
Abstract
The C-H-O-N-S elements that constitute the outgassed atmosphere and exosphere have likely been delivered by chondritic materials to the Earth during planetary accretion and subsequently processed over billions of years of planetary differentiation. Although these elements are generally considered to be volatile, a large part of the accreted C-H-O-N-S on Earth must have been sequestered in the core and mantle, with the remaining part concentrated at the Earth’s surface (exosphere: \(\text{atmosphere} + \text{ocean} + \text{crust}\)). The likely reason for this is that, depending on the prevailing pressure (P), temperature (T) and oxidation state (oxygen fugacity, fO2) in the planet’s interior, the C-H-O-N-S elements can behave as siderophile, lithophile, refractory, magmatophile, or atmophile. It is not clear if these elements might be sequestered in the interiors of planets elsewhere, since the governing parameters of P-T-fO2 during the diverse magmatic processes controlling magmatic differentiation vary greatly over time and from planet to planet. The magma ocean outgassed the first atmosphere, which was probably also the largest in terms of mass, but its nature and composition remain poorly known. Meanwhile, a significant, but unknown, part of the accreted C-H-O-N-S elements was sequestered in the core. These will probably never be liberated into the atmosphere. A secondary atmosphere was then fuelled by volcanism, driven by mantle convection and most likely enhanced by plate tectonics. The Earth still has active volcanism, and the volume and volatile contents of its magma are closely linked to geodynamics. Earth’s volcanoes have long emitted relatively oxidized gases, in contrast to Mars and Mercury. Mantle oxidation state seems to increase with planetary size, although the role of plate tectonics in changing the Earth’s mantle oxidation state remains poorly understood. Water contents of magma from elsewhere in the solar system are not so different from those produced by the Earth’s depleted mantle. Other elements (e.g. N, S, C) are unevenly distributed. A great diversity of speciation and quantity of magmatic gas emitted is found in planetary systems, with the key inputs being: 1 – degassing of the magma ocean, 2 – mantle oxidation state (and its evolution), and 3 – plate tectonics (vs. other styles of mantle convection). Many other parameters can affect these three inputs, of which planetary size is probably one of the most important.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
A variety of processes can affect the composition and structure of planetary atmospheres (Zahnle et al. 2010; Catling and Kasting 2017). Astrophysical, atmospheric, biological and geological processes interact with the surficial planetary sphere and the results of these combined interactions are the subject of ongoing research (Zahnle et al. 2010; Dehant et al. 2019). This chapter focuses on planetary outgassing of the light volatile elements (sometimes called atmophile elements) composing the C-H-O-N-S system. These degassing processes build on a primary atmosphere inherited from solar nebula capture (Zahnle et al. 2010; Olson and Sharp 2019). Planetary atmospheres are initially sourced by planetary degassing, while secondary processes involving fluid-rock reactions (Alt 1995), atmosphere-space exchange (Lammer et al. 2008), photosynthetic life (triggering Earths’ tertiary atmosphere dominated by O2, Catling et al. 2005) and asteroid impacts (Schlichting and Mukhopadhyay 2018) can modify this primary igneous input (Fig. 1).

Flowchart illustrating planetary outgassing as a primary volatile-source for the atmosphere and other secondary processes affecting the C-H-O-N-S surficial reservoirs. The C-H-O-N-S transfer from the planetary interior to the atmosphere uses the magmatic pipeline, which links mantle melting and volcanic outgassing. The outgassed C-H-O-N-S mixture is then affected by secondary processes involving a variety of atmospheric leaks into space (Lammer et al. 2008), impact degassing causing atmospheric blow (Haruka et al. 2019) and many ingassing processes (Alt 1995; Kelemen and Manning 2015; Catling and Kasting 2017; Lammer et al. 2018). These ingassing processes contribute to the burying of C-H-O-N-S elements, and are therefore the opposite to planetary outgassing. C-H-O-N-S ingassing involves both long-term/long-range and short-term/short-range processes. In the long processes (deep ingassing), C-H-O-N-S are irreversibly stored in the planetary core (see Sect. 4) or sent (back) to the mantle from which magma derives via geodynamic transfer, introducing a concept of geochemical cycling (Wallace 2005; Alt et al. 2012; Dasgupta 2013; Kelemen and Manning 2015); in the short process (shallow ingassing), C-H-O-N-S are transferred into the lithosphere (i.e. remain mainly in the crust, e.g. Alt 1995) and may never be recycled in the absence of plate tectonics. The absence of C-H-O-N-S recycling is probably what characterizes planets without plate tectonics like Mars or Venus, while planet Earth undergoes constant renewal in the flow of C-H-O-N-S elements, preventing surficial drying
Planetary outgassing uses the so-called “magmatic pipeline” connecting the planetary interior to its surface. The magmatic pipeline conveys the C-H-O-N-S elements through a process of mantle melting in the deep interior, then upwards transfer of magma, accompanied by degassing during magma ascent towards subsurface conditions. However, the development of a plate tectonics regime, which has not as yet been identified in other terrestrial bodies, must affect the amount and nature of the outgassed mixtures (Noack et al. 2014; Gaillard et al. 2015). Subduction-related volcanism constitutes a different magmatic pipeline, and plate tectonics introduces the concept of geodynamic cycles, in which the outgassed volatile elements can be partly recycled into the deep planetary interior via subduction processes (Sleep and Zahnle 2001; Wallace 2005; Dasgupta 2013; Kelemen and Manning 2015). This concept of geodynamic cycles has geochemical consequences that may be specific to the Earth (Fig. 1). The magmatic pipeline channels planetary outgassing of volatiles, but we will see here that (i) the C-H-O-N-S elements can be expelled with a great variety of speciation, (ii) they are not systematically extracted from the interior via the pipeline but can be retained in the planetary interior, and (iii) some processes can reverse the flow (i.e. transfer material from the planet’s surface to its interior).
These light (C-H-O-N-S) elements, their distribution in the atmosphere, and their involvement in surficial processes, are fundamental because they probably define the limits of planetary habitability (Cockell et al. 2016; Catling and Kasting 2017; Lammer et al. 2018; Dehant et al. 2019). There are several reasons for this: (i) carbon (C), hydrogen (H), oxygen (O), nitrogen (N), and sulfur (S) hereafter (C-H-O-N-S) and a number of metals operate or catalyse biochemical reactions and build the molecules of life (Catling et al. 2005; Cockell et al. 2016); (ii) C-H-O-N-S elements can form species which become concentrated in the atmosphere and affect both surface pressure and the greenhouse effect (Kasting 1988; Halevy et al. 2007; Hu et al. 2015; Wordsworth et al. 2013); and (iii) these elements are also involved in key surficial geochemical reactions such as sulfate-reduction (Lyons and Gill 2010), continental weathering and carbonate precipitation (Berner 2003) controlling surficial and atmospheric composition (Fig. 1). Defining the role of planetary outgassing in the surficial budget of these light volatiles is a central scientific question, of which many aspects remain unclear. In particular, the diversity of outgassing paths may vary greatly from planet to planet.
We review the state of the art on the paths of the C-H-O-N-S elements during the different magmatic stages of planetary differentiation. Though the topic remains debated (Albarède 2009; Wood et al. 2010; Greenwood et al. 2018), C-H-O-N-S elements most likely have a long geological history throughout planetary differentiation (Sarafian et al. 2014; Dauphas and Morbidelli 2014; Marty et al. 2016; Hirschmann 2016; Dauphas 2017; Lammer et al. 2018; Greenwood et al. 2018; Piani et al. 2020). The differentiation of planet Earth, like any terrestrial planet, involved long-term magmatic processes that have separated chemical elements as a function of their geochemical properties and affinity for the prevailing magmatic phases: siderophile elements (iron-loving) were sequestrated into the metallic core (Rubie et al. 2015) while non-siderophile refractory elements remained in the mantle, and non-siderophile incompatible elements (i.e. those concentrated in the melt phase of magma) were stored in the crust; finally, atmophile elements, which are mainly composed of the C-H-O-N-S volatile elements, plus the noble gases, were degassed at the planet’s surface into the atmosphere (Parai and Mukhopadhyay 2012; Moreira 2013; Hirschmann 2016; Lammer et al. 2018). However, two crucial points cast doubt on this generic textbook description:
-
(i)
Various observations indicate that most of the Earth’s carbon, hydrogen, nitrogen and sulfur must have remained stored in the deep interior (i.e. the mantle and the core), with the surficial reservoirs representing only a small portion of these volatiles (Marty 2012; Dasgupta and Grewal 2019). In other words, as shown below, the supposedly volatile C-H-O-N-S elements must behave as siderophile or refractory elements by remaining sequestrated in the planet’s interior. We will look at the magmatic parameters controlling this unexpected behaviour.
-
(ii)
While carbon, hydrogen, oxygen, sulfur, and nitrogen dominate the surficial chemistry on Earth, the surficial abundances of these elements are drastically different on other terrestrial bodies of the solar system (Lammer et al. 2018). Venus’s surface is dry and CO2-rich (Kasting 1988), while Mars was certainly wet but typically lacks both carbon and nitrogen and is covered by sulfate-rich sediments (Edwards and Ehlmann 2015; Bibring et al. 2006; Halevy et al. 2007; Gaillard et al. 2013). Looking at other planetary bodies in the solar system, a remarkable diversity of surface chemistry is noted for some satellites of the giant planets (e.g. the sulfur and hydrocarbon seas of Io and Titan respectively, Spohn et al. 2014), and exoplanets are sure to reveal even more unusual scenarios (Seager and Drake 2010; Pierrehumbert 2013).
We start by defining the magmatic pipeline enabling planetary outgassing and its secular evolution. The very first stage involves the magma oceans that contributed to the primary distribution of the C-H-O-N-S elements between surficial and deep reservoirs, including the core, the mantle and the atmosphere. This section shows that core formation can sequester a large part of the C-H-O-N-S planetary inventory. Therefore, an important portion of the putative atmophile elements supplied to terrestrial planets during accretion has been removed by core formation and segregation. Core formation has a similar impact to degassing: it (irreversibly) removes C-H-O-N-S elements from the surficial environment. The formation of the atmospheres (and their diversity) involves a first stage of core formation with its capacity to extract the C-H-O-N-S elements. Once the magma ocean has solidified, a process which potentially affects the distribution of volatiles in many different ways (Elkins-Tanton 2012; Hier-Majumder and Hirschmann 2017; Lammer et al. 2018; Schaefer and Elkins-Tanton 2018), volcanic outgassing prevails (Gaillard and Scaillet 2014; Ortenzi et al. 2020). We describe and discuss the diverse types of volcanic outgassing found on present-day Earth: mid-ocean-ridge (MOR) magmatism, plume-magmatism and arc-magmatism (i.e. subduction related). For each magmatic type we review the volatile contents and the oxidation states. While the oxidation state appears to be broadly homogeneous and oxidized, there is a great range in the C-H-S content of the Earth’s various magmatic pipelines, which reflects the diversity of the Earth’s mantle sources. The volatile content of magmas elsewhere in the solar system is also reviewed and compared to that on Earth. It is possible that some of the magmas produced on Mars and the Moon are relatively similar to MOR-basalts (MORB) in terms of their H2O contents (i.e. 200–2500 ppm H2O, see Saal et al. 2008; Elkins-Tanton and Grove 2011; Hui et al. 2013; Sarafian et al. 2013; Hauri et al. 2015; McCubbin et al. 2010, 2012a,b, 2015). In contrast, most magmas in the solar system are significantly more reduced than MORB. Regardless of the amount of C-H-O-N-S in the planetary interior, the conditions of degassing, in other words the pressure at the atmosphere-crust interface, appear to be critical, as this distillates the composition of the gas phases: sulfur and water are moderately volatile at high degassing atmospheric pressure (e.g. Venus), while carbon and nitrogen remain volatile under similar conditions to those found on Earth. The final section discusses the geodynamics of planetary outgassing, looking at the effect of plate tectonics and planetary size on the evolution and distribution of surficial volatiles. Plate tectonics enhances volcanic rates (Kite et al. 2009; Noack et al. 2014) and modies the planetary distribution of the C-H-O-N-S elements (Sleep and Zahnle 2001; Dasgupta 2013; Kelemen and Manning 2015; Lammer et al. 2018), thus clearly influences degassing processes. Planetary size affects the outgassing rate and the internal oxidation state (Wade and Wood 2005; Ortenzi et al. 2020). However, before addressing these fundamental points, it is necessary to review the current models on the provenance and the delivery time of C-H-O-N-S in planetary systems.
2 Provenance and Delivery of the C-H-O-N-S Elements on Planet Earth
Despite the obvious presence of water and other volatile elements on Earth, the terrestrial building blocks have long been thought to accrete in “dry” form (e.g. Albarède 2009; Wood et al. 2010; Halliday 2013) within the so-called snowline, where the temperatures were above the condensation temperature of water during planetary formation (Dauphas and Morbidelli 2014; Vacher et al. 2016). As described below, however, the absence of snow or ice in the building blocks of planets does not, from a mineralogical point of view, prevent these blocks from containing large amounts of C-H-O-N-S compounds during accretion (Hallis et al. 2015; Greenwood et al. 2018; Grewal et al. 2019a). Here we introduce the most recent advances that shed new light on the provenance and movement of these building blocks.
It has long been known that the oxygen isotopic signature of the Earth is more akin to that of enstatite chondrites (EC) than any other extra-terrestrial sample analysed to date (e.g. Javoy et al. 2010, Fig. 2). However, the bulk Earth’s chemical composition is very different to EC’s, even in terms of major components such as Si or Mg. Without going into any detail about the controversy as to the nature of the Earth’s building blocks, it can be noted that most recent studies of oxygen, chromium, nickel, titanium, molybdenum and ruthenium isotopes reinforce the close relationship between the isotopic signatures of the Earth and enstatite chondrites. These studies also recognize that their initial common cosmochemical reservoir was already chemically (but not isotopically) fractionated prior to accretion of the Earth and EC parent bodies (Warren et al. 2011; Dauphas 2017).

Bulk oxygen isotopic compositions of different classes of chondrites (Clayton and Mayeda 1999). Each group of chondrites (H, L, LL, CI, CR, CV, CM, CO and enstatite chondrites) probably represents a single asteroidal or planetary body. TFL: terrestrial fractionation line; CCAM: carbonaceous chondrite anhydrous mineral line. The oxygen data shown here correspond to bulk oxygen (oxygen of the phase network) of which the oxygen involved in degassing processes, discussed in this chapter, is only a tiny fraction
The processes that controlled the segregation of such a “dry” cosmochemical reservoir within the protoplanetary disk are not clear, though recently the rapid growth of giant planets has been put forward as a potential controlling factor. These planets are believed to have grown earlier and faster than the terrestrial planets, and may have intercepted the inward drift of icy particles towards the inner Solar System (Morbidelli et al. 2016). This fast growth led eventually to the formation of two isotopically distinct reservoirs in the protoplanetary disk with respect to refractory (Ti, Mo), non-refractory (Ni, Cr), and some volatile components, such as H2O (Warren et al. 2011; Kruijer et al. 2017; Nanne et al. 2019). Within this conceptual framework, relatively water-depleted non-carbonaceous (NC) objects (e.g., the parent bodies of enstatite and ordinary chondrites) represent material formed inside Jupiter’s orbit, whereas the carbonaceous (CC) group originated beyond Jupiter, and included volatile-rich carbonaceous chondrites such as CIs and CMs (Fig. 3, Table 1). Jupiter prevented mixing between the two reservoirs during the first few million years after the formation of calcium-aluminium-rich inclusions (CAIs), and may have formed a dynamic barrier preventing the influx of water-rich material to the inner Solar System for at least 3 to 4 million years (Kruijer et al. 2017). Then, once the nebular gas disk started to disperse, several million years after CAI formation, and during the late stages of planetary accretion, volatile-rich bodies (i.e., CI and CM carbonaceous chondrites) from beyond Jupiter’s orbit could have been loosed into the inner Solar System, either during runaway growth of Jupiter on a fixed orbit or during inward-then-outward migration of Jupiter which destabilized nearby bodies (e.g., Raymond and Izidoro 2017).

