
Abstract
In the present study, five-membered heterocyclic ring systems containing oxygen with one, two, three, and four nitrogen atoms in the ring along with their isomeric forms and their corresponding 1:1 water complexes have been fully optimized at the ab initio molecular orbital theory and Density Functional Theory (DFT) using aug-cc-pVDZ basis set. The optimized geometric parameters and stabilization energies of the complexes are reported. The study suggests that nitrogen of heterocyclic ring is a stronger hydrogen bond acceptor in comparison to oxygen and ability of nitrogen to act as hydrogen bond acceptor increases in the order oxazole (OZ) > oxadiazole (ODZ) > oxatriazole (OTZ) > oxatetrazole (OTTZ). The results are corroborated by Natural Bond Orbital (NBO) analysis, Quantum Theory of Atoms in Molecules (QTAIM), Symmetry Adapted Perturbation Theory (SAPT), and Molecular Electrostatic Potential (MEP) studies. The blue- and red-shifts in the hydrogen bond donors X-H (X = O, C) stretching frequencies have also been analyzed. Hydrogen bond ability has also been governed in the presence of reactivity descriptors including chemical potential (μ), global hardness (η), and electrophilicity index (ω).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Five-membered heterocycles with O and N heteroatoms are among the privileged structures that play pivotal role in many biologically natural products and useful pharmaceutical agents [1,2,3,4]. The oxazole moiety is a valuable precursor in many biological potent natural products such as phenoxan, pimprinethine, pimprinaphine, calyculins, pimprinine, and rhizoxin [5,6,7,8]. In nature, the oxazole ring is created by microorganisms in post-translational modifications from serine or threonine residues in peptides. Oxazole derivatives display antiinflammatory [9], analgesic [10, 11], antibacterial [12], antifungal [13, 14], antimicrobial [15,16,17], antituberculosis [18, 19], antidiabetic [20], antiviral [21], antiproliferative [22,23,24], and anticancer [25,26,27] activities. Within the drug discovery and development, a number of compounds containing an oxadiazole moiety are in late stage clinical trials including zibotentan [28] as an anticancer agent, ataluren for the treatment of cystic fibrosis [29], and raltegravir [30] as an antiretroviral drug for the treatment of HIV infection. Attributing to these facts, heterocyclic ring systems with O and N heteroatoms have allured attention in the field of medicinal research.
The hydrogen bonding interactions are important because these interactions play an influential role in determining the shapes, properties, and functions of number of biomolecules. Heterocycles with O and N heteroatoms display interesting hydrogen bond (HB) acceptor properties. Studies have shown that hydrogen bonding ability of oxygen covalently bonded to sp 2 hybridized carbon atoms are weaker relative to nitrogen in the equivalent situation. This is in contrast to the common expectation, based exclusively on electronegativity, that both are good acceptors. In biological systems, the prevalence of N and O suggests that substituting an oxygen with a nitrogen acceptor in a ligand could retain the hydrogen bonding in the protein and yet produces a chemically distinct ligand. This requires knowledge of the directionality and relative strengths of the HB acceptors. The water is known as an active constituent in biochemical processes [31, 32]. Thus, the intermolecular hydrogen bonding between the water and molecules of biological interest is significant in rationalizing the mechanism to regulate the biochemical processes. HB interactions involving oxygen and nitrogen (as HB acceptors) and C-H (as HB donors) of heterocyclic molecules with water play an important role in drug-receptor interactions, protein structure, crystal structure arrangement, and conformational preferences of drug molecules in the ligand pocket. The theoretical study of non-classical C-H···O HBs with C-H of heterocycles as hydrogen donors has significance in structures of biopolymers and in the living processes like enzyme catalysis or molecular recognition.
The aim of the present work is focused on studying hydrogen bonding ability of family of oxazole members with water through 1:1 complex formation. The HB donor and acceptor abilities of various sites of heterocycles have been studied. The energetic, topological, electronic, and structural factors affecting the stability of hydrogen bonded complexes of heterocycles with water have been analyzed and compared. By investigating HOMO-LUMO energy gap, the chemical stability against electronic excitation also has been studied. The global molecular descriptors including chemical potential (μ), hardness (η), and electrophilicity index (ω) have also been calculated from the optimized geometries of heterocycles with water.
Computational methods
The geometries of the monomers and the corresponding hydrogen bonded complexes were optimized using second-order Møller-Plesset perturbation theory (MP2) with aug-cc-pVDZ basis set using Gaussian 09 software [33]. The density functional theory calculations at B3LYP level were also performed using aug-cc-pVDZ basis set for comparison purposes. The stabilization energy (ΔE BSSE) was estimated as the difference between the total energy of the complex and the sum total of the monomers. Calculated interaction energies (ΔE BSSE) for the hydrogen bonded complexes were corrected for the basis set superposition error (BSSE) employing counterpoise (CP) method of Boys and Bernadi [34]. Natural Bond Orbital (NBO) analysis was performed at B3LYP/aug-cc-pVDZ level via the method incorporated within the Gaussian 09 package [35]. The topological and energetical properties at the bond critical points (BCP) were analyzed using the QTAIM program [36]. The Molecular Electrostatic Potential (MEP) was computed using WFA surface analysis suite [37], using 0.001 au (electrons/bohr3) contour on the molecule’s electron density [38]. To analyze the electrostatic, induction, dispersion, and exchange contributions to the total stabilization energy, Symmetry Adapted Perturbation Theory (SAPT) analysis was performed at the MP2/aug-cc-pVDZ level with the use of GAMESS package linked to the SAPT 2012.2 code [39,40,41].
