spectrochim Acta Part A 67:944
Chen S, Fan RQ, Sun CF, Wang P, Yang YL, Su Q, Mu Y (2012). Cryst Growth Des 12:1337
Yang LB, Wang HC, Dou AN, Rong MZ, Zhu AX, Yang Z (2016). Inorg Chim Acta 446:103
Zhang L, Dang LL, Luo F, Feng XF (2016). J Mol Struct 1106:114
Roy B, Mukherjee S, Mukherjee PS (2013). CrystEngComm 15:9596
He W, Wang B, Bai X (2008). Transit Met Chem 33:399
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 21371151, 21461029, and 21561033).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, DW., Wang, T., Du, L. et al. Four supramolecular transition metal(II) complexes based on triazole-benzoic acid derivatives: crystal structure, Hirshfeld surface analysis, and spectroscopic and thermal properties. Struct Chem 29, 1013–1023 (2018). https://doi.org/10.1007/s11224-018-1084-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1084-6