Comparison of (A) sulphur, (B) carbon, (C) nitrogen, and (D) H contents of non-carbonaceous [(\(\text{NC} = \text{enstatite}\) chondrites (EH, EL) and ordinary chondrites (H, L, LL)], and carbonaceous chondrites [\(\text{CC} = \text{CM}\), CR, CO, CV]. These meteorites represent the three major chondrite classes. All volatile abundances are normalized to those of CI carbonaceous chondrites, whose bulk elemental compositions are similar to that of the solar photosphere. Data from Wasson and Kallemeyn (1988), Kallemeyn et al. (1994), Alexander et al. (2012), Robert (2003) and Vacher et al. (2020)
What is the contribution of these CC-type objects to the Earth’s atmophile budget? Focussing now more specifically on the provenance of these elements delivered to the Earth, a rapid survey of the C-H-O-N-S content and speciation in chondritic samples reveals several salient features (Fig. 3, Table 1). First, all chondrites have relatively high C-H-O-N-S contents in comparison to differentiated planetesimals (i.e., achondrites) and the terrestrial planets. However, volatile abundances vary significantly between the different chondrite groups (Fig. 3). Second, the data compiled in Fig. 3 paint a contrasting picture, where some NCs show similar or even higher S and N concentrations than CCs (Fig. 3). Enstatite chondrites, for instance, are more volatile-rich than CO and CV carbonaceous chondrites and contain enough H to account for the terrestrial hydrogen content without invoking any additional sources (Fig. 3; Piani et al. 2020). This highlights a pivotal concept: the volatility of most so-called volatile elements depends on the prevailing thermodynamic and mineralogical conditions during their interaction with solids in the disk. In enstatite chondrites for instance, nitrogen is accommodated in nitrides and sulfur in sulphides, both being stable at very high temperatures provided that redox conditions are sufficiently reducing (Rubin and Choi 2009; Rubin and Ma 2017). In addition, H and C are stabilized in chondrites that travelled well beyond the snow line. In this respect, soluble and insoluble organic matter, hydrous amorphous silicates and clay minerals commonly observed in the matrices of most chondrites can preserve “water” molecules and adsorbed C-rich compounds well above 300 K, in regions of the disk where no ice could survive (Le Guillou et al. 2015). In terms of H and C abundance, NCs are relatively similar to estimates for the Earth (Marty et al. 2016; Piani et al. 2020). Although the isotopic compositions of H and N for ECs match those of the Earth’s mantle (Piani et al. 2020), Earth’s surficial zones (oceans and atmosphere) are enriched in D and 15N relative to ECs. For this reason, the contribution of CCs to the volatile budget need to be quantified because even minor amounts of these materials could greatly affect the Earth’s carbon and hydrogen content and isotopic signatures. Further, the CR (Renazzo-type) chondrites are characterized by large D-enrichments relative to the Earth’s oceans and mantle water, leaving CI and CM chondrites with the closest bulk H and N isotopic compositions to the terrestrial planets (Alexander et al. 2018; Füri and Marty 2015; Vacher et al. 2020; Piani et al. 2020). Based on this, it is proposed that 4 wt% of CI material was added to a proto-Earth made of H-rich EC-type bodies during accretion (Piani et al. 2020). Interestingly, existing geochemical data indicates that this CI/CM volatile addition to a proto-Earth did not occur during the late veneer, but during the last stage of accretion (Marty 2012; Lammer et al. 2018). The contribution of more distant water sources (e.g. interstellar water, Cleeves et al. 2014) is still debated. From the data collected during the Rosetta mission, it seems that a significant contribution of cometary xenon could explain the enigmatic signature of this element in the Earth’s atmosphere (Marty et al. 2016). In this context, about 22% of the Earth’s atmospheric xenon would be of cometary origin (Marty et al. 2016) but this in turn would have a very limited effect on the water and carbon budget of the Earth (Cleeves et al. 2014).
In conclusion, Earth was mainly accreted from material which was chemically fractionated but isotopically akin to sampled enstatite and ordinary chondrites. These meteorites contain enough H, C, N and S to explain the Earth’s endowment. However, a scenario involving the late arrival of carbonaceous materials cannot be excluded, and is apparently supported by isotopic constraints and by N-body simulations. Other alternatives have also been put forward recently. For instance, a large proportion of the volatiles could have been delivered to Earth via the Moon-forming impact of a Mars-sized differentiated planetesimal, which itself contained volatiles from a carbonaceous chondrite-like source (Grewal et al. 2019a). This is another potential origin for volatiles on terrestrial planets. In addition, recent work on HEDs (i.e., the howardite-eucrite-diogenite class of meteorites originating from the asteroid Vesta) and angrites containing traces of water and nitrogen show them to have CI/CM-like isotope compositions (Sarafian et al. 2014, 2017a,b; Barrett et al. 2016). The H and N isotopic signatures of Mars also resemble a CI/CM-like source (Alexander et al. 2018). These observations may suggest that terrestrial planets, as well as the earliest generation of differentiated planetesimals from the inner Solar System, accreted volatiles right from the beginning, sampling a single cosmochemical reservoir.
3 The Magmatic Pipeline for Volatiles and Its Secular Evolution
The C-H-O-N-S elements, derived from chondritic-like materials, have evolved throughout the various stages of planetary differentiation from early accretion to the present-day via magmatic processes (Gaillard and Scaillet 2014; Moreira 2013; Tucker and Mukhopadhyay 2014; Hirschmann 2016). We define here the concept of a magmatic pipeline, which connects C-H-O-N-S volatiles from the planet’s interior to its surface. This magmatic pipeline enables an unknown proportion of volatile elements to remain sequestrated in the planet’s interior, while the remainder is outgassed at the surface. This pipeline combines melting reactions at depth, causing chemical dissolution of a fraction of the mantle’s C-H-O-N-S elements into melt, with volcanic degassing, permitting the release of C-H-O-N-S elements into a vapour phase, as the magma rises towards the atmosphere. Different phases within this magmatic pipeline can host the C-H-O-N-S elements (Fig. 4) and the buoyancy of the dominant host phase determines the efficiency of the outgassing; these phases, in which volatiles can be dissolved at concentration levels varying from ppm to wt%, are melt phases, (i.e. silicate, carbonate, salt, metallic and sulfide melts), solid mineral phases, (i.e. nitrides, graphite, diamond, alloys, carbides, carbonates, sulfate, silicate crystals) and fluid phases (i.e. bubbles, gases, supercritical fluids). Silicate and carbonate melts and fluids are highly efficient outgassing phases, while the other hosts prevent outgassing and sequester volatiles either in the deep interior of the planet or in the magma.

The volatile zoo in the magmatic pipeline. The deep and shallow Pipelines involving the C-H-O-S-N elements and various host phases (boxes) in the magmatic processes (yellow arrows) ruling planetary differentiation. The shallow Pipeline involves the “degassing” at surficial conditions, while the deep Pipeline involves the “ingassing” of the C-H-O-N-S elements in the magmatic liquids at depth
Figure 4 illustrates the zoo of host phases for the C-H-O-N-S elements in the magmatic pipeline. This diversity explains why the C-H-O-N-S elements can be refractory (e.g. C hosted in graphite/diamond/carbonate, see Blundy et al. 1991; Holloway et al. 1992; Hirschmann and Withers 2008; Rohrbach and Schmidt 2011), siderophile (e.g. in metals, carbides, nitrides, see Roskosz et al. 2006, 2013; Speelmanns et al. 2018; Grewal et al. 2019a,b; Dasgupta and Grewal 2019), chalcophile (in sulphides, see Baker and Morretti 2011), magmatophile (in silicate melt, see Iacono-Marziano et al. 2012a,b; Namur et al. 2016) and they behave as volatiles only if they are involved in the fluid phases (Gaillard and Scaillet 2014). Therefore it is not always the case that melting and eruption is conducive to outgassing of the mantle, as there are a variety of melting/degassing paths that may lead to the sequestration at depth of the C-H-O-N-S elements and thus restricted degassing of them at surficial conditions.
What controls the preference of the C-H-O-N-S elements for a given phase in the pipeline? At every stage of a planet’s evolution, the following parameters operating in the magma source control the behaviour of the C-H-O-N-S elements: pressure (P, ranging from a few millibars to several tens of kbar, i.e. surficial degassing to deep melting), temperature (T, related to the age of the planet), and oxidation state (fO2, oxygen fugacity varies by more than 10 orders of magnitude in the known solar system, Herd 2008; Righter et al. 2016; Namur et al. 2016). Oxygen fugacity, fO2, is a measure of the oxidation state of the system, where high fO2 values mean oxidizing conditions. The definition of redox processes in magmatic rocks, and broadly speaking in the planet’s interior, and their relationship with oxygen fugacity (fO2) are detailed elsewhere (Frost 1991; Frost and McCammon 2008; Gaillard et al. 2015) These P-T-fO2 parameters evolve from (i) the deep magmatic pipeline, which enables C-H-O-N-S elements to dissolve in the melt at the depth of partial melting, to (ii) the shallow magmatic pipeline, enabling the degassing of these C-H-O-N-S elements into the atmosphere (Fig. 4). Capturing how P-T-fO2 evolves from depth to surface and whether this evolution has undergone secular changes is an important and much debated issue.
The magmatic pipeline is highly varied and evolutive throughout a planet’s history (Figs. 5–6), implying changing P-T-fO2 conditions. The depth of melting and the age of the planet are related: the younger the planet, the hotter the mantle, and the deeper the melting. There is a continuous evolution in the melting regime from a high-degree of deep melting in the hot-young planet to the low-degree of shallow melting in the cold-older planet. It is noteworthy that there are two distinct regimes of melting: (i) thermal melting occurs when the temperature exceeds the volatile-free solidus, while (ii) incipient melting can occur below the volatile-free solidus in the presence of trace amounts of CO2 and H2O. Figure 5 helps visualize these two melting regimes in P-T space. Incipient melting refers to small degrees of mantle partial melting producing small-volume magmas that are very rich in volatiles (Hirschmann 2010; Dasgupta 2013; Okumura and Hirano 2013; Tappe et al. 2018; Gaillard et al. 2019), while thermal melting can produce high degrees of partial melting and yield large volumes of magma that are volatile-poor. The magma ocean (Fig. 6a) is the end-member of the high degree of mantle melting, while alkaline magma and carbonatites (being CO2-rich magma) constitute the low degree melting end-member. In between, as shown by the magmatic rock library recorded by planet Earth as it cooled, are different types of magmas: (hot) komatiites that were predominant during the Archean, picritic basalts, and modern (cooler) basalts (Herzberg et al. 2010). Alkaline magmas and carbonatites are rare in the Precambrian (Blichert-Toft et al. 1996) and became increasingly abundant as the planet cooled. This simple thermal evolution is probably a planetary law that captures the age-distribution relationships for magmas on Earth (Herzberg et al. 2010; Keller and Schoene 2012). The discovery of alkaline magma on Mars (Edwards et al. 2017) probably indicates that the recent volcanic activity had its source in the cool, shallow mantle, while “komatiite-like” magmas, formed long ago in a hot Martian mantle at great depth, are more similar to some of the shergottite meteorites (Ding et al. 2015). If we extrapolate this cooling planetary rule, then future volcanic activity on Earth should be increasingly dominated by alkaline magmas, probably carbonatites, formed in a cool mantle.

Melting in the deep Magmatic Pipeline: Upper Mantle (peridotite) solidus for dry (grey curve) vs. H2O-CO2-saturated system (grey curve after Hirschmann 2000, blue curve after Wallace and Green 1988; see Gaillard et al. 2019) and the evolution of mantle melting in a cooling planet. Young planets are hot and have a magma ocean with surface \(\text{T}>1700~^{\circ}\text{C}\). Komatiite and Archean lavas are typical of mantle temperatures close \(1650\text{--}1750~^{\circ}\text{C}\) (5–7 GPa, 160–230 km), while modern arcs and plume-related magma reveal mantle temperatures of \(1100\text{--}1400~^{\circ}\text{C}\) (2–5 GPa, 60–160 km). In the coolest mantle regions, where \(\text{T}\sim 1000\text{--}1300~^{\circ}\text{C}\), carbonatite melts of alkaline composition are expected. This last end-member is the volcanism of the Earth’s future and this is probably a planetary law

The evolutive magmatic pipeline. (a) Sketch of the volatile distribution and magmatic processes throughout planetary differentiation and the construction of the successive atmospheres. Time evolves clockwise and the increase in planetary diameter illustrates the ongoing accretion. We term the successive atmospheres nebular (capture of the solar nebula, Olson and Sharp 2019), Magma Ocean (degassing of the MO, Fegley et al. 2020), Volcanic (produced by volcanic degassing, Gaillard and Scaillet 2014), and biological or mineral (buffered by biological or chemical reaction in the soil). (b) The T-fO2 (oxygen fugacity) diagram of typical terrestrial magmas ranges from the reduced Magma ocean to the oxidized arc-magmas that are associated with subduction. Magmatic conditions on Mercury, Mars and Earth are shown here and discussed in the text and references are given in Table 2. The solar Nebula fO2 is taken from Jurewicz et al. (1995) at \(T=1900~^{\circ}\text{C}\). With cooling, it should follow a line parallel to the 1‰ H2O dashed lines. The solid buffers (IW, NNO, HM) stand for iron-wustite, nickel-nickel oxide, and magnetite-hematite respectively and are taken from O’Neill and Pownceby (1993). EPM stands for Earth’s primitive mantle, that is to say the fO2 of an Fe-saturated magma containing 8 wt% FeO (see Behrens and Gaillard 2006). The domains of shergottites and Mercury are after Herd (2008) and Namur et al. (2016) respectively. Archean basalts are bracketed by Aulbach and Stagno (2016) and Canil (1997). The domain of Arc magma is after Kelley and Cottrell (2009) and Behrens and Gaillard (2006), while MORB is bracketed by Christie et al. (1986) and Cottrell and Kelley (2011)
Therefore, the first order modifications of the magmatic pipeline are associated with mantle temperatures, which decrease from the hot magma ocean stage to the cool incipient melting regime. However, it has been suggested that the mode of mantle convection might change this (Herzberg et al. 2010; Noack et al. 2014; Korenaga 2018), and that the mantle may have actually increased in temperature during a putative early stagnant lid stage (Noack et al. 2016, see Sect. 7.1). The initiation of plate tectonics would then have caused a rupture in the Earth’s thermal evolution (Noack et al. 2014; Korenaga 2018). Geochemical evidence for such a thermal rupture have been documented via compilation of geochemical data on magmatic rocks (Keller and Schoene 2012). The thermal evolution of planet Earth is relatively well-known and the theory ruling heat exchange in the planet’s interior is well established (Noack et al. 2016). This contrasts with the redox evolution of the planetary interior: it remains difficult to link secular thermal changes to alterations in redox conditions in the Earth’s interior (Berry et al. 2008; Canil 1997, 2002; Keller and Schoene 2012; Gaillard et al. 2015; Aulbach and Stagno 2016).
On planet Earth, the oxidation state of magmas must have evolved from the highly reduced magma oceans during the first few million years to the strongly oxidized magmatic products emitted at modern subduction zones (Fig. 6a). Figure 6b clearly illustrates this redox evolution which includes, from the most reduced to the most oxidized object: the solar nebula, the very first condensed calcium-alumina-rich inclusions, magma oceans, Mercurian magmas, Martian shergottites, komatiites, MOR-Basalts (MORB), Ocean Island basalts and arc-magma. The nature and the origin of this redox evolution remains enigmatic since its cause(s) and its timing are not well constrained and some results are conflicting (Canil 1997; Frost and McCammon 2008; Keller and Schoene 2012; Aulbach and Stagno 2016; Schaefer and Elkins-Tanton 2018; Dehant et al. 2019). However, acknowledging this redox dynamic is essential. To illustrate its importance, we show in Fig. 6b (top) the fraction of water in the fluid phase at equilibrium with the different types of magma in a T-fO2 space: the fraction of H2O in a fluid composed of H2-H2O increases from ca. 1‰ in the solar nebula to 999‰ in arc fluids. Oxidized magma appears to be crucial for delivering water by outgassing, while reduced magma (i.e. \(\text{fO}_{2}<\text{IW}\)) would outgas more H2 than H2O.
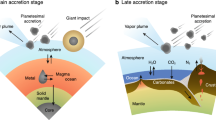
A cartoon illustrating the evolution of the magmatic pipeline is shown in Fig. 6a. The magma ocean(s) (MO) magmatic pipeline, a highly simplified system determining core-mantle differentiation, may have outgassed the first atmosphere. This MO stage is complex, and there may have been several magma ocean stages from planetesimals to large planet (Greenwood et al. 2005). The earliest magma ocean stages on planetesimals, during the first few million years of the solar system, most likely captured a nebular atmosphere with a high content of H2 and He (Moreira 2013). Once the solar nebula dissipated, a secondary magma ocean atmosphere was outgassed. These early MO stages defining the overall starting point of the volatile distributions between the atmosphere, the mantle and the core are fundamental (see Sect. 4), but research into this topic is still in its infancy. After the MO solidification stage leading to the formation of the mantle, further (partial) melting occurs by decompression-melting in upwelling mantle regions (Noack et al. 2014). This type of pipeline includes MORB and hotspot magmatism. Finally, oxidized and volatile-rich magmas are produced in the mantle wedge of subduction zones (Kelley and Cottrell 2009). Each of these magmatic pipelines can have very different degrees of efficiency in terms of redistributing and outgassing the C-H-O-N-S elements because the prevailing parameters P-T-fO2 are highly variable (Fig. 6b).
4 Volatiles in the Magma Ocean, the Core and the Primary Outgassed Atmosphere
As illustrated in Fig. 7, the planetary body at a magma ocean stage could be seen as a vast lava lake, most likely very reduced, in which Fe-rich metal droplets and diamonds tend to sink (Suzuki et al. 1995; Rubie et al. 2015), while gas bubbles and graphite tend to float in a convective magma ocean (Elkins-Tanton 2008, 2012; Lebrun et al. 2013; Massol et al. 2016; Hirschmann 2016; Keppler and Golabek 2019). Magma ocean stages were certainly diverse, operating for various reasons on planetesimals, planetary embryos and large planets like Earth (Greenwood et al. 2005). While magma oceans have been intensively studied by high-pressure surveys focusing on metal-silicate partitioning, the volatile behaviour of the C-H-O-N-S elements implies that significant partitioning into the fluid phase, in other words the proto-atmosphere, must also have occurred. On the other hand, the C-H-O-N-S elements can dissolve into Fe-metal droplets implying that the core may constitute a potentially vast reservoir for these elements that are usually considered atmophile. The fate of the C-H-O-N-S elements during the magma ocean stages is thus a three-body problem: How do the C-H-O-N-S elements partition between the core, mantle and atmosphere?