Results and discussion
Stabilization energies and geometrical parameters
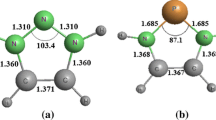
The five-membered heterocycles oxazole (OZ), oxadiazole (ODZ), oxatriazole (OTZ), and oxatetrazole (OTTZ) along with their isomeric forms have been optimized at B3LYP/aug-cc-pVDZ (L1) and MP2/aug-cc-pVDZ (L2) theoretical levels (Fig. 1). The relative energy order of isomeric forms of OZ, ODZ, OTZ is as follows: OZ13 > OZ12, ODZ134 > ODZ124 > ODZ123 > ODZ125, and OTZ1234 > OTZ1235. The 1:1 complexes of these heterocycles with water have also been optimized at the abovementioned levels. The stabilization energy (ΔE BSSE) of hydrogen bonded complex is an important criterion in diagnosing the presence of HBs. The presence of HBs in these complexes has been explored by analyzing the distances and respective angles between the probable HB donors and acceptors and is cited in Table 1. The ∆r values that represent the difference in sum of van der Walls radii of atoms involved in hydrogen bonding and the distances computed between the atoms in the complex are also included in Table 1. The optimized geometrical parameters for the complexes are using L1, and L2 theoretical levels are summarized in Tables S1–S13. It is apparent from Table 1 that the stabilization energies of these complexes are computed with in a range of −1.56 to −5.72 kcal/mol at L2 theoretical level, while the range at L1 level falls in −1.30 to −5.42 kcal/mol. The IUPAC report agrees on important geometric feature that the angle D-H···A (D is HB donor and A is HB acceptor) should preferably be above 110° and close to 180°. The noncovalent bonded distance between H···A is usually observed to be lower than sum of van der Waals radii of H and A, but in some cases, it is found to be longer also. Furthermore, the length of D-H bond usually increases on HB formation. As can be seen from Table 1, the angle D-H···A in all these complexes is above 110°, i.e., they tend towards linearity except in case of C-H···O interactions. As can be seen from Table 1, the HB angles in the complexes range between 101.81° and 178.74°. Interestingly, the HB angles in the complexes that are stabilized by single HB are more linear while the presence of second HB leads to the formation of quasi five-membered ring structure along with heterocyclic ring forcing the HB angle to be less than 180°.

Optimized geometries of five-membered heterocycles along with their isomeric forms at MP2/aug-cc-pVDZ level
Table 1 displays the change in D-H bond length and shifts of stretching frequencies (Δʋ in cm−1) of the HB donor group upon complex formation relative to monomers. Red-shift in the O-H stretching frequency has been traditionally considered as one of the main fingerprints of HBs, assuming that formation of a HB weakens an O-H single bond. Upon complex formation, the O-H bond of water (HB donor) in all the complexes is elongated and accompanied by a red-shift in the stretching frequency which spans over a range of −9.43 to −154.50 cm−1 for this bond. Blue-shift in ʋ C-H is reflected with a range of +2.52 to +12.69 cm−1 in complexes involving C-H as HB donor and O-H of water acting as HB acceptor. But it is worth emphasizing that the C-H stretching is found to be red-shifted for complexes involving single C-H···Ow interaction. For such complexes, the increase in C-H bond length ranges between 0.001 and 0.004 Å and stretching frequency shift for this bond lies in the range −8.49 to −49.54 cm−1.
The 1,2 and 1,3 positioning of O and N in the heterocyclic ring represents the two isomeric forms of OZ. The complexation between the OZ12 and water resulted in three energy minimum structures, out of which OZ12-W1 is the highest in stabilization (ΔE BSSE = −4.85 kcal/mol) and is supported by single O10-H9···N2 HB. The O9···H10-O1 and C5-H8···O9 HBs in OZ12-W2 stabilize the complex by −3.68 kcal/mol which is next to OZ12-W1. The HB distances and HB angles both suggest O9-H10···O1 HB to be more stable relative to C5-H8···O9. The stabilization energy of −2.29 kcal/mol associated with C4-H7···O9 single HB in OZ12-W3 supports the importance of nonconventional HBs. The OZ13-W1 with N of heterocyclic ring as HB acceptor to water has stabilization −5.72 kcal/mol which is 0.87 kcal/mol higher relative to that of OZ12-W1 having similar HB. The C5-H8···O9 hydrogen bonded OZ13-W2 (single HB) has ΔE BSSE value of −2.46 kcal/mol which is higher relative to C4-H7···O9 hydrogen bonded OZ12-W3 (single HB). The difference in HB strength can be rationalized in view of difference in HB strength.