The Magma ocean is a three-body problem comprising the core-mantle-atmosphere, where ingassing and outgassing processes involve the C-O-H-N-S life-forming elements. While the community has largely focused on metal-silicate partitioning associated with core formation during this stage, the fate of the C-O-H-N-S elements during this process has only recently been addressed (see text). Yet, the fact that these elements behave in a volatile manner and can therefore be segregated into the early atmosphere, has rarely been quantitatively calculated or linked to the deep processes operating in the magma ocean. The way C-H-O-N-S partitions between the metallic core, the solid mantle, the magma ocean and the atmosphere is currently unknown and requires future work
Noble gas constraints have provided ample evidence for primordial mantle degassing (Moreira 2013; Tucker and Mukhopadhyay 2014) associated with the magma ocean stage, but this powerful approach does not address the degassing of the major C-H-O-N-S life-forming elements. At the same time, the mantle abundances of various metals have shed light on the removal of the most siderophile elements into the core, but it has become increasingly clear that a large proportion of the C-H-O-N-S elements were also sequestered in the core (e.g., Dasgupta and Grewal 2019; Fischer et al. 2020; Li et al. 2020). Below, we review the state of the art on this deep sequestration.
4.1 Carbon
The amount of carbon constituting the surficial reservoir (carbonates, organic carbon, and the whole crustal inventory) corresponds to about 23 ppm C of the bulk Earth mass (Marty 2012; \(\text{BE} = 5.98\times 10^{27}~\text{g}\)). The amount of carbon stored in the mantle (upper + lower mantle) is much less well known but all estimations indicate that the mantle constitutes the largest C-reservoir (i.e. 80–700 ppm C of the BE, Marty 2012; Dasgupta and Grewal 2019). In detail, it is considered that the upper mantle (and probably, an unknown part of the lower mantle) is a C-depleted reservoir, in that it contains, on average, \(38\pm 14~\text{ppm}\) C (7 ppm C BE, after LeVoyer et al. 2017), while the deeper mantle is thought to contain significantly more C (ca. 500 ppm BE after Marty 2012; or more, according to Miller et al. 2019). The size of this deep mantle enriched C-reservoir is unknown (Miller et al. 2019). It should be noted that there is no evidence that the entire lower mantle is homogeneously C-rich. A C-rich primordial mantle is sampled in oceanic island basalts (Miller et al. 2019), indicating a deep source, but this may represent isolated mantle regions that have survived 4.5 Ga of mantle convection (Marty 2012; Tucker and Mukhopadhyay 2014; Ballmer et al. 2017).
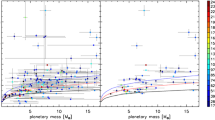
An increasingly accepted hypothesis is that these concentrations are inherited from the core-magma-ocean equilibration, which, at C-saturation (i.e. graphite/diamond), would produce silicate mantle containing 10–4000 ppm C (Fig. 8). These are maximum numbers since they represent the carbon content dissolved in silicate melts at C-saturation. Whether the magma ocean was saturated in graphite or diamonds is still under debate. The most abundant form of C in the present-day mantle is most likely diamonds (Frost and McCammon 2008), but it is not known if diamonds were present there from the beginning. At high pressure (\(>10~\text{GPa}\)), the silicate magma ocean seems to favour ferric iron over dissolved CO or CO2 (Hirschmann 2016; Schaefer and Elkins-Tanton 2018; Armstrong 2019), which implies that enhanced diamond precipitation is expected at transition zone and lower mantle depths (Armstrong 2019). This localized precipitation of deep C may well constitute a recipe for producing a deep mantle C-rich region. It is thought that graphite saturation occurs at surficial pressures (Keppler and Golabek 2019). Graphite-saturation prevailing at surficial conditions in the magma ocean of Mercury has been suggested to account for the presence of several wt% C at its surface (Charlier and Namur 2019). So, the dissolved carbon content of the magma ocean at C-saturation shown in Fig. 8 may well represent the minimum carbon content in the magma ocean, with graphite flotation at the surface of the magma ocean and diamonds sinking down to depths of 500 km.

The carbon contents in silicate melts at metal and C-saturation as a function of oxygen fugacity. The data points are compiled from laboratory experiments conducted in the P-T range 1–20 GPa, \(1400\text{--}2400~^{\circ}\text{C}\) (Malavergne et al. 2019; Duncan et al. 2017; Li et al. 2016; Stanley et al. 2014; Wetzel et al. 2015; Armstrong et al. 2015; Li et al. 2015; Chi et al. 2014; Dasgupta et al. 2013; Kadik et al. 2014). The coexisting Fe-metal contains \(6\pm1\) wt% C revealing the siderophile behaviour of carbon. The line with opposite (positive and negative) slopes illustrates the controls on carbon solubility in silicate melt by the solubilisation of various species following reactions that are labelled on the curves. High pressure and high temperature conditions yield the highest C-contents in silicate melts at metal-saturation. The boxes on the RHS on the figure show the C-content of various geochemical reservoirs on Earth (Mantle and surface) expressed in wt%. The surface wt% C is expressed relative to the Bulk Silicate Earth (after Marty 2012)
Returning to the chondritic materials described in Sect. 2, and assuming that they were the building-bricks for planets at the magma ocean stage, their C-content (4,000–32,000 ppm C) could largely account for the C-saturation level of the upper and lower mantles and could therefore explain the present-day C-concentration in the mantle. Enstatite chondrites, supposedly the most important contributor to the Earth’s accretion (e.g. Javoy et al. 2010; Dauphas 2017), contain 4,000 ppm C (Table 1). This could still account for the present-day mantle-C value. However, we do not know the amount of C in the Earth’s core. Not surprisingly (as shown by studies into the metallurgy of steels), carbon can be highly siderophile and the metal/silicate partitioning coefficient of C ranges from 10 to \(10^{5}\) (Malavergne et al. 2019; Dasgupta and Grewal 2019). The reason for such a large range of C metal-silicate partitioning is partly illustrated in Fig. 8: a combination of variations in P-T-fO2 and hydrogen contents of the system can produce this large range of carbon affinity for the core or for silicate melts (see discussion in Dasgupta and Grewal 2019). In addition, the solubility of C in iron-based liquid alloys can be reduced by the presence of sulphur or silicon (Li et al. 2015). We will probably gain more insight into the many combinations of parameters through research programmed over the next decade. At this stage, what we know for certain is that carbon is siderophile, implying that if ca. 100 ppm C were preserved in the Magma ocean from metal silicate equilibration, \(>1000~\text{ppm}\) C must have been sequestered into the developing core (Malavergne et al. 2019). This would make the Earth’s core the main C-reservoir (Dasgupta and Grewal 2019). Thus in terms of atmosphere formation, this carbon becomes unavailable from the point at which differentiation begins.
From the vast range of dissolved carbon contents in the silicate magma ocean shown in Fig. 8, a picture emerges: increasing H2O, T and P increases C-content in the magma ocean; while dry MO under moderate P and T conditions displays low C-solubility. This allows us to speculate that core formation on small planetary systems like Mars must have drastically removed C from the magma ocean, because of the weak affinity of C for the Martian silicate magma ocean. In contrast, on larger planets (Earth, Venus), a deeper and hotter magma ocean must have limited the carbon depletion in the magma ocean during core segregation. These are working hypotheses for future work, but a robust and predictive model of C behaviour during core – magma ocean processes is clearly needed. Such a model should, in principle, be able to account for the variety of P-T-fO2 conditions in the different terrestrial magma oceans. Empirical approaches have so far been proposed (i.e. Li et al. 2015; Malavergne et al. 2019), but in order to try and interpret the parameter space and the diversity of Magma ocean conditions, we need a sound theoretical basis founded on thermodynamics.
4.2 Hydrogen
Hydrogen is one of the more likely light elements in the Earth’s core due to its high abundance in the solar system and its solubility in metal iron at high pressure (Poirier 1994). Some experiments have been carried out to investigate hydrogen behaviour during core-mantle segregation, and demonstrate hydrogen partitioning at high pressure between hydrous iron (FeHx) and hydrous silicates, solid or molten (Okuchi 1997; Iizuka-Oku et al. 2017; Clesi et al. 2018 and Malavergne et al. 2019). These experiments are relevant in the determination of hydrogen partitioning between metal and silicate (\(\text{D}_{\mathrm{H}}^{\text{metal-silicate}}\)) during core-mantle segregation. In Fig. 9, we report the experimental results of these studies. This figure shows that the \(\text{D}_{\mathrm{H}}^{\text{metal-silicate}}\) depends on pressure and to a lesser extent on the oxygen fugacity or temperature. The \(\text{D}_{\mathrm{H}}^{\text{metal-silicate}}\) coefficients calculated from the data of Okuchi (1997) are slightly above unity at 7.5 GPa and \(1500~^{\circ}\text{C}\) while the data of Clesi et al. (2018) and Malavergne et al. (2019) are always \(<1\) but tend to increase with pressure to reach 1 above 20 GPa. The reason for this discrepancy could be the result of the interactions between carbon and hydrogen. In fact, in the experiments of Clesi et al. and Malavergne et al. the iron is saturated with carbon, while in Okuchi’s experiments carbon is absent. Percent-level concentrations of carbon would likely decrease \(\text{D}_{\mathrm{H}}^{\text{metal-silicate}}\) by about one order of magnitude owing to the limiting factor of carbon on hydrogen solubility in metallic iron melt. Another explanation, which is not mutually exclusive with the carbon interactions with hydrogen, is the much higher water content in Okuchi’s experiments compared to those of Clesi et al. and Malavergne et al. By taking all these parameters into account, and also the limiting factors of both S and Si (two major light elements of the Earth’s core) on hydrogen solubility in iron, it was found that hydrogen core concentration is limited to 64 ppm (Clesi et al. 2018) while Malavergne et al. (2019) suggested a maximum H content in the core of ca. 600 ppm based on high pressure partitioning data approaching unity. All these values are consistent with the range of geochemical estimations reported by Rubie et al. (2015) and those derived from a first-principles molecular dynamics study by Zhang and Yin (2012). This is also consistent with cosmochemical constraints suggesting 600 ppm hydrogen in the core (McDonough 2003). From these studies it appears that hydrogen is only a minor element in the Earth’s core, which is also consistent with isotopic constraints (Shahar et al. 2016), but its concentration level is similar to the range of silicate Earth H contents (e.g. Marty 2012; Halliday 2013). It is therefore important to retain that a significant fraction of the Earth’s hydrogen is in its core. In contrast to the hydrogen (in the form of water) stored in the mantle, this deeply sequestered hydrogen is no longer available for the surficial environment via outgassing.

Hydrogen partitioning between metal and silicate melts determined by experiments at high pressure-high temperature. The data have been collected in the T-fO2 ranges \(1500\text{--}2300~^{\circ}\text{C}\) and IW-5 to IW-1. The data points of Clesi et al. (2018) and Malavergne et al. (2019) correspond to C-saturated conditions while the experiments of Okuchi (1997) were obtained in C-free systems. See text for details
4.3 Nitrogen
In the past decade, significant efforts have been paid to determine the behaviour of nitrogen during metal/silicate partitioning in the magma ocean. Somewhat paradoxically, the fate of the most abundant component of the present-day atmosphere of the Earth had previously been poorly characterized in comparison to carbon, sulfur and hydrogen, in spite of its crucial importance in unravelling the formation and evolution of the atmosphere. In particular, it has been suggested that, although representing 78 %vol of our atmosphere, nitrogen is anomalously depleted on Earth compared to other volatile elements (Marty 2012). Yet, the origin of this depletion and the exact distribution of nitrogen among mantle minerals, melts, fluids and metallic alloys were until recently mostly unknown despite some early efforts (Kadik et al. 2011).
This lack of first order information concerning nitrogen behaviour at depth motivated the first experimental study carried out at high pressure on the molten metal/silicate partitioning of nitrogen (Roskosz et al. 2013). This initial study revealed the evolution from highly siderophile to moderately siderophile at increasing pressure. It was then proposed, as a likely origin of the deficit in nitrogen in the accessible Earth, that nitrogen may be favourably accommodated in core forming materials.
Since then, a large corpus of data has been collected by different groups, almost uniquely at low pressures (up to 6 GPa) and essentially at carbon saturation (e.g. Armstrong et al. 2015; Dalou et al. 2019a,b; Kadik et al. 2017; Speelmanns et al. 2018; Grewal et al. 2019b). These studies revealed an increasingly complex behaviour and mitigated the initial results. They confirmed a moderately siderophile behaviour, and noted an almost lithophile behaviour of nitrogen depending on the P-T-fO2 conditions. Though absolute values may differ slightly from one study to another, most studies are in agreement (Fig. 10) that with more reducing conditions, nitrogen becomes progressively lithophile because nitrides are formed and stabilized within the structure of molten silicates. For instance, Speelmanns et al. (2018) and Grewal et al. (2019b) show that below a fO2 of \(\Delta\text{IW-3}\), the metal-silicate partitioning for nitrogen (\(\text{D}_{\mathrm{N}}\)) varies between 0.003 and 0.5 whereas in more oxidizing conditions, at \(\Delta \text{IW-0.8}\), the partition coefficient is as high as 52. Further details on the empirical quantitative solubility and partitioning models can be found in Grewal et al. (2019b) and Speelmanns et al. (2018). These multiparameter fits of a growing body of data have also been linked to accretion models that take into account the evolution of the redox conditions from the very reduced early accretion to the late oxidation of the mantle. The outputs of these models are still disputed, as to whether the core is a significant reservoir of nitrogen at a global scale (Roskosz et al. 2013; Grewal et al. 2019a,b; Speelmanns et al. 2018). Because of the strong effect of oxygen fugacity on nitrogen’s behaviour, the conclusions so far seem highly dependent on the nature of the material accreted and the timing of the subsequent oxidation of the mantle.

Nitrogen metal-silicate partition coefficients for the silicate melt-(\(\text{Fe-Ni-C-N}\pm\text{S}\pm\text{Si}\)) alloy melt equilibria as a function of oxygen fugacity, pressure and temperature (after Grewal et al. 2019b)
4.4 Sulfur
S is an element which has long been known to be siderophile (e.g. Li and Agee 2001; Chabot and Agee 2003; Rose-Weston et al. 2009; Malavergne et al. 2014; Boujibar et al. 2014; Suer et al. 2017). However, as can be seen in Fig. 11a, showing the partition coefficients of S between metal and silicate melt \(\text{D}^{\mathrm{S}}_{\text{metal-silicate melt}}\) obtained between 0.001–91 GPa and \(1000\text{--}3830~^{\circ}\text{C}\) for the last 20 years, there is a large domain within this parameter space where sulfur becomes lithophile during core formation. For the redox conditions required to form the Earth’s core (IW-2) or under more oxidizing conditions, S is highly siderophile with partition coefficients that can exceed 600 beyond IW-1 (e.g. Asahara et al. 2004). At the reducing conditions necessary for terrestrial core formation, it has been proposed (e.g. Gaillard et al. 2013; Boujibar et al. 2014) that S is present as \(\text{S}^{2-}\) anions in silicate melts and that S can substitute for O following the reaction:
Under such conditions, S would favour the metal species at the time of core segregation. Pressure appeared to be a factor which favoured the affinity of S for metal (e.g. Rose-Weston et al. 2009; Boujibar et al. 2014). However, the role played by pressure on the siderophile nature of S was cast into doubt by a recent study (Suer et al. 2017, carried out at up to 91 GPa) which showed that S becomes less siderophile than expected at lower mantle pressures with \(\text{D}^{\mathrm{S}}_{\text{metal-silicate melt}}\) values not exceeding 55. Finally, under more reducing conditions (\(\text{fO}_{2}<\text{IW-3}\)), it was observed that S becomes less siderophile and even lithophile (e.g. Malavergne et al. 2014; Namur et al. 2016). These results are explained by the fact that S dissolves in the silicate melts at the expense of the metal. This behaviour is similar to that of nitrogen seen in the previous section. During the cooling of a highly reduced magmatic ocean such as on Mercury, containing several wt% of S, Mg-Ca-rich sulfide could form (e.g. Avril et al. 2013; Malavergne et al. 2014; Namur et al. 2016) in a planetary mantle. In an Earth-like Magma ocean, which is moderately reduced, the majority of sulfur would dissolve in the core, leaving a mantle with ca. 100–900 ppm S present as FeS. Finally, on Mars, which is much more oxidized than Mercury, the S content of the mantle would not be able to reach very high values. Indeed, under such conditions, S would remain siderophile (even more so than on Earth), implying greater storage of S in the core at the expense of the Martian mantle (e.g. Rivoldini et al. 2011). The pressure would reinforce this effect by promoting the presence of S in the core as shown in Fig. 11b.

Sulfur partitioning between metal and silicate melts determined by experiments at high pressure-high temperature. (a) as a function of fO2 (relative to IW); (b) as a function of P. The 0.5 slope along which the data are aligned in panel b corresponds to the stoichiometry in Eq. (1)
4.5 Interactions, Speciation and Evolving P-T-fO2 Conditions
This short review illustrates that the first geochemical event paving the way for habitable worlds is probably the magma ocean stage but this is not always considered in the state of the art discussions (e.g. Dehant et al. 2019). Each of the C-H-O-N-S elements can be significantly, even predominantly, sequestered into the core depending on the conditions of core – silicate magma ocean equilibration. This deeply sequestered C-H-O-N-S is then no longer available for surficial processes, including the formation of the atmosphere. There is still the possibility that a small portion of core-mantle boundary materials might be involved in mantle plumes (e.g. Heron 2018; Rizo et al. 2019), but this does not alter our conclusion that the core is probably the greatest C-H-N-S planetary reservoir.
However, there are further complications:
-
(i)
A few major questions, currently poorly explored, may strongly modify the above conclusions drawn from the behaviour of individual elements in the magma ocean. This may also reconcile the community regarding the possible relative fractionation between N, S, C and H on a planetary scale: What are the interactions resulting from the multicomponent nature of the C-H-O-N-S system? This aspect is particularly important in the literature since most studies on N partitioning were performed at C saturation (with the exception of Roskosz et al. 2013). Similarly, partitioning data on hydrogen reveals differences between C-saturated (Clesi et al. 2018; Malavergne et al. 2019) and C-absent conditions. The presence (and interactions) of both carbon and sulfur has been shown to mutually reduce their solubility in the metal phase (Li et al. 2015). Not to mention the fact that O, Si, Ni and other elements, once accommodated in the metal reservoir, can profoundly change the solubility of C-H-O-N-S in the core. Likewise, the influence of the speciation of these elements in the silicate melts implies that multicomponent systems must behave differently: for instance, the solubility of C in silicate melts is enhanced at low fO2 in the presence of H since it allows the solubilisation of CH4 components in the silicate melt. Our current understanding of this complex aspect of the C-H-O-N-S system is still at an early stage, but given the current research efforts conducted by the community on this topic, significant advances are expected within the next decade.
-
(ii)
Core formation is not a single-stage process and a range of evolving P-T-fO2 conditions occur from the earliest to the latest stage of accretion. What drives this evolution and what the P-T-fO2 path of the Earth was during accretion remains poorly understood for the moment, and goes well beyond the scope of this review paper. At this stage, the take home message is that a significant part of the planetary C-H-O-N-S endowments are retained in the core of our planet, and this conclusion likely holds for other terrestrial worlds. The ratio of C-H-O-N-S elements retained in the core to that remaining in the silicate planet will vary with the P-T-fO2 conditions of core – Magma ocean equilibration. We do not yet have the conceptual tools and the experimental database to look at this diversity in a comparative planetology perspective, but major international research projects are currently underway.
4.6 Outgassing the Magma Ocean and Its Redox Evolution
Surprisingly, while the species forming from the C-H-O-N-S are expected to be volatile, their volatility and speciation in the conditions of the magma ocean are poorly known. The most advanced outgassing models for the magma ocean simulate a H2O-CO2 mixture equilibrating between an atmosphere and a silicate magma ocean (Elkins-Tanton 2008; Massol et al. 2016; Lebrun et al. 2013; Nikolaou et al. 2019). This means a 3-component (C-H-O) system in which each species can take a single oxidation state, that of the most oxidized one. The chemical complexity arising from the multicomponent nature of the C-H-O-N-S system and the fact that the magma ocean is a highly reduced and warm environment have not yet been tackled. The speciation of the gas phase at such extreme conditions has however been addressed by the thermodynamic MAGMA code (Zolotov and Fegley 1999; Schaefer and Fegley 2007; Fegley and Schaefer 2012). It mainly involves the following species: H2-H2O-CO-CH4-CO2-H2S-S2-SO2-COS-N2-NH3 and other minor species. Additional unanticipated species may exist at the extreme temperatures of the MO (\(>1800~^{\circ}\text{C}\)) but no experimental research is currently planned to address this question. Another major issue is the vaporisation of non-volatile elements such as some metals (Sossi and Fegley 2018) that may vaporize from the magma ocean in significant quantity. This point has also been poorly addressed by experiments so far. On the silicate melt side, the speciation of the C-H-O-N-S elements is currently being intensively studied. So far the following species have been identified (Armstrong et al. 2015; Dalou et al. 2019a,b): \(\text{CO}_{3}^{2-}\), CO, CH4, \(\text{OH}^{-}\), H2O, H2, \(\text{S}^{2-}\), \(\text{S}^{6+}\), N2, \(\text{NH}_{\mathrm{x}}\). This remains work in progress and no thermodynamic description of these species in the silicate melts has so far been done. Consequently, no model of melt-gas equilibration under the hot, reduced and multicomponent nature of the magma ocean has been constructed. Nonetheless, generic calculations by Gaillard et al. (2013), Hirschmann (2016) and Gaillard et al. (2015) indicate that H2 and CO should dominate the composition of the prevailing atmosphere during the Magma ocean stage on Earth and elsewhere. No sulphur should vaporize under such conditions. In contrast to assumptions, CH4 occurs only as a minor gas species, in addition only being present when atmospheric pressure is high enough. However, upon cooling in the upper part of the atmosphere, H2 and CO should react to form CH4 and H2O.
The redox state of the magma ocean is much reduced because the metal-silicate equilibria that determine the formation of the Fe-core indicate fO2 values as low as IW-5, which progressively evolve towards more oxidizing conditions (Wade and Wood 2005; Cartier et al. 2014; Schaefer and Elkins-Tanton 2018). The final fO2 of metal silicate equilibria is IW-2. This is deduced from the FeO content of the Earth’s mantle, i.e. 8 wt% (Rubie et al. 2015). Two popular models can account for this increase in fO2 during the magma ocean stages on Earth: (i) Javoy et al. (2010) proposed the enstatite model, where ultra-reduced materials were the highest contributors during the Earth’s accretion; in the latest accretion stage, at the extreme P-T conditions prevailing at the base of the MO, silicon increasingly dissolved into the core causing the following reaction that increased the MO FeO content:
(ii) The alternative model (which is not mutually exclusive of the previous model) reappraises the ancient heterogeneous Earth accretion model (Rubie et al. 2011): it involves a series of impacts of smaller differentiated bodies originating from various regions of the early solar system. These impactors re-equilibrated with the Earth’s MO and the equilibrated metallic portion merged with the Earth’s core. The change in provenance of these differentiated bodies throughout accretion, from the innermost to the outermost region of the solar system, implies that the fO2 of these bodies increased through time (Rubie et al. 2011).
However, once the core had segregated, the metal-silicate equilibration came to an end, and the associated constraints on the magma ocean fO2 vanished. How, then, did the magma ocean fO2 evolve once the core had segregated? The answer to this could help define the composition of the equilibrium atmosphere. It has recently been proposed (Armstrong 2019) that the Earth’s magma ocean could have been as oxidized as the present-day shallow mantle as a result of the dependence on pressure of the solubility of ferric iron in silicate melts (see also Schaefer and Elkins-Tanton 2018 for a prediction of these effects). At great depth (\(>500~\text{km}\), or high pressure), the fraction of ferric iron in equilibrium with Fe-metal tends to increase, while this fraction is negligible at shallower conditions. This has two consequences: it generates a vertical fO2 gradient in the magma ocean and it offers a mechanism to enhance carbon sequestration in the form of diamonds at depths equivalent to the present-day lower mantle or transition zone, where diamonds in a magma ocean would gravitationally tend to segregate (Suzuki et al. 1995). In terms of atmospheric composition at the MO stage, if this model is correct, the simplified H2O-CO2 system may well be closer to reality than the above. On the other hand, such an oxidized magma ocean should have testable consequences with respect to other elements such as sulphur or nitrogen. We therefore conclude that this scenario constitutes a hypothesis that should be investigated, since it relies on the assumption that the vertical profile in mantle fO2 is controlled by \(\text{Fe}^{3+}/\text{Fe}^{2+}\). However, the buffering capacity of the ferric-ferrous iron in a silicate melt in the shallow mantle is limited, particularly at low fO2, and it is very likely that other, more efficient, redox reactions buffering the fO2 were simultaneously in operation. In particular, in the presence of carbon, graphite-saturation and the C-CO equilibria can have a significant buffering capacity, imposing fO2 and buffering the partial pressure of CO and CO2 in the atmosphere as suggested by Keppler and Golabek (2019).
In conclusion, there is not yet a degassing model for the magma ocean which takes into account the complex interactions at work in the C-H-O-N-S system and this is needed in order to predict the composition and structure of the magma ocean atmosphere. In particular, noble gas geochemistry indicates that the outgassing event operating during the magma ocean phase defined most of the present-day Ar, Ne, He distribution between the mantle and the atmosphere. The question remains as to whether this was also the case for the C-H-O-N-S elements?
5 Magmatic Volatiles on Present-Day Earth & Other Terrestrial Worlds
5.1 The Depleted Mantle and the Bulk Mantle
Geochemical analyses provide indications as to the volatile contents and the oxidation of present-day magmatism on Earth (Table 2) and elsewhere. We review here the information to date on the distribution of volatiles among the various types of magmas so far found in the solar system. On Earth, it appears that there is a strong link between the geodynamic setting and the volatile content of magmas (Fig. 12). This link is due to the conditions of melting (source) and degassing (subaerial/submarine). The sampling and available observations for other terrestrial worlds are clearly more limited, but some important conclusions and comparisons with the dominant types of magmatism on Earth can be drawn.