The hydrogen bonding between ODZ and water resulted in 12 optimized complexes, considering the four isomeric forms of ODZ. The optimized structures are shown in Fig. 2. In all the isomeric forms of ODZ, the most stable 1:1 complex with water is named with W1 at the suffix. As can be viewed from Fig. 2, in all these four complexes, water acts as an HB acceptor towards C-H of heterocyclic ring and HB donor towards the N of the ring, resulting in quasi-five-membered ring. The stabilization energy of these four complexes increases in the order ODZ125-W1 < ODZ123-W1 < ODZ124-W1 < ODZ134-W1. The wO-H···N is the major contributing interaction in ΔE BSSE in comparison to C-H···Ow as suggested by geometrical parameters. The geometrical parameters reflect that the wO-H···N HB distance increases in the order of decreasing stabilization energy. The complex ODZ125-W2 is unique in the sense that both the lone pairs present on oxygen of water act as an HB acceptor towards two C-H bonds of heterocyclic ring. The complexes ODZ123-W2, ODZ124-W2, and ODZ134-W2 have ΔE BSSE values −3.74, −3.66, and −3.08 kcal/mol, respectively, and have similar HB donors and acceptors. The ΔE BSSE values for ODZ123-W3 complex are −1.38 kcal/mol lower than that of ODZ124-W3 though both are stabilized through WO-H···N HB. The C-H···OW hydrogen bonded ODZ123-W4 complex with ΔE BSSE of −2.84 kcal/mol highlights the importance of such interaction in drug target binding. The geometrical parameters of ODZ134-W2 also reflect C-H···OW strong HB interactions along with weak support from O-H···OW HB. The weakly bound ODZ125-W2 complex with two unconventional C-H···OW interactions where C-H bonds of heterocyclic ring are donor to oxygen of the water has O···H distance exceeding over 2.6 Å. The ∆r H···A values for these weak interactions are negative with C-H···OW angles nearly equal to 108°, and the hydrogen-donating bonds are shortened by 0.001 Å as shown by Δd values in Table 1.


Optimized geometries of 1:1 hydrogen-bonded complexes of five-membered heterocycles with water at MP2/aug-cc-pVDZ level
The two isomers of OTZ are OTZ1234 and OTZ1235, each containing one oxygen and three nitrogens at 1,2,3,4 and 1,2,3,5 positions, respectively. Four complexes each of the two isomers with single water have been optimized and are shown in Figs. 2 and 3. The most stabilized complex in the two isomeric forms (OTZ1234-W1 and OTZ1235-W4) have stabilization energy of −5.35 and −4.60 kcal/mol, respectively. Three complexes OTZ1234-W1, OTZ1235-W1, and OTZ1235-W4 are adorned by two WO-H···N and C-H···OW HBs. The difference in ΔE BSSE values results from difference in HB donor and acceptor sites in the presence of three nitrogens and one oxygen in the heterocyclic ring. The W2 complexes of OTZ1234 and OTZ1235 involve single HB in which water is acting as HB donor towards N of heterocyclic ring and ΔE BSSE values are −2.46 and −2.37 kcal/mol, respectively. The lower ΔE BSSE values for these two complexes indicate that electronegativity of N atom involved in HB is decreased significantly by the electronegative atoms in adjacent position to it and the linear approach being hindered.


Molecular graphs of complexes of five-membered heterocycles with water at the MP2/aug-cc-pVDZ level. Small green balls indicate bond critical points and small red balls indicate ring critical points
Complexes which involve single HB formation with WO-H···N interaction are OZ12-W1 (2.044 Å), OZ13-W1 (1.994 Å), ODZ123-W3 (2.114 Å), ODZ124-W3 (2.073 Å), OTZ1234-W2 (2.169 Å), OTZ1235-W2 (2.164 Å), and OTTZ12345-W1 (2.324 Å). The order of stabilization energies in these complexes is OZ13-W1 (−5.72 kcal/mol) > OZ12-W1 (−4.85 kcal/mol) > ODZ124-W3 (−4.76 kcal/mol) > ODZ123-W3 (−3.38 kcal/mol) > OTZ1234-W2 (−2.46 kcal/mol) > OTZ1235-W2 (−2.37 kcal/mol) > OTTZ12345-W1(−1.56 kcal/mol) which support the fact that N of heterocyclic ring act as strong HB acceptor and the ability increases in the order OZ > ODZ > OTZ > OTTZ. Complexes which involve single HB formation with C-H···OW interactions are OZ12-W3 (2.272 Å), OZ13-W2 (2.241 Å), ODZ123-W4 (2.236 Å), OTZ1234-W3 (2.103 Å), and OTZ1235-W3 (2.152 Å). The order of stabilization energy in these complexes is OTZ1234-W3 (−4.33 kcal/mol) > OTZ1235-W3 (−3.69 kcal/mol) > ODZ123-W4 (−2.84 kcal/mol) > OZ13-W2 (−2.46 kcal/mol) > OZ12-W3 (−2.29 kcal/mol) which support the fact that C-H HB donor ability decreases in the order OTZ > ODZ > OZ.
NBO analysis
Charge analysis
NBO has been used to determine the extent of charge transfer and predict the HB acceptor ability of various sites in a molecule. Table 2 illustrates the values of charge transfer from heterocyclic unit to water (CTH-W) for the complexes under study at B3LYP/aug-cc-pVDZ level. The complexes OZ12-W2, ODZ123-W1, ODZ124-W1, ODZ124-W4, ODZ125-W1, ODZ134-W1, OTZ1234-W1, OTZ1235-W1, and OTZ1235-W4 involve HBs with both heterocyclic ring and water acting as donor as well as acceptor; therefore, charge transfer occurs from heterocyclic unit to water and vice versa. The CTH-W values of these complexes being positive indicate that charge transfer from the heterocyclic unit to water is higher than that from water to heterocyclic unit, so the direction of net charge transfer is from heterocyclic ring to water.