Geodynamics of magmatism on Earth. MORB stands for Mid-Ocean-Ridge basalts, OIB for Oceanic-Island basalts, SRB/ARC stands for Arc-related basalts that are formed during subduction, PSB stands for Petit-spot basalts and IB means intraplate basalts. OIB and IB are related to hotspot upwellings, which sample lower mantle material, while MORB, Petit-spots and SRB sample material from the upper mantle (sometimes called the asthenosphere)
Broadly speaking, the mantle fuelling the Earth’s magmatism is a two-layer structure: the upper mantle which acts as the source for Mid-Ocean-Ridge magmatism is volatile-depleted (e.g. \(150\pm 42~\text{ppm}\) H2O and \(137\pm 54~\text{ppm}\) CO2), while hotspot magmatism appears to contain more volatiles because it derives from upwellings of lower mantle material, which are much more volatile-rich (\(>500~\text{ppm}\) of both H2O and CO2) (Marty 2012; Miller et al. 2019; and see Fig. 12). Estimations of the volatile contents of the depleted mantle (DM) and of a deep, less well-defined mantle suffer from great uncertainties but the various methods used, i.e. 4He/C and 40Ar/C after Marty (2012) or the Nb/C and Ba/C ratios after Miller et al. (2019), reach consistent conclusions as to the presence of a deep, highly-enriched C-H reservoir. The depletion of the DM with respect to the lower mantle is usually linked to the extraction of incompatible elements during the development of the crust but this is not necessarily linked to volatile depletion. The origin of the volatile depletion remains poorly known and is still much debated (e.g. Tucker and Mukhopadhyay 2014). Arc-volcanoes are sourced from the so-called mantle wedge, which is enriched in volatiles by the subduction of oceanic lithosphere. The mantle source for magmas elsewhere in the solar system, including the Moon, resembles the depleted mantle on Earth; this is particularly true for H2O.
Before we go on to a detailed description of these diverse magmatic volatiles on the scale of the solar system, we briefly define the link between volcanic gases, magma and silicate melts and how their interactions evolve as the magma ascends to the surface.
5.2 Volcanic Gases, Silicate Melts, and Mantle Sources: Lessons from Volcano Monitoring
What is termed magma by volcanologists is a three-phase mixture composed of silicate melt, crystals and bubbles. The C-H-O-N-S species outgassed into the atmosphere correspond to the chemical components hosted in the bubbles. However, there is a continuous mass transfer of these chemical elements between the three magmatic phases as the magma ascends to the surface.
Volcanic gases are emitted from lava lakes, volcanic craters, and volcano flanks. They are produced on a massive scale during eruptions, and can also occur during non-eruptive periods (including long term quiescent periods or brief interruptions between two eruptions). Volcanologists have long been trying to interpret the changes in volcanic gas compositions observed in monitored volcanic systems. Variations in gas compositions emitted by a volcano are frequently observed and do not necessarily reveal a change in the composition of the magma, but are rather related to the depth of magma ponding and the dynamics of the eruption (e.g. Edmonds and Gerlach 2007). This interpretation also involves the use of melt inclusions, which represent deep magmatic liquids trapped in crystals, and which preserve the deep volatile contents contained within the magma (Metrich and Wallace 2008). It became clear that magmas reaching the Earth’s surface are mostly degassed with respect to the volatile contents revealed by melt inclusions: degassing must therefore occur during magma ascent.
These observations have been widely reproduced using experimental petrology, and the experiments show that the solubility of volatiles in silicate melts increases with pressure. Clearly, the ascent of magmas from the depth at which they were produced or stored towards the surface causes a reduction in the solubility of C-H-O-N-S species in the silicate melt. This decrease in melt C-H-O-N-S content is compensated by the formation of a gas phase, taking the form of bubbles growing in the magmatic liquid, and ultimately forming a coalesced network. The gases then escape from the magma via this connected network. Escape can occur at depth or at the surface depending on the dynamics of the magma ascent. The gas composition therefore reflects the pressure of fluid-melt equilibrium. Monitoring of volcano degassing is essentially used for the purpose of estimating the depth of the degassing magma. In the case of deep storage or submarine eruptions, the equilibration pressure can reach several hundred bars, while the pressure is close to atmospheric pressure during subaerial degassing. This means that magma from a given source can have very different gas compositions depending on the changes undergone during gas-melt equilibration at various pressures. This pressure control on the composition of the outgassed C-H-O-S mixture has been made clear and its role in a solar system perspective is important (Gaillard and Scaillet 2014). Pressure control on the degassing of various magmas is illustrated below, and explained in Sect. 6.
5.3 Mid-Ocean-Ridge Basalts
Lavas
MORBs constitute more than 70% of Earth’s magma. They erupt on the seafloor at an average depth of ca. 2900 m below sea level. MORBs are produced by melting of the so-called depleted mantle, DM (i.e. the upper mantle plus a non-constrained part of the lower mantle). Typical H2O content of MOR basalts is 1,200 ppm-wt H2O, though a number of variations from depleted (350 ppm H2O) to enriched (5000 ppm H2O) end-members have been detected (Saal et al. 2002). On average, this indicates a mantle source containing ca 150 ppm H2O (Metrich and Wallace 2008). Sulfur content in primitive MORBs is ca. 1000–1200 ppm (Metrich and Wallace 2008). These ranges in H2O and S contents correspond to direct measurements in melt inclusions. Because of its great volatility in magmatic systems, CO2 content estimates in MORBs are indirectly determined and are much debated (LeVoyer et al. 2017). At the pressure of MORB emplacement (ca. 300 bar), analyses of CO2 in the majority of MORB glasses indicate that it is no longer present, and must have been degassed from the melt at deeper conditions. The “undegassed CO2 MORB content” must then be reconstructed using a variety of proxies (either He/C, Ar/C ratio e.g. Marty 2012 or Nb/C, Ba/C ratio e.g. LeVoyer et al. 2017) from data collected using melt inclusions. An average MORB CO2 content has been proposed (1400 ppm, LeVoyer et al. 2017) but the range of estimated contents is extremely large: from 200 to 12,000 ppm CO2 (Saal et al. 2002; Cartigny et al. 2008; LeVoyer et al. 2017). These variations may reflect the contribution of lower mantle enriched material/or subducted slab material that has mixed with the upper mantle, before being sampled by MOR magmatism (Donnelly et al. 2004; Edmonds and Gerlach 2007). Alternative views suggest that the carbon content in MORBs reflects the oxygen fugacity at its source, being graphite-saturated (Holloway et al. 1992; Stagno et al. 2013). The reaction buffering the CO2 content in basalts would be:
Such a buffering of CO2 content in basalts is believed to be a planetary law (Blundy et al. 1991; Righter et al. 2008), that is to say, planetary mantles with a low fO2 produce basalts with low CO2 contents (e.g. Hirschmann and Withers 2008). Returning to MORB, a correlation between MORB CO2 and H2O contents is observed, but the range is narrower for water: 350 ppm to \(\sim 5000~\text{ppm}\) H2O (Danyushevsky et al. 2000; Saal et al. 2002). No such variation is observed for the sulfur content in MORB since this value is fixed by the sulphur content at sulphide saturation via the reaction (Baker and Morretti 2011):
The sulphur content (\(\text{S}^{2-}\)) in basalt on the Earth that is saturated in sulphide (FeS) is about 800–1200 ppm for basalts having ca. 8 wt% FeO (Baker and Morretti 2011).
Fluorine and chlorine, which are not really dealt with in this review, have been worked on and treated by some workers as volatile elements (Aiuppa et al. 2009). Their contents in MORB are low (Table 2, see Hanley and Koga 2018). Fluorine contents are about 10 times greater than Cl.
The oxygen fugacity of MORB has been debated. The first estimation by Christie et al. (1986) indicated reduced conditions (i.e. FMQ-1; Fig. 5), while measurements in Cottrell and Kelley (2011) indicated a relatively oxidized MORB, higher than the FMQ buffer (see Fig. 5). Recent data (Berry et al. 2018) suggested that MORBs have fO2 close to or slightly below FMQ.
Volcanic Gases
The composition of MORB volcanic gases has never been sampled because of the problems of accessing them 3000 meters below sea level. Estimations can be made using gas-melt equilibria codes (Gaillard et al. 2011). Illustrations of these calculations are shown in Fig. 13.

Volcanic gases from MORBs expressed in ppm of species degassed from the melt vs. pressure of emplacement. The undegassed magma contains 1200 ppm H2O, 1100 ppm CO2, 1100 ppm S (see Saal et al. 2002) and the Y-axis gives the mass of outgassed C-H-O-S components (in ppm-wt of magma). Temperature is \(1300~^{\circ}\text{C}\). On average, present-day MORB is emitted at oceanic ridges 3000 meters beneath sea level (i.e. 300 bar total pressure), but this average depth has likely changed through time (see Gaillard et al. 2011). The nature and amount of volatile species expelled by MORB magma is then plotted for variable pressure (multiply by 10 to get depth below seawater or by 3 to get depth beneath Earth’s surface). Oxygen fugacity is FMQ. Full lines show oxidized volcanic species, dashed lines show reduced ones. CO2 degasses first, at high pressure, and by the depth of MORB eruption (3000 meters), 80% of the CO2 initially dissolved in the magma has already degassed via bubbles. Due to their much lower density and the overall low viscosity of the magma, these bubbles tend to escape the magmatic system, making it difficult to estimate the CO2 content of the undegassed magma. Note, at the depth of MORB emplacement, H2O remains undegassed, and only tiny amounts of sulphur have started to outgas
Figure 13 illustrates the primary control that pressure has on the composition of the gas phase. The melt compositions are an average MOR-basalt with the content in H2O, CO2, S and the fO2 described above. This figure clearly illustrates that basalts containing 1200 ppm CO2 will have degassed most of their CO2 at \(>1000~\text{bar}\) (10,000 meters below sea level). We also see that MORBs do not degas H2O at all, and only about 50–100 ppm S is outgassed by the depth of emplacement (300 bar). To summarize, MORBs essentially emit CO2, as the other volatiles are not able to degas at this pressure. The CO2 outgassed by MORBs beneath seawaters rises as bubbles in the ocean. Broadly speaking, the mass of outgassed CO2 is equivalent to the mass of CO2 deposited via hydrothermal reactions in the oceanic crust (Kerrick 2001). Nitrogen degassing is not addressed here since no solubility law has been validated at magmatic pressure.
5.4 Oceanic Island Basalts (OIB)
Lavas. In contrast to MORB, which have been well studied over a long period (Saal et al. 2002), the C-H-O-S contents of primitive OIBs and their oxygen fugacity are poorly documented (Gaillard et al. 2015). Clearly OIBs are volatile-richer (Aubaud et al. 2005; Miller et al. 2019) and more oxidized than MORBs (Brounce et al. 2017). Broadly speaking, OIBs have volatile contents close to that of enriched-MORBs (E-MORB) (Marty 2012) but some OIBs can have a much greater quantity of volatiles than E-MORBs (Table 2). The discovery of massive amounts of sulphur (6000 ppm S) in the melt inclusions of primitive alkaline magma from the Canary Islands is a good example (Longpré et al. 2016). These very high amounts of magmatic sulfur, mainly present as sulfate (\(\text{S}^{6+}\)), leave no doubt as to their strongly oxidized nature. In addition of being rich in S, it has been inferred that the alkaline basaltic magma of the Canary Islands contains several wt% of CO2, which is at least 10 times greater than that found in MORBs (Longpré et al. 2016). The amount of H2O in these alkaline melts also greatly exceeds that of MORB (i.e. 4,000 to 30,000 ppm H2O). Such high abundances of CO2, H2O and S, together with their oxidized nature, may be related to the small degree of mantle melting. This is also suggested by the highly alkaline composition of the magmatic rocks at the Canary Islands (e.g. Basanites, Longpré et al. 2016). However, the mantle source still seems to be different to that of the MORB source (Marty 2012). Noble gases and C/Nb ratios of the mantle source for OIB indicates an enriched mantle, probably related to deep convection cells sampling an unmixed reservoir (Miller et al. 2019; Labidi et al. 2020) and/or recycling of subducted slabs (Marty 2012; Cabral et al. 2014). At Hawaii, the magma is much less alkaline (i.e. high degree of melting), but recently Brounce et al. (2017) showed that these melts are also more oxidized than MORBs. The CO2 content of primary melts at Hawaii is close to 1 wt% (390–10,000 ppm CO2, Tucker et al. 2018), while the H2O contents seem moderately high and similar to E-MORBs (max. of 8,000 ppm H2O). Intraplate volcanic rocks, which are the surface expression of hotspots in the mantle, are enriched in C-H-O-S with respect to MORB. The element most affected by this enrichment is probably carbon (e.g. Miller et al. 2019). Strongly alkaline OIB are much more volatile-enriched (i.e. several wt% of CO2, H2O and S) than mildly alkaline OIB, probably reflecting variable degrees of mantle melting. It remains unclear whether a variable degree of mantle melting also affects oxygen fugacity (i.e. a low degree of melting may yield high fO2, Gaillard et al. 2015). With planetary cooling, alkaline C-H-O-S-enriched melts increasingly dominate planetary magmatism: they are smaller-volume, but richer in volatiles. From a planetary perspective, it is important to note that these alkaline magmas can contain massive amounts of halogens with respect to conventional basalts (Longpré et al. 2016; Hanley and Koga 2018), though the concentration range varies greatly (Table 2). If the last volcanic activities on cooling planets consist of such alkaline magmas, we could expect them to transfer important quantities of sulfur and halogens to the surface as a sort of late varnish. The surface of Mars, rich in sulphur, and but also containing a lot of halogens, may have preserved such a late volcanic event, though alternative views will be discussed in Sect. 7.
Volcanic Gases
Sampling and in situ analysis of subaerial volcanic gases has greatly advanced over the last 10 years (Aiuppa and Gaillard 2016). At Hawaii, Nyiragongo, Erta Ale, and the Vanuatu islands, as well as in numerous active volcanic systems, field measurements reveal high emission rates of CO2 and SO2 (Burton et al. 2013; Werner et al. 2019). However, when you look in detail, the gas composition fluctuates, which is related to the degassing dynamics of the magma (eruption explosiveness and surficial vs. deep magma degassing, e.g. Edmonds and Gerlach 2007). In Fig. 14 we attempt to filter these fluctuations and relate magma eruption to volatile emissions. This figure shows the gases emitted vs. pressure (bar) by a melt of average OI-basalt composition (Hawaiian basalt) for H2O, CO2 and S. The temperature is \(1300~^{\circ}\text{C}\) and the fO2 is FMQ \(+0.8\) (Brounce et al. 2017; see Fig. 14). As shown for the MORB in Fig. 13, the gas compositions are controlled by the pressure of degassing. However, the amount of outgassed C and S is higher than for MORBs. If we consider alkaline basalts such as the basanites from the Canary Islands, even greater C and S contents would be expected.

Volcanic gases emitted by OIBs. The nature and amount of volatile species expelled by OIB magma is plotted as a function of pressure in bars. Full lines show oxidized volcanic species, dashed lines show the reduced ones. One sees that CO2 degasses first, at high pressure, and that as pressure of degassing decreases, the fraction of CO2 in the degassed mixture is diluted by other components
5.5 Subduction-Related Arc Basalts
Volcanic degassing at subduction zones, where arc volcanoes expel massive amounts of the C-H-O-N-S elements, constitutes a specific pipeline (Fig. 15). Subduction processes drag the oceanic lithosphere (including the crust) into the mantle, and this buried material is enriched in C-H-O-S(-N) due to seafloor water-rock interactions. These interactions involve serpentinization, leading to massive water storage in the lithosphere, associated with moderate carbonate and deep sulfide/shallow sulfate precipitation (Alt 1995; Alt et al. 2012). This process releases reduced hydrothermal fluids in the ocean containing variable amounts of H2 and CH4. This process also concentrates Cl in the percolating fluids (Debure et al. 2019), because water is absorbed and locked into the mineral serpentine (which contains 13 wt% H2O) by the serpentinization process. Where subductions occur, this volatile-bearing lithosphere sinks into the warm mantle. The temperature increase as the material descends into the mantle causes devolatilisation reactions (serpentine or carbonate breakdowns) and may also cause direct melting of the subducted basaltic crust and sediments since both H2O and CO2 facilitate melting of silicate rocks (Fig. 5). Overall, most of the volatiles involved in arc magmatism derive from seafloor processes. As such, outgassing from arc volcanoes does not constitute planetary outgassing in the sense that the volatiles released are not internal species but are simply processed through a subduction cycle illustrated in Fig. 15 (e.g. Wallace 2005).