The CTH-W values of complexes that are stabilized through single HB with WO-H···N interaction fall in the order OZ13-W1 > OZ12-W1 > ODZ123-W3 ~ ODZ124-W3 > OTZ1234-W2 ~ OTZ1235-W2 > OTTZ12345-W1. The CTH-W values of these complexes being positive indicate that charge transfer occurs from heterocyclic unit to water and indicates that N of heterocyclic ring acts as better HB acceptor in the order OZ > ODZ > OTZ > OTTZ. The C-H···Ow single hydrogen bonded complexes OZ12-W3, OZ13-W2, ODZ123-W4, OTZ1234-W3, and OTZ1235-W3 have negative values of CTH-W, as net charge transfer occurs from water to heterocyclic ring. The order of CTH-W values suggest that C-H HB donor ability increases in the order OTZ > ODZ > OZ.
The CTH-W values of complexes ODZ123-W1, ODZ124-W1, ODZ124-W4, ODZ125-W1, ODZ134-W1, OTZ1234-W1, OTZ1235-W1, and OTZ1235-W4 (WO-H···N as much stabilizing HB interactions along with weak C-H···OW interactions) being positive and the CTH-W values for complexes OZ12-W2, ODZ123-W2, ODZ124-W2, and ODZ134-W2 (with WO-H···O as main stabilizing interactions along with weak C-H···OW interactions) being negative indicate that N of ring act as better HB acceptor as compared to O of ring.
The largest charge transfer with CTH-W value of +0.011 and −0.011 is reflected for OZ13-W1 and OTZ1235-W3. Both the complexes are bonded through single HB. The former has charge transfer associated with in nN3 → σ* O-H, while the latter reflects nO7 → σ* C4-H6. Both the charge transfers are in opposite directions and clearly indicate that charge transfer away from heterocyclic ring contributes more towards stability.
Analysis based on second-order delocalization energy [E (2)]
The information about the electron delocalization is reflected by the E (2) values, second-order perturbation energies associated with the orbital interaction. The complexes OZ13-W1, OZ12-W1, ODZ123-W3, ODZ124-W3, OTZ1234-W2, OTZ1235-W2, and OTTZ12345-W1 possess nN → σ* O-H charge transfer interactions only. The order of E (2) values in the complexes OZ13-W1 (8.38 kcal/mol) > OZ12-W1 (6.01 kcal/mol) > ODZ124-W3 (5.43 kcal/mol) > ODZ123-W3 (5.38 kcal/mol) > OTZ1234-W2 (4.11 kcal/mol) > OTZ1235-W2 (4.03 kcal/mol) > OTTZ12345-W1 (1.36 kcal/mol) again strengthens the fact that N of heterocyclic ring acts as strong HB acceptor in the order OZ > ODZ > OTZ > OTTZ and the HB acceptor ability of N decreases with increasing number of N atoms in the ring. This order matches with ΔE BSSE and charge transfer values.
Complexes OZ12-W3, OZ13-W2, ODZ123-W4, OTZ1234-W3, and OTZ1235-W3 possess nO → σ* C-H charge transfer interactions only. The E (2) values in these complexes OTZ1234-W3 (8.37 kcal/mol), OTZ1235-W3 (7.15 kcal/mol), ODZ123-W4 (6.16 kcal/mol), OZ13-W2 (5.68 kcal/mol), and OZ12-W3 (4.82 kcal/mol) which strengthens the fact that the C-H of heterocyclic ring act as HB donor in the order OTZ > ODZ > OZ and this ability increases with increasing number of heteroatoms in the ring. This order matches with ΔE BSSE and charge transfer values.
The E (2) values of orbital interaction nN → σ* O-H for complexes ODZ123-W1 (4.36 kcal/mol), ODZ124-W1 (3.28 kcal/mol), ODZ124-W4 (3.61 kcal/mol), ODZ125-W1 (3.07 kcal/mol), ODZ134-W1 (5.20 kcal/mol), OTZ1234-W1 (2.28 kcal/mol), OTZ1235-W1 (1.18 kcal/mol), and OTZ1235-W4 (1.81 kcal/mol) with N of ring as HB acceptor are comparatively higher than bond orbital interaction nO → σ* O-H for complexes OZ12-W2 (1.29 kcal/mol), ODZ123-W2 (0.42 kcal/mol), ODZ124-W2 (0.34 kcal/mol), and ODZ134-W2 (0.16 kcal/mol). Thus, the strength of N···H HB has edge over O···H because of stronger covalent component inspite of the fact that O being more electronegative than N favors stronger electrostatic interactions with HB donors. The nO → σ* C-H orbital interaction in complexes OZ12-W2 (0.76 kcal/mol), ODZ123-W1(0.38 kcal/mol), ODZ124-W1 (0.37 kcal/mol), ODZ124-W4 (0.32 kcal/mol), ODZ125-W1 (0.49 kcal/mol), ODZ125-W2 (0.29 kcal/mol), ODZ134-W1 (0.36 kcal/mol), OTZ1234-W1 (0.81 kcal/mol), OTZ1235-W1 (0.87 kcal/mol), and OTZ1235-W4 (0.82 kcal/mol) have E (2) values with negligible to small magnitude only as the angle at bridging hydrogen disfavors the CT.
Molecular electrostatic potential
MEP is useful in understanding the interactive behavior as well as determining sites for electrophilic and nucleophilic attack. Electrostatic potential is increasingly becoming a regularly used tool in the basic research of molecular behavior in the design and synthesis of potent and safer molecules of medicinal interest. The importance of electrostatic potential in the research of molecular reactivity is assumed to grow. Figures 4 and 5 depict the contour maps of MEP of heterocyclic molecules on the 0.001-au electron density isosurface where red and blue regions indicate positive and negative MEP regions, respectively. As evident, the MEP of these monomers exhibit positive region on the outermost portion of hydrogen atom, along the C-H bond vector and large region of negative MEP can be observed on the outer surface of oxygen and nitrogen atoms. The most negative-valued points in the MEP topography usually indicated with the notation V min is widely used to gauze the electron rich site of the molecule, while the most positive-valued denoted by V max is used to indicate the electron-deficient site of the molecule.

Molecular electrostatic potentials on the 0.001 au contour of molecular electron density of five-membered heterocycles. The black dot represents the location of V max and blue dot represents the location of V min

HOMO-LUMO of five-membered heterocycles and their energy gap calculated at the MP2/aug-cc-pVDZ level
The electronic changes that accompany the bond formation can be clearly understood by comparing V min values of isolated heterocyclic molecules with V min values of heterocyclic molecules in the complex (designated as V′min). Hence, the electronic reorganization during the bond formation can be quantified as ΔV min = V′min − V min and these MEP parameters are summarized in Table 2. For the complexes OZ12-W1, OZ13-W1, ODZ123-W3, ODZ124-W3, OTZ1234-W2, OTZ1235-W2, and OTTZ12345-W1, involving single N-H···OW interaction, ΔV min is positive indicating that amount of electron density from heterocyclic ring has been transferred to water during the formation of complex. The complex OZ12-W3, OZ13-W2, ODZ123-W4, OTZ1234-W3, and OTZ1235-W3, involving single C-H···OW interaction, value of ΔV min comes out to be negative indicating the gain in electron density by heterocyclic ring at the expense of water molecule upon complexation with it. In complex ODZ125-W2, ΔV min is more negative which indicate a greater shift of electron density towards heterocyclic ring from water. The E (2) and charge transfer values also corroborate the same as evident from Table 2.
Topological analysis
QTAIM specifies an atom as an open system and provides a substantial method for the study of hydrogen bonding using the electron density distribution ρ(r). The critical points can be recognized in the electron density distribution where ρ(r) = 0 and classified according to the properties of the Hessian matrix. There are two types of topologically stable critical points in three dimensions, which are designated as (3, −1) and (3, +1) called bond critical points (bcps) and ring critical points (rcps), respectively. Figure 3 shows the molecular graphs of complexes of five-membered heterocycles with water. The most studied topological properties at the critical points are the electron density (ρ c), its Laplacian (∇2 ρc), the total electron energy density (H c), and its two components potential electron energy density (V c) and kinetic electron energy density (G c). The QTAIM has been broadly and successfully applied to the study of the properties of HBs. The main topological properties at the (3, −1), (3, +1) critical points were computed and displayed in Table 3. The values of the Laplacian ∇2 ρc, the energy density H c, and the balance between the kinetic electron energy density and the potential electron energy density −G c/V c display the nature of the interaction. ∇2 ρc provides information about either the charge concentration (∇2 ρc < 0) or the charge depletion (∇2 ρc > 0) of the electron distribution. The negative ∇2 ρc indicates that there is a shared interaction as in a covalent bond. The positive ∇2 ρc concerns the interaction of closed-shell systems: ionic interaction, van der Waals forces or HBs. The positive ∇2 ρc and the negative H c mean that the interaction is partly covalent in nature. If the ratio −G c/V c is greater than 1, then the interaction is noncovalent. If the ratio is between 0.5 and 1, then the interaction is partly covalent in nature, and when −G c/V c is less than 0.5, the interaction is a shared covalent one.