The cycling of C-H-O-S volatiles through subduction zones. Most of the subducted H2O appears to be efficiently cycled back during arc-volcanism. A significant portion of carbon and sulfur may well be buried in the mantle, but uncertainties are very large (Wallace 2005; Kelemen and Manning 2015). The cycling of nitrogen during subduction is not shown here for the sake of clarity. Nitrogen is present in sediments and the serpentinized oceanic lithosphere (Mysen 2019) that are subducted. Subducted nitrogen seems to be efficiently recycled in arc volcano gases, as observed in some settings (Fischer et al. 2002; Busigny et al. 2003) but this may not occur everywhere since poor N recycling has also been suggested (e.g. Mitchell et al. 2010; Zerkle and Mikhail 2017)
Because of this intensive recycling, the amount of water released during subduction is enormous. Primitive arcs basalts are believed to contain on average 4 wt% H2O (Plank et al. 2013): this is far more than MORBs and about 2 to 4 times more than OIB. Large amounts of sulfur can also be emitted by arc-volcanoes, with a major climatic impact since some of the gas can be injected into the upper atmosphere. However, this last type of volcanic event is due to the eruption of differentiated, felsic, magma rather than basalts. Their occurrence is linked to basalt production in the mantle wedge created by subduction, followed by long-term storage and cooling of the magma in the continental crust. Estimations of C-H-O-S contents in primitive arc basalts are: 1–5 wt% H2O (Plank et al. 2013); 0.2–0.3 wt% S (Metrich et al. 1999; Mironov and Portnyagin 2018); Estimated CO2 content ranges from 1800 μg/g (analysed in melt inclusions) to 6000 μg/g (gas flux estimations) (Wehrmann et al. 2011). Such CO2 contents, together with the isotopic constraints, indicate that most of the CO2 emitted by volcanoes comes from the breakdown of carbonates from the subducted lithosphere (Wallace 2005; Hammouda and Keshav 2015). Yet, this does not mean that all subducted carbonates undergo thermal breakdown in the mantle wedge. An unknown but significant amount of the subducted carbonate may be buried in the lower mantle (Kelemen and Manning 2015). Oxygen fugacity calculated for arc magmas reveals oxidized conditions (Kelley and Cottrell 2009; Gaborieau et al. 2020), though it is not clear whether primary arc magmas and their mantle source are oxidized (Gaillard et al. 2015). Existing constraints on the fO2 of primary arc basalts are unclear and hardly distinguishable from those of MORBs (Li and Lee 2006). The fate of nitrogen during the subduction processes is not shown in Fig. 15 for the sake of clarity. There are many unknowns, leading to contradictory conclusions: nitrogen seems to be efficiently recycled during subduction (what goes down, comes back up) according to some researchers (e.g. Fischer et al. 2002; Busigny et al. 2003) while others argue that ca. 50% of the subducted nitrogen is not degassed in volcanic arcs (e.g. Mitchell et al. 2010). High-pressure experiments and theoretical calculations (Mikhail and Sverjensky 2014) to simulate the passage of nitrogen through subduction reaction zones (e.g. Foerster et al. 2019) should provide an explanation for these geochemical inconsistencies.
Volcanic gases emitted by arcs basalts are predominantly composed of water (\(>70\%\)), while SO2 and CO2 are present in similar quantities (\(\text{SO}_{2}+\text{CO}_{2}<20\%\)) (Symonds et al. 1994). For the calculation of the fluid phase in this specific arc-magma system, the reader is referred to Burgisser et al. (2015).
5.6 The CO2 Enigma Linked to Unconventional Types of Degassing
Petit-Spot Volcanoes
In 2006, a new class of volcanoes was discovered (Hirano et al. 2006), which are unrelated to hotspot, arc or MOR magmatism. These were called Petit-spot volcanoes and they represent km-size submarine volcanic cones composed of alkaline magma containing high quantities of volatiles (several wt% of CO2). Okumura and Hirano (2013) suggested that this new type of volcanism significantly affected the planetary magmatic CO2 emissions. These volcanoes are found in front of almost all subduction zones, but the geodynamic triggers for such magmatism and its associated eruptions remain enigmatic. It is likely that these volcanic rocks are composed of the small fraction melts deriving from the low velocity region at the base the oceanic lithospheres (Schmerr 2012). At Petit-spot volcanoes and in other settings where alkaline magmatism occurs, this melt erupts, while elsewhere it remains trapped in the mantle (Sifré et al. 2014; Gaillard et al. 2019) and contributes to so-called mantle metasomatism (Aulbach et al. 2017). The process of mantle metasomatism stores volatile-rich melts at the base of the lithosphere. These volatile-rich magma zones can be reheated, remelted and outgassed if geodynamic modifications, such as continental rifting, occur.
Rift
Possibly related to these metasomatised alkaline magma sources, regions of continental breakup (rifting) also produce alkaline lavas associated with massive CO2 outgassing (De Moor et al. 2014; Lee et al. 2016). The CO2 content measured in some alkaline magmas of the East-African rift regions is similar to that of the “Petit-spot” lavas (several wt%, De Moor et al. 2014). In the Antarctic rift, similarly elevated CO2 contents have been found in the primitive alkaline magmas (Moussallam et al. 2014). The breakup of continents associated with tectonic plate movements may therefore have an impact on the amount of outgassed CO2 and on the global carbon cycle.
Non-volcanic Degassing
In many regions on planet Earth, diffuse degassing of CO2 is observed even in regions where no volcanic activity occurs. Italy is prime example, where massive CO2 outgassing occurs in the absence of eruptive activity (Burton et al. 2013; Werner et al. 2019). This type of non-eruptive degassing is also observed on a massive scale in the East-African rift (Lee et al. 2016). The probable cause for this type of degassing is metamorphic reactions at depth (Iacono-Marziano et al. 2009) and/or crustal ponding of magma that can release CO2 (because of its low solubility in silicate melts). This leads to the question of the degassing of intrusive magma (magma that does not make it to the surface). In general, the volume of intrusive magma (plutons) is believed to largely exceed that of extrusive magma (i.e. volcanic rocks). There are examples of large volumes of magma which have ponded in deep crustal levels (Laumonier et al. 2017) and there is no doubt that these deep magmas contain large amounts of volatiles (Laumonier et al. 2017). Yet, the contribution of these deep magmas to planetary outgassing remains poorly quantified (Werner et al. 2019). Furthermore, in specific regions, such as Italy, such non-eruptive CO2 degassing occurs where deep magma is hosted in sedimentary carbonates (Iacono-Marziano et al. 2009), which may indicate a non-mantle origin for this CO2. Overall, the degassing yield of deep magma and metamorphic processes remains difficult to quantify on a planetary scale but existing estimations reveal that it may be equivalent to the CO2 emitted by volcanic eruptions (Werner et al. 2019).
5.7 Magmatic Volatiles on Other Terrestrial Worlds
In addition to the Earth, active volcanoes have also been found on Io, Jupiter’s moon which may be the only other volcanically active body in the solar system (Spencer et al. 2000). The gas plumes emitted by volcanoes on Io are dominated by sulfur species. Volcanic activity on Venus has been suggested based on fluctuations in SO2 content in the atmosphere (Marcq et al. 2013; Encrenaz et al. 2019). If SO2 outgassing from Venusian volcanoes (100 bar of atmospheric pressure) is confirmed, such findings could provide information on the oxidation state of magma formed in the mantle of Venus.
Below, we review the estimations of the C-H-O-N-S contents of sampled basaltic magmas from the solar system. This covers the Earth’s Moon, Mercury, Mars, Vesta and Io. An important unresolved issue is to establish to what extent magmas in the solar system are markedly depleted in volatiles with respect of MORBs. The distribution of H2O content in planetary basalts remains under debate, with no clear evidence of dry volcanism on the Moon or Mars (e.g. Filiberto et al. 2019; Hauri et al. 2015). Other light volatiles, such as carbon and sulfur show major variations in magmas throughout the solar system.
Moon
The discovery of indigenous H-bearing species and other highly volatile elements (C, F, S, Cl; Saal et al. 2008; Sharp et al. 2010; Wetzel et al. 2015) in various lunar samples brought back by the Apollo missions – together with the detection of water ice in polar regions at the lunar surface – has led to a paradigm shift in our knowledge and understanding of the volatile inventory of the Moon. Advances in analytical techniques have made it possible to detect trace amounts of “water” (H, OH and/or H2O) in lunar volcanic glasses (Saal et al. 2008), olivine-hosted melt inclusions (Hauri et al. 2011), apatite (McCubbin et al. 2010), and highland plagioclase (Hui et al. 2013), thus completely reversing the notion that the Moon is anhydrous. However, estimating bulk Moon H2O abundances is highly challenging because 1) some of the targeted samples (i.e., pyroclastic glasses) may be derived from water-rich heterogeneities in the lunar interior (Albarède et al. 2015), 2) mineral-melt partition coefficients required to assess parent melt compositions are not well-known for lunar magmatic conditions (Potts et al. 2015), and 3) pervasive degassing has led to extensive volatile loss from all melts sampled in the form of extrusive lunar rocks (Hauri et al. 2015). Nonetheless, Hauri et al. (2015) argued that existing data imply that the bulk silicate Moon is only moderately depleted in highly volatile elements compared to the BSE, and may contain between 133 and 292 ppm H2O, a value matching the estimation of H2O content in the Earth’s upper mantle. Furthermore, results from melt inclusions within volcanic rocks indicate that the carbon, fluorine, sulfur, and chlorine content of lunar magmas is comparable to those of melts derived from the Earth’s upper mantle (Hauri et al. 2011; Wetzel et al. 2015), suggesting that the Earth and the Moon are characterized by a similar volatile abundance signature. This interpretation is disputed by some people (e.g. McCubbin et al. 2015) and it remains unclear as to whether the wet Moon volcanic rocks are representative of the bulk Moon volatile content.
Mercury
This is the closest planet to the Sun, and shows evidence of major and turbulent volcanic activity up to 3.5 Gy (Thomas and Rothery 2019). Inferred from surface measurements, it appears that Mercury’s lavas have peculiar characteristics compared to others observed in the Solar System, with compositions ranging from alkali-rich komatiite to bonite, corresponding to moderate-to high effusion rates. They also show very low Fe content (i.e., almost FeO-free) and high S and alkali concentrations up to 11 wt% and 7 wt%, respectively (Charlier and Namur 2019; Namur et al. 2016; Nittler et al. 2017). Locally, graphite-rich regions (i.e. containing several wt% C) have been identified (Charlier and Namur 2019). MESSENGER also revealed the abundant presence in Mercury’s surface lavas of putative volatile elements such as chlorine and sulfur (Weider et al. 2016). Such volatile species could have been the cause of the unexpectedly explosive volcanism recorded on Mercury, with the proposed mechanism requiring graphite oxidation to generate volatile exsolution from an already reduced magma. This is coherent with oxygen fugacity findings, as on the basis of the solubility of S in reduced silicate melts, oxygen fugacity conditions in Mercury’s magma are inferred to have been IW \(-5.4 \pm 0.4\) (Namur et al. 2016). Other estimations show a range of oxygen fugacity for Mercury’s interior of between IW-3 to IW-7, based on Fe and S data inferred from the MESSENGER mission (McCubbin et al. 2012a,b; Zolotov et al. 2013). Hence, Mercury’s volcanism is different to that on Earth, its explosive nature being powered not by H2O, CO2 or SO2 but by oxidation reactions involving S and C during (i) magma ascent through the crust or (ii) interaction between reduced lavas and oxide-bearing magmas (Weider et al. 2016).
Mars
Most estimations of the C-H-O-S content in Martian magmas come from the analysis of meteorites, which are mostly interpreted as crystal-dominated cumulate rocks that once coexisted with molten Martian lavas. From the analysis of amphibole and apatite, it is concluded that the water and halogen contents of Martian lavas are heterogeneous, with both enriched and depleted magmas, like on Earth. The range of estimated magma water contents is ca. 100–2,000 ppm H2O (Filiberto et al. 2016, 2019), which overlaps with the estimations for MORBs (particularly true if we consider the depleted MORBs described in Saal et al. 2002). In addition, Chassignite enriched rocks may have contained up to 8,000 ppm (McCubbin et al. 2012a,b; Filiberto et al. 2019), which is equivalent to the enriched MORBs described in Sect. 4.1. The carbon content of the Martian lavas has long been estimated based on the CO2 content of magma saturated in graphite calculated via the equilibrium equation (1), based on the assumption that the Martian mantle is graphite saturated. While graphite has been found in Allan Hills 840001 meteorites, a mantle source region being C-saturated remains unproven (Steele et al. 2012). This calculation also assumes that the oxygen fugacity of the Martian mantle is known. Estimates based on mineralogical assemblages yield an oxygen fugacity of about IW (Righter et al. 2008; Hirschmann and Withers 2008; Herd 2008). The likely CO2 content of magma deduced from such constraints is 73–1,100 ppm from the most reduced to the most oxidized magma. Finally, the interior of Mars is believed to be enriched in Cl with respect to planet Earth while the F contents of both planetary systems are seemingly identical (Filiberto et al. 2016, 2019). Chlorine contents of primary Martian basalts may then be expected to range from 250 to nearly 1,000 ppm for the most enriched mantle sources (Filiberto et al. 2016).
Vesta
The asteroid 4 Vesta, the second largest object in the asteroid belt, can be considered to represent the smallest terrestrial planet because it is the only known existing asteroid with a basaltic crust, an ultramafic mantle, and a metallic core (Keil 2002). Remote observations by NASA’s Dawn mission (McSween et al. 2013) have strengthened the notion that 4 Vesta is the parent body of the howardite-eucrite-diogenite (HED) meteorites, a group of achondrites (Drake 2001). Although the low K/Th ratio recorded by HED meteorites implies that Vesta is more volatile-depleted than Earth and Mars (Prettyman et al. 2015), the discovery of OH-bearing apatite (Sarafian et al. 2013), together with the detection of water in pyroxene (Sarafian et al. 2019), in several eucrite samples suggests that indigenous water is present in Vesta’s interior. In addition, secondary silicates observed within fractures of some eucrites may have been deposited by aqueous fluids (Barrat et al. 2011). Thus, although Sarafian et al. (2019) concluded that Vesta’s bulk water content is lower than that of Mars and Earth, these findings suggest that the Vestan mantle contains a significant amount of volatiles, lower but comparable to Mars or Earth’s (depleted) upper mantle.
Io
Discovered in 1610 by Galileo Galilei, Io is the innermost Galilean satellite of Jupiter. This planetary body is one of the most unusual objects in the Solar System due to a high level of active volcanism driven by Jupiter- and Europa-induced tidal heating. Thanks to the Galileo mission, several types of volcanic eruption styles have been identified: flow-dominated volcanism, explosion-dominated volcanism and caldera-like activities (the so-called intra-patera eruptions). The nature of the volcanic activity had been controversial with both silicate and sulfur volcanism having been proposed (Williams and Howell 2002). Based on high-temperature surface lavas (\(>700~\text{K}\)), the Galileo mission provided unambiguous support for widespread silicate volcanism, with more than 100 active sources (McEwen et al. 1998). On the contrary, only a few lava flows consisting of sulfur and SO2 have been reported, though sulfur represents the primary constituent of Pele-type plumes (Spencer et al. 2000). Io is believed to be generally volatile-depleted due to its higher density relative to other Galilean satellites. Volcanic gases on Io are mainly composed of sulfur and oxygen compounds such as SO2, S, S2 and SO with smaller amounts of sodium, chlorine and potassium compounds (Na, K, and NaCl). The molecular composition and oxygen fugacity of the gas plumes most likely reflect the source conditions in Io’s interior (Zolotov and Fegley 1999). Calculated fO2 varies from (i) Ni-NiO to hematite-magnetite buffers for hot SO2 and (ii) Ni-NiO to a few log fO2 units lower for Pele-type plume volcanic gases (Zolotov and Fegley 1998). However, this fO2 range is strongly dependant on the (last) equilibration pressure reached during eruption, which can be very low for Io (e.g. \(<10^{-4}~\text{bar}\)). It is known that at low pressure, SO2 can be the dominant gas species, even at very low fO2 (Gaillard and Scaillet 2014), making the fO2 estimations tentative. In addition, the degassing of SO2 from a magma containing sulfur in its reduced form (\(\text{S}^{2-}\)) causes a significant decrease in fO2 as degassing proceeds, implying evolving fO2s. Nevertheless, if this high fO2 range is validated, we can conclude that Io must be differentiated with an iron core segregated from the silicate mantle (Zolotov and Fegley 1999).
6 Planetary Controls on Magmatic Outgassing
6.1 The Importance of the Degassing Pressure
Gaillard and Scaillet (2014) made clear the overwhelming control of pressure on the composition of volcanic gases. This study implies that the volatile contents and the oxidation state of the source mantle, which determines the C-H-O-N-S content in the emitted basalts, play a secondary role in controlling the composition of the volcanic gases. Broadly speaking, water-rich basalts will tend to emit gases which are also water-rich, but the main control remains the pressure of degassing, which acts as a sort of filter determining the sequence of volatility. Figure 16 illustrates that this is effective if the degassing pressure varies over several orders of magnitude. This is the case on the scale of the solar system since the Venusian atmosphere is ca. 100 bar, while the volcanoes on Io, emit gaseous sulphur-rich mixtures in a vacuum. The sequence of degassing follows with a sequence of carbon-dominated gases at high pressure, water-rich gas at pressure close to the that of the atmosphere on Earth and sulphur-dominated gases at the lowest pressure. The changes in gas composition with pressure reflect certain trends in the systematics of volcanic degassing on a planetary scale. For an exoplanet, atmospheric pressure could exceed thousands of bars for a massive planet. In that case, even CO2 (and CO) might become soluble in the melt and therefore less volatile. This allows an upper limit to be placed on the atmospheric pressure because, at extremely high pressure, all gases would dissolve into the magma. This also implies that, for a high-pressure atmosphere, the gas phase should accommodate the most volatile species, i.e. noble gases (Ar, Ne, He) and N2 if conditions are not excessively reduced, or H2 in the case of very reduced conditions (Kite et al. 2019).

The composition of magmatic gases in equilibrium with basaltic melts as a function of pressure. The subaerial pressure of degassing on several planets is shown at the top of the diagram for reference (modified after Gaillard and Scaillet 2014)
Temperature has no identified effect of the solubility of gas species in silicate melts, so changing the temperature does not modify the scenario described above. However, gas speciation can be modified by significant changes in temperature. The most sensitive elements are carbon and sulfur. The speciation of carbon in reduced magmatic gases (i.e. IW) is dominated by CO coexisting with H2. However, magmatic mixtures emitted by low temperature magmas (e.g. \(800~^{\circ}\text{C}\), see Yang et al. 2014) or cooled high temperature magmatic gases (Iacono-Marziano et al. 2012a,c) can form abundant amounts of CH4 at the expense of CO and H2. Similarly, sulfur speciation in volcanic gases on Earth is mostly dominated by SO2, but H2S becomes increasingly dominant on cooling. These speciation changes for cooling of magmatic gases and their consequences on atmospheric processes have been poorly investigated (e.g. Ganino and Arndt 2009; Iacono-Marziano et al. 2012c).
6.2 The Importance of Oxygen Fugacity
Oxygen fugacity controls not only the speciation in the gas phase of multivalent elements but it also rules some of the gas-melt equilibria. Redox (homogeneous) equilibria in the gas phase are:
The melt-gas exchanges (heterogeneous) reactions being redox dependant are:
It is the interplay between homogeneous and heterogeneous redox reactions that determine the chemical aspect of magmatic degassing and its fO2 controls. Examples of computations of such interplays are available in Moretti and Papale (2004), Burgisser et al. (2015), Gaillard et al. (2011, 2015) and Ortenzi et al. (2020), showing the effect of redox conditions on the nature, abundance and speciation of the gas mixtures released by magmas. Using such models here, we illustrate the gas composition in equilibrium with MORB at \(1400~^{\circ}\text{C}\) vs fO2 at two different pressures (100, 1 bar). This is shown in Fig. 17. It can be seen that the oxidized gas speciation typical of present-day volcanism on Earth (CO2, H2O, SO2) requires \(\text{fO}_{2}> \text{IW+2}\). Below this value, the reduced gas compositions prevail (CO, H2, no S-species). By comparing calculations at two different pressure, it is possible to appreciate the combined effect of P and fO2 on controlling the volcanic output: with a few tens of bars of pressure, water does not outgas, implying H-poor gas at low fO2 (i.e. linked to moderate amounts of H2) and H-free gas at high fO2. Therefore both pressure and fO2 play a major role during magmatic degassing. There is also a feedback process since the atmospheric pressure is partly controlled by the amount of outgassed C-H-O-N-S and the composition of the outgassed mixture is controlled by pressure (Gaillard and Scaillet 2014). This aspect has recently been investigated by Ortenzi et al. (2020), providing insight into the interplay between magmatic fO2, amount of outgassed volatiles, planetary size and atmospheric pressure.

The effect of fO2 (relative to the IW buffer) of the gas composition in equilibrium with basalt at 1 and 100 bars, and \(1400~^{\circ}\text{C}\). The basalt has the MORB composition given in Table 2. The grey area shows the domain where graphite is stable. Additional numbers such as the fraction of volatiles remaining in the magma or the mass of emitted gas can be obtained via an email to the corresponding authors
Figure 18 (after Ortenzi et al. 2020) shows the effect of magmatic fO2 on the structure of an outgassed planetary atmosphere. The system is restricted here to the C-O-H elements, ignoring sulfur and nitrogen since the redox behaviour of these elements is not sufficiently well constrained. This plot shows the thickness of the generated atmosphere for an oxidized case (the FMQ buffer, e.g. Earth, Fig. 6b) and a reduced case (IW, e.g. Moon or Mars; Fig. 6b). The variations in atmospheric thickness are caused by a release of mostly H2 plus CO (minor H2O) in the reduced state, and H2O plus CO2 under oxidised conditions (Ortenzi et al. 2020). After 4.5 Gyr of mantle convection, the corresponding outgassed atmospheres show that in the reducing scenario the atmospheric thickness is greater and the pressure at the surface is lower compared to those in oxidised atmospheres (Ortenzi et al. 2020). In conclusion, the simulated atmospheres are affected by the oxygen fugacity of the mantle in terms of both composition and radial extent. There is thus a link between the redox state of a planetary interior and its atmospheric properties which could be used to help interpret present day and future exoplanet observations.

Atmospheric thickness versus planet radii and corresponding planet mass in Earth masses after 4.5 Gyr mantle convection and degassing. Each dot represents a simulation with different initial parameters comprising volatile and radioactive heat source contents of the mantle (see Ortenzi et al. 2020 for details). The plot shows that under reducing conditions (IW) the atmospheric thickness is greater than that of the oxidising (QFM) cases
6.3 Mantle Redox State and the Lower Mantle Oxygen Pump
How does the oxygen fugacity of planetary mantles vary throughout the solar system and are there identifiable trends and common processes? Figure 6 shows a vast range of fO2 in the solar system, with Mercury’s mantle being the most reduced and planet Earth being the most oxidized. Venus, if volcanic outgassing of SO2 is confirmed by future remote measurements of its atmosphere (Marcq et al. 2013; Encrenaz et al. 2019), may well also be oxidized to some degree. Mars lies in between the two end members, with Vesta probably similar to Mars (Herd 2008; Righter et al. 2016). Despite the limited number of samples and observations on these objects, this description indicates that smaller bodies may have more reduced mantles than larger bodies. How could this come about? The most accepted scenario for this is that the lower mantle and the transition zone contain minerals that can integrate a lot of ferric iron with respect to the minerals of upper mantle, in which ferric iron is rare. The process by which ferric iron is accommodated in perovskite and in minerals of the transition zone is named disproportionation (Frost and McCammon 2008): \(3\text{Fe}^{2+} = \text{Fe}^{\circ} + 2\text{Fe}^{3+}\). This process allows a large amount of ferric iron to be accommodated even at highly reduced conditions in the deep mantle while, under otherwise similar conditions, the upper mantle contains only ferrous iron. This implies that as the planet grows by accretion, it begins with a mantle which is only composed of Earth’s upper mantle mineralogy (dominated by olivine). It is therefore poor in ferric iron. Once it reaches a given size and is able to develop a transition zone and a lower mantle, it can produce ferric iron from ferrous iron following the disproportionation reaction mentioned above. Once convection sets in, it allows the ferric-iron-rich material to mix with the upper mantle, which triggers an increase in the ferric iron content of the upper mantle. The transition zone and lower mantle is suggested to act as an oxygen pump (Frost et al. 2004; Wade and Wood 2005) affecting the oxygen fugacity of the upper mantle of large planets, while small planets would remain unaffected by this process. In order to be efficient, this process relies on getting rid of the metal iron (\(\text{Fe}^{\circ}\)) produced by the disproportionation reaction, by having it sink, at least partly, into the core in order to leave behind an oxygen enriched mantle. It should be noted that the amount of oxygen the upper mantle needs to gain to evolve from IW (e.g. Mars) to IW+4 (Earth) is very small, making the overall process feasible.
The lower mantle oxygen pump (Frost et al. 2004; Wade and Wood 2005) remains the only sound hypothesis relating the size of a planetary body and the redox state of its mantle. This hypothesis implicitly distinguishes small planets without lower mantle (reduced) from large planets with a lower mantle (oxidized). It does not conflict with existing data and can be used to make predictions for planets like Venus, which should have an oxidized mantle like that of the Earth. Spatial observations have revealed large rocky exoplanets, well exceeding Earth’s size. This implies that they must have grown a huge lower mantle, possibly maximizing its functioning as the oxygen pump. Would we then expect these planetary mantles to be much more oxidized than Earth’s one? If convection is able to mix the mantles, the answer is yes. The implication is that mantle outgassing on these planets must produce an abundance of oxidized gases, dominated by SO2 and possibly containing trace amounts of O2. The production of such gases has never been demonstrated, but it is theoretically possible from oxidized magma degassing SO2. In such melts, the form of oxidized S in the magma is SO3. Upon degassing, these oxidized magmas react as follows:
Degassing of (oxidized) sulfur therefore causes an increase in magmatic fO2 (see Gaillard et al. 2015). The stability of O2 (supposedly produced from reaction 13) depends on the presence of other redox sensitive elements. In a magmatic environment, only iron can be expected to consume O2. It is therefore possible that oxidized magma with a low iron content and containing SO3 could outgas a mixture containing appreciable fractions of O2, e.g. \(\text{X}_{\text{O2}}>10^{-4}\).
6.4 The Importance of Mantle Oxygen Fugacity in Carbon Outgassing and O2 Production
The oxygen pump involves chemical processes occurring at great depth in the mantle (\(> 400~\text{km}\)). The regions where partial melting and basalt production occur are much shallower (Fig. 5), typically in the range of 50–200 km depth. In this “shallow” mantle, carbon is believed to play an important role in controlling fO2 during melting via the equilibrium:
This equilibrium process operates for a mantle oxygen fugacity in the range FMQ-5/FMQ-1.5 (Rohrbach and Schmidt 2011; Stagno et al. 2013; Hirschmann and Withers 2008; Righter et al. 2008; Li et al. 2015). Moreover, it tells us that if there is enough ferric iron in the solid peridotite, basaltic melts can efficiently take up CO2 during mantle melting. Otherwise, the abundance of ferric iron in the mantle may limit the outgassing of CO2. This process it thought to operate in the Earth’s mantle by some researchers (Holloway et al. 1992; Stagno et al. 2013), although the fO2 of most basalts reaching the surface is close to, or higher than, FMQ. Furthermore, this equilibrium is shifted to the right as pressure decreases, in other words mantle fO2 must decrease as pressure or depth increases. This scenario is well accepted in the community and it must have implications regarding the secular evolution of mantle fO2 (i.e. Gaillard et al. 2015), though the geochemical proxies so far used have not reached firm conclusions on the possible variations of the Earth’s upper mantle oxidation state (Canil 1997; Delano 2001; Li and Lee 2006; Berry et al. 2008; Keller and Schoene 2012; Yang et al. 2014; Aulbach and Stagno 2016).
In mantle which is more reduced than that of Earth, such as Mars, it has also been suggested that carbon acts as a redox buffer (Righter et al. 2008). The C-rich mantle nature revealed by Ureilites is thought to come from large planetary bodies, where carbon redox buffering and smelting may be important processes (Barrat et al. 2017). Carbon may play an important role in buffering mantle fO2, but in turn, the ferric iron-rich mantle must enhance the outgassing of the mantle C as CO2.
The outgassing of mantle CO2 has long been linked to the deployment of cyanobacteria and photosynthesis (Holland 2002). It is accepted that \(\sim80\%\) of the outgassed mantle CO2 is taken up by continental weathering and ultimately yields carbonate precipitation in seawaters (Holland 2002). The remaining 20% is used directly by cyanobacteria to produce O2 (Holland 2002). Adding the photosynthesis reaction to Eq. (14) reveals a global process linking mantle fO2 (or ferric iron contents) and the appearance of O2 in the atmosphere:
Of course, this is a simplification since other multivalent elements play a role here (e.g. sulfur and nitrogen, Holland 2002; Lyons and Gill 2010; Gaillard et al. 2011; Wordsworth 2016) and we note that only 20% of the outgassed C is involved in photosynthesis, while the remaining 80% is directly taken up by continental weathering. From Eq. (15), we can conclude that planetary mantle which does not contain enough Fe2O3, or C, or is unable to outgas H2O, could not evolve photosynthesis.
Many workers have tried to link the Great Oxidation Event (the appearance of O2 in the Earth’s atmosphere, 2.5 Ga, Holland 2002) to a change in the ferric iron content of the mantle (Kasting et al. 1993a,b; Aulbach and Stagno 2016; Andrault et al. 2018) or a change in the C outgassing/ingassing rates (Eguchi et al. 2020). All these mechanisms involve changes in mantle source processes. While a change in tectonic style (i.e. mantle temperatures, crustal production) may have marked the late Archean (Keller and Schoene 2012; Tang et al. 2016; Andrault et al. 2018), there is no clear change in mantle fO2 at 2.5 Ga (Keller and Schoene 2012) and the large uncertainties/heterogeneities in published estimations cast doubts on certain claims made (e.g. Keller and Schoene 2012 vs. Aulbach and Stagno 2016). Enhanced CO2 degassing via recycling of oceanic carbonates at subduction zones could represent a mechanism which would have boosted photosynthesis without requiring an increase in mantle ferric iron (Eguchi et al. 2020). It may also match the geodynamic changes of the late Archean if one accepts that this period marks the onset of subduction (e.g. Tang et al. 2016). Two alternative models consider the O2 sinks rather than the sources. Gaillard et al. (2011) suggested that the enhanced outgassing of SO2 in the late Archean (recorded by mass-independent fractionation of sulfur; see Holland 2002) was caused by the emergence of continental surfaces (subaerial volcanism). This would have triggered the onset of sulfate reduction processes. In turn, this would have permitted the cyanobacteria-produced O2 to invade the atmosphere instead of being titrated by iron at hydrothermal vents. Overall, the appearance of O2 in the atmosphere would have accompanied the disappearance of banded iron formations and their replacement by massive sulphide deposits (Gaillard et al. 2011). Sulfate reduction also involves the seafloor oxidation of organic carbon (into CO2) and the burial of pyrites (Lyons and Gill 2010). This sulfur-outgassing scenario matches geochemical records in sediments of the late Archean (Olson et al. 2019). Another O2 sink model involves the transition from mafic (i.e. basaltic) to felsic (granitic) crust marking the late Archean (Smit and Metzger, 2017). In this model, the Fe-rich basaltic crust consumes O2 by a variety of weathering processes. The emergence of the Fe-poor felsic crust at the end of the Archean (Tang et al. 2016) reduced the O2-sink effect of the basaltic crust, which overall, facilitated the accumulation of O2 in the atmosphere. It is worth mentioning here that the rate of CO2 uptake by continental weathering and marine carbonate precipitation (see Eq. (15)) is much less marked on felsic than on basaltic crust. Consequently, the appearance of a felsic crust should also be accompanied by an accumulation of CO2 in the atmosphere, therefore enhancing photosynthesis and O2 production.
The debate over the trigger of Great Oxidation Event is far from finished. Future modelling will probably connect changes in the planetary interior to biogeochemical surficial models (e.g. Ozaki et al. 2019). There is an underlying link between mantle and crustal geodynamics, mantle oxidation state, carbon and sulfur outgassing and photosynthetic activity. All in all, the change in mantle temperature regime (linked to a shift in the global geodynamic styles) which marked the end of the Archean (3–2.5 Ga, Keller and Schoene 2012; Tang et al. 2016; Aulbach and Stagno 2016; Andrault et al. 2018) may have been the trigger for surficial biogeochemical changes (Gaillard et al. 2011; Smit and Mezger 2017; Olson et al. 2019; Ozaki et al. 2019). However, the link between the planetary interior and surficial processes remains unclear.
7 Plate Tectonics and Volatile Fluxes from Mantle to Surface and Vice Versa
The C-H-O-N-S elements were initially distributed after the magma ocean stage between the various geochemical reservoirs and then likely redistributed between the Earth’s interior and surface throughout the Earth’s history via geodynamic mass transfer. The volatile geodynamic cycles on Earth involve a chain of processes which we first touched on in the introduction (Fig. 1): 1) the mantle volatile endowment determining mantle rheology, plate-like behaviour of the lithosphere, rock melting temperatures and outgassing volumes; 2) chemical reactions at the surface (e.g. carbonate formation; serpentinization; sulfate reduction); 3) atmosphere volatile losses (either sinks at the surface or losses to space); and 4) crustal evolution and erosion (influencing sediment production and evolution of plate tectonics). Finally, plate tectonics links all these processes by operating/modifying the mass transfers between surficial and deep geochemical reservoirs.
7.1 Magmatic Outgassing in the Presence or Absence of Plate Tectonics
It is not easy to estimate the exact influence of plate tectonics on volatile outgassing fluxes and to predict/compare fluxes for a hypothetical Earth stagnant-lid planet. Our two neighbour planets (Mars and Venus) are both in the stagnant-lid mode (though plate tectonics on Venus has been discussed, e.g. Schubert and Sandwell 1995) and show no signs of subduction or other recycling zones, though the surface of Venus is in general rather young and there are no observable traces of the planet’s history beyond about 1 Gyr at the surface. Still, present-day volcanic activity seems to be much stronger on Earth than on Venus (Smrekar et al. 2010), while Mars has been volcanically inactive in its younger past (Hauber et al. 2011). On the other hand, plate tectonics includes the subduction of cold surface material into the mantle, and therefore cools the mantle much more efficiently than in a stagnant-lid scenario (Breuer 2009). Based on the mantle temperature, one would therefore expect deeper volcanic activity for a stagnant-lid planet than for a planet experiencing plate tectonics, which is at odds with the comparative observations of the Earth, Venus and Mars. The explanation lies within the material that is subducted back into the mantle. Figure 19 (calculated using the code of Noack et al. 2014; see also Noack et al. 2016) shows the comparative evolution of the inner mantle temperature as well as the crust production rate for three different cases. In the stagnant-lid scenario, the mantle temperature increases due to radiogenic heating in the mantle and cooling from the core, but crust production is limited due to the thick lithosphere that forms, separating the mantle (and melt region) from the surface. For the plate tectonics case, it is assumed from the beginning of the simulation (i.e. 4.5 Ga) that efficient subduction dominates the entire surface. In this scenario, crust does not stay at the surface, but is always immediately recycled into the mantle. This leads to an efficient refertilization of the mantle and an enhanced convection rate implying continuous, strong volcanic activity. Similarly, we investigate a model where we assume that the mantle is efficiently hydrated by the subduction of wet oceanic crust. In that case, the mantle convects more vigorously and the melting temperature of hydrated rocks is reduced, leading to strong volcanic activity especially in the first few hundred Myrs, where latent heat is consumed upon melting resulting in an even colder mantle than for the dry plate tectonics case.