In the present work, the wave functions of hydrogen bonded molecular geometries obtained from MP2/aug-cc-pVDZ have been employed to characterize the topological properties. As seen in Table 3, all the HBs in complexes under study satisfy Koch and Popelier’s criteria with ρ c and ∇2 ρc of (3, −1) values well within the respective ranges 0.0060–0.0231 au and 0.0300–0.0884 au. The ρc and ∇2 ρc of (3, −1) values for the HBs involving N of ring as HB acceptor in complexes OZ13-W1, OZ12-W1, OTZ123-W3, OTZ1234-W2 ~ OTZ1235-W2 > OTTZ12345-W1 reflect N as a remarkable HB acceptor. The ρ c and ∇2 ρc of (3, −1) values of these complexes lie in range 0.0113–0.0231 and 0.0419–0.0884 au, respectively. The values of ρ c and ∇2 ρc in these complexes follow the order OZ13-W1 > OZ12-W1 > ODZ124-W3 > ODZ123-W3 > OTZ1234-W2 ~ OTZ1235-W2 > OTTZ12345-W1 which is same as stabilization energy, second-order delocalization energy, and charge transfer order. The ρ c and ∇2 ρc of (3, −1) values for HBs involving C-H of heterocyclic ring as HB donor lie in the ranges 0.0116–0.0163 and 0.0420–0.0613 au, respectively. The order of ρ c and ∇2 ρc of (3, −1) values in these complexes follow the order OTZ1234-W3 > OTZ1235-W3 > ODZ123-W4 > OZ13-W2 > OZ12-W3 which is same as stabilization energy, second-order delocalization, and charge transfer order. Complex ODZ125-W2 shows the smallest values for the sum of ρ c and ∇2 ρc of (3, −1) bcps among all complexes. Though lower the value of sum of ρ c and ∇2 ρc of (3, −1) bcps, these values are large enough to state that HBs in ODZ125-W2 complex fulfill the Koch and Popelier criteria. The ρ and ∇2 ρc values for C-H···OW interactions in quasi-five-membered ring structure are seen to be relatively smaller which indicates them to be weak HBs. Figure 6 depicts good correlation between the sum of ρ c values and ΔE BSSE for complexes under study. Also, there exists linear relationship between sum of ∇2 ρc values and ΔE BSSE (Fig. 7).

A plot of the relation between stabilization energy (ΔE BSSE in kcal/mol) and ρ c (au) at MP2/aug-cc-pVDZ level

A plot of the relation between stabilization energy (ΔE BSSE in kcal/mol) and ∇2 ρ c (au) at MP2/aug-cc-pVDZ level
SAPT analysis
Decomposition of the stabilization energy by means of the SAPT method is useful for unveiling the nature of noncovalent interactions stabilizing the complexes. The method decomposes the stabilization energy arising from the intermolecular interactions into physically meaningful components like electrostatic (E els ), induced (E ind ), dispersion (E disp ), and exchange (E exch ) interactions. The SAPT components for the complexes of five-membered heterocycles with water at MP2/aug-cc-pVDZ basis set are reported in Table 4. The E els , E ind , and E disp components are stabilizing and the E exch component is destabilizing. The E exch component arises mainly from the antisymmetry requirement of the wave function and the large magnitude of this component is due to the shorter binding distance. In all the complexes, E els , E ind , and E disp contributions are 54.26–68.73, 13.79–27.05, and 14.94–20.96%, respectively, towards stabilization. The comparison highlights the importance of increasing electrostatic interactions involving permanent dipoles and interactions of the induced moments. Electrostatic component accounts for 67.11–68.73% of the total attractive interactions in complexes involving single C-H···OW interaction, while it accounts for 53.58–59.68% in complexes involving single WO-H···N interaction. The complexes involving single C-H···OW interaction have E els , E ind , and E disp components in the range of −4.04 to −6.95, −0.83 to −1.70, and −1.15 to −1.48 kcal/mol, respectively, while the ranges evaluated for the respective components are −2.87 to −11.89, −1.35 to −4.85, and −1.03 to −3.57 kcal/mol for the complexes involving single WO-H···N interaction. It is observed that the percentage stability due to electrostatic component is higher in complexes involving single C-H···OW interaction (67.11–68.93%) relative to that in complexes involving single WO-H···N interaction (53.38–59.68%). The percentage contribution of the E ind (24.05–27.05%) and E disp (14.47–19.62%) components in complexes involving single WO-H···N interaction suggests the relative importance of the E ind term over the E disp term, whereas the percentage contribution of the E ind (13.79–16.91%) and E disp (14.36–19.10%) components in complexes involving single C-H···OW interaction are nearly the same.
HOMO-LUMO analysis
In molecular interaction perspective, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) interact with each other and they are accepted as the most significant energy levels since they are considered for the determination of reactivity and nature of molecule. For heterocycles and their complexes, the HOMO and LUMO energies and their energy difference (ΔE = E LUMO − E HOMO) are elucidated and the band gap energies are summarized in Tables 5 and 6. It is well known that the energy of HOMO characterizes the ionization potential, whereas the energy of LUMO is related to the electron affinity. The energy gap reflects the chemical reactivity of molecules. Global molecular descriptors including chemical potential (μ), hardness (η), and electrophilicity index (ω) are calculated from the optimized geometries obtained in gas phase. The chemical potential μ, which measures the escape tendency of an electron from equilibrium, is defined as μ = (E HOMO + E LUMO) / 2. The global hardness, η, that outlines important properties to measure the stability and reactivity of molecules is defined as η = (E LUMO − E HOMO) / 2. The electrophilicity index (ω) that outlines the capacity of a species to accept electrons is defined as ω = μ 2 / 2η. A hard molecule has a large energy gap and a soft molecule has small energy gap. The pictorial representation of HOMO and LUMO orbitals of five-membered heterocycles is shown in Fig. 5. Since energy gap between HOMO and LUMO orbitals (ΔE = E LUMO − E HOMO) is believed to be index of kinetic stability, a large HOMO-LUMO gap implies high kinetic stability and low chemical reactivity. The order of ΔE in the selected five-membered heterocycles is as follows: OTTZ (13.17 eV) > OTZ (11.29–11.56 eV) > ODZ (9.74–10.86 eV) > OZ (8.46–9.22 eV). This order reflects that with an increase in number of heteroatoms, energy gap increases and stability decreases. The calculated μ, ω, and η values of five-membered heterocycles in gas phase range from −5.16 to −7.40, 3.15 to 4.15, and 4.23 to 6.59 eV, respectively. The values of μ, ω, and η for monomers decrease in the order OTTZ12345 > OTZ1234 > OTZ1235 > ODZ125 > ODZ 124 > ODZ134 > ODZ123 > OZ12 > OZ13 in gas phase which show that OTTZ has the highest and OZ has the lowest escape tendency of electrons, capacity to accept electrons, and stability. In the complexes involving single WO-H···N interaction, μ, ω, and η values increase upon complexation except OTTZ12345-W1 which show that escape tendency of electrons, capacity to accept electrons, and stability of these complexes increase whereas these features decrease in complexes involving single C-H···OW interaction as indicated by the decrease in the values of μ, ω, and η upon complexation.
The HOMO-LUMO gap of water is higher than that of OZ, ODZ, and OTZ but lower than that of OTTZ indicating its lesser reactivity than OZ, ODZ, and OTZ but higher reactivity than OTTZ and hence predicting its hydrogen bonding ability to be lower than OZ, ODZ, and OTZ but higher than OTTZ. The lower HB acceptor ability of OTTZ than that of water is evidenced through the ΔE BSSE value of OTTZ12345-W1 predicting its HB acceptor ability to be lower than that of water.
Conclusions
The hydrogen bonding ability of five-membered heterocyclic oxygen containing molecules with one, two, three, and four N atoms has been studied. The stabilization energies for all complexes under study fall in the range −1.56 to −5.72 kcal/mol. By selecting the most stable complex of each isomeric form of the five-membered heterocyclic ring system, the range of stabilization energies display is −4.17 to −5.72 kcal/mol. All these complexes have at least one HB with N of heterocyclic molecule as an HB acceptor. The study also reflects that nitrogen is a significantly better HB acceptor than oxygen. The WO-H···N noncovalent distances are markedly shorter by 0.192–0.208 Å relative to the values for WO-H···O noncovalent distance. The study also suggests that N at position β to O in the heterocyclic ring is a better HB acceptor relative to that in case of α-positioned N. Comparing stabilization energies associated with ODZ123-W1, ODZ124-W1, ODZ125-W1, and ODZ134-W1 highlights the important relative position of multiple heteroatoms leading to highest stabilization in ODZ134-W1. The stabilization energy indicates that in complexes which involve single HB formation with WO-H···N interaction, N of heterocyclic ring acts as a strong HB acceptor in the order OZ > ODZ > OTZ > OTTZ, whereas in complexes which involve single HB formation with C-H···OW interactions, HB donor ability of C-H decreases in the order OTZ > ODZ > OZ as indicated by ΔE BSSE values, NBO, AIM, and SAPT analysis. Significant frequency red-shifts for the O-H bond (−9.43 to −154.50 cm−1) are predicted. The C-H stretching is found to be blue-shifted in the complexes involving C-H···OW interaction along with WO-H···N interaction, whereas in the complexes involving single HB formation with C-H···OW interaction, C-H stretching is found to be red-shifted. MEP analysis indicates that in complexes involving single WO-H···N interaction, ΔV min is positive indicating that the amount of electron density from five-membered heterocyclic ring has been transferred to water during the formation of complex, and in complexes involving single C-H···OW interaction, the value of ΔV min is negative indicating the gain in electron density by heterocyclic ring at the expense of water molecule upon complexation with it. The HOMO-LUMO analysis indicates that upon complexation, escape tendency of electron, capacity to accept electrons, and stability increase in complexes involving single WO-H···N interaction, whereas these features decrease in the complexes involving single C-H···OW interaction.
References
Kumar D, Sundaree S, Patel G, Rao VS (2008). Tetrahedron Lett 49:867–869
Naik SR, Harindran J, Varde AB (2001). J Biotechnol 88:1–10
Lewis JR (1995). Nat Prod Res 12:135–163
Wipf P (1995). Chem Rev 95:2115–2134
Wei Y, Fang W, Wan Z, Wang K, Yang Q, Cai X, Shi L, Yang Z (2014). Virology 11:95
Partida-Martinez LP, Hertweck C (2007). Chem Bio Chem 8:41–45
Fagerholm AE, Habrant D, Koskinen AMP (2010). Mar Drugs 8:122–172
Loper JE, Henkels MD, Shaffer BT, Valeriote FA, Gross H (2008). Appl Environ Microbiol 74:3085–3093
Chobanian HR, Guo Y, Liu P, Chioda MD, Fung S, Lanza TJ, Chang L, Bakshi RK, Dellureficio JP, Hong Q, McLaughlin M, Belyk KM, Krska SW, Makarewicz AK, Martel EJ, Leone JF, Frey L, Karanam B, Madeira M, Alvaro R, Shuman J, Salituro G, Terebetski JL, Jochnowitz N, Mistry S, McGowan E, Hajdu R, Rosenbach M, Abbadie C, Alexander JP, Shiao L-L, Sullivan KM, Nargund RP, Wyvratt MJ, Lin LS, DeVita RJ (2014). ACS Med Chem Lett 5:717–721
Boyd RE, Press JB, Rasmussen CR, Raffa RB, Codd EE, Connelly CD, Bennett DJ, Kirifides AL, Gardocki JF, Reynolds B, Hortenstein JT, Reitz AB (1999). J Med Chem 42:5064–5071
Gürsoy A, Demirayak Ş, Çapan G, Erol K, Vural K (2000). Eur J Med Chem 35:359–364
Stokes NR, Baker N, Bennett JM, Chauhan PK, Collins I, Davies DT, Gavade M, Kumar D, Lancett P, Macdonald R, Macleod L, Mahajan A, Mitchell JP, Nayal N, Nayal YN, Pitt GRW, Singh M, Yadav A, Srivastava A, Czaplewski LG, Haydon DJ (2014). Bioorg Med Chem Lett 24:353–359
Bull JA, Balskus EP, Horan RAJ, Langner M, Ley SV (2007). Chem Eur J 13:5515–5538
Ryu C-K, Lee R-Y, Kim NY, Kim YH, Song AL (2009). Bioorg Med Chem Lett 19:5924–5926
Tomi IHR, Tomma JH, Al-Daraji AHR, Al-Dujaili AH (2015). J Saudi Chem Soc 19:392–398
Hemavathi SN, Kumar Vishu BK, Lokanatha Rai KM (2011). Int J Pharm Pharm Sci 3:110–114
Reddy AB, Hymavathi RV, Swamy GN (2013). J Chem Sci 125:495–509
Moraski GC, Chang M, Villegas-Estrada A, Franzblau SG, Möllmann U, Miller MJ (2010). Eur J Med Chem 45:1703–1716
Sasahara K, Shimokawa Y, Hirao Y, Koyama N, Kitano K, Shibata M, Umehara K (2015). Drug Metab Dispos 43:1267–1276
Kumar A, Ahmad P, Mauryara RA, Singh AB, Srivastava AB (2009). Eur J Med Chem 44:109–116
Zhong Z-J, Zhang D-J, Peng Z-G, Li Y-H, Shan G-Z, Zuo L-M, Wu L-T, Li S-Y, Gao R-M, Li Z-R (2013). Eur J Med Chem 69:32–43
Wu C, Liang Z-W, Xu Y-Y, He W-M, Xiang J-N (2013). Chin Chem Lett 24:1064–1066
Liu X-H, Lv P-C, Xue J-Y, Song B-A, Zhu H-L (2009). Eur J Med Chem 44:3930–3935
Zhou J, Jin J, Zhang Y, Yin Y, Chen X, Xu B (2013). Eur J Med Chem 68:222–232
Kumar D, Kumar NM, Sundaree S, Johnson EO, Shah K (2010). Eur J Med Chem 45:1244–1249
Choi MJ, No ES, Thorat DA, Jang JW, Yang H, Lee J, Choo H, Kim SJ, Lee CS, Ko SY, Lee J, Nam G, Pae AN (2013). J Med Chem 56:9008–9018
Yang WS, Shimada K, Delva D, Patel M, Ode E, Skouta R, Stockwell BR (2012). ACS Med Chem Lett 3:35–38
James ND, Growcott Zibotentan JW (2009). Drugs Future 34:624–633
Jones AM, Helm JM (2009). Drugs 69:1903–1910
Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M (2008). J Med Chem 51:5843–5855
Chaplin M (2006). Nat Rev Mol Cell Biol 7:861–866
Ball P (2008). Chem Rev 108:74–108
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision B01. Guassian, Inc, Wallingford,
Boys SF, Moran D, Radom L (2007). J Phys Chem A 111:11683–11700
Reed AE, Curtiss LA, Weinhold F (1988). Chem Rev 88:899–926
Keith TA (2012) AIMALL, Version 12.06.03., Overland Park
Bulat FA, Toro-Labbe A, Brinck T, Murray JS, Politzer P (2010). J Mol Model 16:1679–1691
Politzer P, Truhlar DG (1981) Chemical applications of atomic and molecular electrostatic potentials. Plenum, New York,
Jeziorski B, Moszyński R, Szalewicz K (1994). Chem Rev 94:1887
Moszyński R, Heijmen TGA, Jeziorski B (1996). Molec Phys 88:741–758
Bukowski R, Cencek W, Jankowski P, Jeziorska M, Jeziorski B, Korona T, Kucharski SA, Lotrich VF, Misquitta AJ, Moszyński R, Patkowski K, Podeszwa R, Rob F, Rybak S, Szalewicz K, Williams HL, Wheatley RJ, Wormer PES (2013) Żuchowski PS. University of Delaware and University of Warsaw, Newar,
Acknowledgements
The authors are highly thankful to University Grants Commission (UGC) New Delhi, India, for the financial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOC 493 kb)
Rights and permissions
About this article

Cite this article
Chopra, N., Kaur, D. & Chopra, G. Hydrogen bonded complexes of oxazole family: electronic structure, stability, and reactivity aspects. Struct Chem 29, 341–357 (2018). https://doi.org/10.1007/s11224-017-1032-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-1032-x