Average mantle temperature and crust production rates calculated over time with a 1D thermal evolution model (Noack et al. 2016) applied to an Earth-size planet assuming either a stagnant-lid scenario (SL, red), a plate-tectonics scenario without (PT, blue) or with hydration effect on the mantle (PT wet, blue dashed)
This simple example shows that even though plate tectonics leads to much cooler mantles than observed for a stagnant-lid planet, continuous melting will still be much higher at all times. Melting can also efficiently cool the mantle, as observed in the beginning of the plate tectonics cases as well as during the main melt-production phase observed in the stagnant-lid case. The observed cases here are end-member scenarios where either the entire surface is stagnant or the entire surface actively recycles into the mantle (which are the only two scenarios that can be investigated with the simple 1D thermal evolution model used here). On Earth, a large part of the surface (the continental crust and a large portion of the oceanic crust) is stagnant and not (immediately) recycled back into the mantle. Earth’s present-day crust production rate is therefore in-between these two end-member scenarios.
7.2 Planetary Size Controls on Magmatic Outgassing
Here the focus is the degassing volume and the influence of the planetary mass on the amount of outgassed species and the implications for the atmospheres formed. The planet size affects a variety of aspects during the volatile pathway from the mantle to the atmosphere, starting from melt production and mantle convection to the final atmospheric thickness and composition. In general, an increase in planet size produces an increase in surface gravitational acceleration and in the pressure gradient within the mantle. The consequence is that the solidus temperature of the mantle is shifted to higher values and the production of melt is reduced or absent compared to that at lower pressures. Furthermore, higher values of the pressure gradient lead to a higher mantle viscosity causing a less intense mantle convection, which has a negative impact on the final outgassing volume. On the other hand, a more massive planet has a larger reservoir of volatiles and radioactive heat sources that facilitate melt production in the mantle. Thus, considering 4.5 Gyr of mantle convection and cumulative outgassing (Noack et al. 2017; Dorn et al. 2018; Ortenzi et al. 2020), melt production and volatile outgassing is favoured for planets within the range of 2–4 Earth masses (\(\text{M}_{\mathrm{E}}\)) for stagnant-lid rocky planets. As shown in Fig. 20, outgassing is most efficient below a threshold of \(4~\text{M}_{\mathrm{E}}\). The redox state drives the composition of the outgassed atmospheres and both in reducing or oxidising conditions the pressure at the surface is highest for \(2\text{--}4~\text{M}_{\mathrm{E}}\). Above \(4~\text{M}_{\mathrm{E}}\), outgassing becomes weaker, which is reflected by a decreasing atmospheric pressure. For cases with high degassing rates, another factor to consider is that increased atmospheric pressure also increases the solubility threshold that the volatiles need to cross to outgas. However, within the 2 and \(4~\text{M}_{\mathrm{E}}\) range, the volatile content of the melt remains high enough to produce outgassing.

Atmospheric thickness and partial pressure of outgassed volatiles for different mantle redox states and volatile contents. The three cases correspond to different mantle volatile contents in ppm as follows (in wt%): dry case is for \(0.005^{\mathrm{a}}\) of H2O and 0.018 of CO2, low water content is for 0.015 of H2O and 0.0022 of CO2, and high water content is for 0.045 of H2O and 0.005 of CO2 (data from Elkins-Tanton 2008. aHere this is considered a very small fraction of water). Figure adapted from Ortenzi et al. (2020)
7.3 C-H-O-N-S Fluxes at Subduction
As shown in Fig. 15, subduction transports volatiles (bound into hydrated rocks and sediments as well as carbonates) back down into the mantle. At different depths, these minerals may become unstable, releasing volatiles into the near-surface mantle and mantle wedge. Hydrated mantle rocks start to melt at much lower temperatures than dry rock (Katz et al. 2003), so plate tectonics triggers strong volcanic activity. Most of the volatiles that were transported into the mantle during subduction are thus brought directly back to the surface by volcanic activity, leading to a near-surface, shallow volatile cycle (Wallace 2005). The net influx of volatiles into the mantle crucially depends on the mineral destabilization and remelting processes in the mantle wedge.
It is not clear if burial and subduction of carbon leads to net sequestration of carbon, or if carbon is in general remobilized and returned to the surface (Kelemen and Manning 2015). The efficiency of carbon recycling influences the deep carbon cycle of Earth (e.g., Dasgupta 2013; Orcutt et al. 2019). The subduction of carbon is highly dependent on surface chemical reactions. At the present day, carbonates are formed at the Earth’s surface mainly by the carbon-silicate cycle. Carbonic acid forms from carbon dioxide reacting with water. Silicate weathering on continents leads to the formation of calcium cations (\(\text{Ca}^{2+}\)), which are consumed by oceanic carbonate precipitation which creates carbonates. This silicate weathering reaction, of extracting carbon from the atmosphere and converting it into sediments, can be expressed simply as the Urey reaction, assuming the availability of water as a catalyst (see e.g. Kasting et al. 1993a,b):
Carbon can then be recycled to the interior in the form of carbonates. Before the rise of continents, seafloor weathering would be expected to be the dominant carbonate formation process. A simple formula for the efficiency of the process was derived based on temperature and atmospheric partial pressure PCO2 (Brady and Gíslason 1997). Using this approach, simple estimates show that at least during the early Earth stages, seafloor weathering would have been an important mechanism for extracting carbon from the oceans and hence from the atmosphere (Sleep and Zahnle 2001; Foley 2015). In a more recent study, Krissansen-Totton and Catling (2017) developed a more sophisticated seafloor-weathering model incorporating the dependence on ocean chemistry using pH- and temperature-dependent kinetics based on laboratory and empirical studies. They found that already 100 Myr ago, the efficiency of seafloor weathering would have been almost comparable to continental weathering.
Water return fluxes into the mantle depend on various factors such as thickness of sedimentary layer, igneous crust alteration, temperature in the mantle wedge and serpentinisation reactions (Parai and Mukhopadhyay 2012; Höning et al. 2014; Schmidt and Poli 2003; Li and Lee 2006), which are not very well constrained for the present-day, and not at all for early Earth conditions. Parai and Mukhopadhyay (2012) estimated the average water return flux to correspond to 2–5% serpentinisation in 10 km of global lithospheric mantle, which would lead to a net intake of water into the mantle. Other geochemical estimates indicate that the subducted water goes back to arcs (Wallace 2005). Höning et al. (2014) conducted numerical models on the subduction of water including shallow and deep water release from the subducting slab due to shallow dewatering of sediments, compaction and bending of the oceanic crust, dehydration by metamorphic reactions and finally destabilization of hydrous minerals such as amphiboles and serpentines. They coupled the water fluxes with the feedback of volatiles on convective behaviour and found a self-regulating system leading to almost steady-state water concentrations in the mantle and ocean thickness. Water transport into the Earth’s mantle seems to be possible to depths of at least 100 km as suggested from seismic evidence (e.g., Kawakatsu and Watada 2007) or maybe even deeper. However, there is a still on-going debate as to the current mantle water concentrations (especially in the transition zone; Karato 2011; Houser 2016), both inherited from the magma ocean phase (Elkins-Tanton 2008) and their evolution during the Hadean and Archean aeons.
Plate-tectonics, as it occurs on present day Earth, may not have been possible in the early Archean (Davies 1992), or possibly by flat subduction (that is to say, subduction of lithosphere into the mantle at a low angle, see Van Hunen and Moyen 2012). However, other recycling processes such as for example sagduction of crust and crustal delamination due to formation of denser minerals such as eclogite (e.g. Rozel et al. 2017) or addition of new crust on top of the hydrated crust, pushing the hydrated minerals deeper into the mantle (Kamber et al. 2005), may have contributed to early Earth’s volatile cycles and may have changed the upper mantle’s redox state, which would have influenced outgassing products (e.g., Mikhail and Sverjensky 2014; Yang et al. 2014).
In general, subduction of volatiles and oxidized material into the mantle can affect the redox state and hence the speciation of gases during arc outgassing. The relationship between subduction and oxidation of the mantle wedge is still under discussion.
During the subduction process, oxidised components are transported with the slab into the mantle. Hydrothermal alteration of ferrous iron (\(\text{Fe}^{2+}\)) in olivine (Fe2SiO4) in the oceanic crust occurs at low temperatures (\(200\text{--}350~^{\circ}\text{C}\)) and produces magnetite (Fe3O4) containing ferric iron (\(\text{Fe}^{3+}\)). This essentially increases the local oxidation state (Lécuyer and Ricard 1999; Shanks et al. 1981). The subduction of oxidised volatile species is another important aspect for the redox evolution of the upper mantle. Kelley and Cottrell (2009) analysed the oxidation state of undegassed basaltic samples that are representative of different plate tectonic settings (e.g. MORB or back-arc basin basalts). They show that the oxygen fugacity increases linearly with water content. This result would indicate that water subducted into the upper mantle would lead to a more oxidized mantle. However, it has been suggested that H2O is not directly linked to a higher oxidation level of the Earth’s upper mantle (Hirschmann 2009; Gaillard et al. 2015). For the present-day level of oxygen fugacity, water is a stable oxidised volatile (together with CO2) down to 5 GPa and therefore it is not expected to affect the redox state of the upper mantle (Frost and McCammon 2008). However, as a possible explanation for the linear increase in oxidation with water content, Kelley and Cottrell (2009) suggested that water released from the subducting slab into the mantle wedge might partly dissociate into H2 and O. Removal of H2 from the mantle would then lead to an increase in oxygen fugacity, which would become significant if H2O was introduced into the mantle wedge at subduction zones in large quantities (i.e. high water/rock ratio). Alternative scenarios suggest that sulfur may be the oxidizing element in the mantle wedge (Metrich et al. 1999; Prouteau and Scaillet 2013; Alt et al. 2012; Gaillard et al. 2015). Most likely, SO2 released from the subducting slabs reacts upon ascent through the mantle wedge with ferrous iron and raises the fO2 via the reaction (Alt et al. 2012; Gaillard et al. 2015):
However, this reaction can only operate once a significant amount of sulfur has been produced on the seafloor before being subducted (Alt et al. 2012). It should be noted that the Archean ocean is believed to have been sulphur-poor, and that the water at the depth of the seafloor was probably euxinic for most of the Earth’s history (Stolper and Bucholz 2019). A recent geochemical survey suggests that the most ancient subduction magmas were not that oxidized and that it was only when seafloor processes became significantly oxidizing that arc-magmas, in turns, became significantly oxidized (Stolper and Bucholz 2019). Based on various redox proxies, Stolper and Bucholz (2019) suggested that arc-magmas became more oxidized 400–800 Ma.
7.4 Subduction and Secular Evolution of Mantle fO2
The exact influence of subduction on the mantle redox state is still strongly debated, but as summarized above, several links exist between the oxidation state of the mantle and subduction of volatiles as well as oxidized minerals, leading to the conclusion that plate tectonics plays a key role in the geodynamic redox evolution of a rocky mantle. Has subduction modified the global mantle volatile contents and oxidation state? If the Stolper and Bucholz (2019) model is correct, and subduction only recently became efficient in terms of redox transfers, the answer is clearly that it is not likely to have had much effect. The oxygen fugacity of the uppermost mantle seems to have remained unchanged from about 3.9 Gyr ago (Canil 1997; Delano 2001), which corroborates Stolper and Bucholz’s proposal. But these conclusions are disputed by Aulbach and Stagno (2016) and one must recognized that there are still large uncertainties for the oxidation state of the Archean mantle, partly because of sampling issues (Aulbach and Stagno 2016). During the Hadean aeon, for which we only have zircon crystals as redox proxy, the Earth’s mantle may have been more reduced (Yang et al. 2014), though Trail et al. (2011) suggested a constant mantle fO2 based on analyses of zircons dating back to 4.4 Gyrs.
Overall, the issue remains controversial and it has not been proven that subduction could have modified the oxidation state of the whole mantle. Oxidizing seafloor processes seem too recent to have affected the mass and the buffering capacity of the mantle. In addition, the difference in mantle fO2 between Arc magmas and MORBs is not that large (Table 2). Finally, enriched MORBs that possibly correspond to recycled subducted slabs are not more oxidized than more depleted (normal) MORBs (Cottrell and Kelley 2013). If it is confirmed that 4.5 Ga redox geodynamics of planet Earth have not significantly affected the mantle oxidation state, then the cooling, outgassing, mixing and crystallization of the magma ocean seems to be next focus for research efforts in order to identify these possible redox fluctuations of the mantle from the stage of core formation.
7.5 No Subduction, no Recycling, Runaway Drying of Planetary Surfaces?
In the absence of plate tectonics, the surficial waters and other volatiles (CO2, SO2) would still react with the lithosphere via seafloor processes producing hydrothermal deposits. The difference is that these volatiles would not be returned to the atmosphere by arc-volcanoes. We present here a simple consequence of the absence of return processes linked to plate tectonics that should be tested in future research.
Where (i.e. at what depth) does hydrothermal reaction and volatile deposition in the lithosphere stop? On Earth, serpentinisation cannot occur at temperatures exceeding \(350\text{--}400~^{\circ}\text{C}\) (Alt 1995), which is \(<25~\text{km}\) depth for most oceanic lithosphere. The reason for this is the location of the \(1300~^{\circ}\text{C}\) isotherm at ca 100 km in the oceanic basin (McKenzie et al. 2005). In contrast, in the absence of plate tectonics, a thick stagnant lid with a slow temperature increase with depth (e.g. Plesa et al. 2018) allows for serpentinization to occur at higher depths. This implies that, depending on the surficial water volume, oceans situated above a stagnant lid may well dry out by lithospheric suction caused by serpentinisation and other types of hydrothermal deposits affecting the fluids (e.g. Alt 1995). This must also cause an increase in water salinity, possibly creating brines, as recently proposed (Debure et al. 2019). If this process is not compensated by subduction which releases the hydrothermal C-H-O-S deposits via arc magmatism, it seems inevitable that the surficial volatile inventory would be reduced with time and cooling of the lid. The S-Cl-rich deposit on Mars’s surface (Keller et al. 2006; Gaillard et al. 2013) and serpentinisation processes (Ehlmann et al. 2010) may be partly due to such drying processes.
Subduction, high heat flow in oceanic basins and recycling of the subducted C-H-O-N-S elements by arc-volcanoes therefore appear to be important steps in maintaining the surficial reservoir of volatiles and sustaining (if not enhancing) volatile fluxes between the deep and surficial reservoirs.
8 Take-Home Messages & Perspectives
The C-H-O-N-S elements are considered to as volatiles and as such, they should preferentially partition into the atmosphere. However, we have seen here that they can be siderophile, refractory, chalcophile, magmatophile, or atmophile depending on the prevailing oxygen fugacity and pressure. Figure 21 summarizes these changes in behaviour as a function of fO2. The magma ocean and post-magma ocean magmatic stages for some planets are also shown in this figure.

The behaviour of C-H-O-N-S elements in magmatic systems as a function of oxygen fugacity expressed in log-units relative to the iron – wustite buffer (IW, see Fig. 6). The fO2 domains of planetary magma oceans and those of the present-day mantle of Mercury, Mars and Earth are indicated. This summary figure illustrates the diverse behaviour of the C-H-O-N-S elements (plus noble gases), as fO2 varies. Some elements (sulfur and nitrogen) become increasingly magmatophile (i.e. less siderophile) as conditions becomes more reducing. This is because they are soluble in silicate melts as anions. Carbon, in contrast, becomes increasingly siderophile as fO2 is reduced. This variety of behaviour implies that the ratio of C-H-O-N-S elements to noble gases must change with fO2, and should be used to guide future interpretations of geochemical data on the chemical diversity planetary fluids
Because of this diversity of behaviour, which is essentially controlled by oxygen fugacity, we conclude that the largest reservoirs of C-H-O-N-S elements are likely to be the core and the mantle. The C-H-O-N-S distribution in the mantle is probably heterogeneous (LeVoyer et al. 2017; Marty 2012; Miller et al. 2019) with some regions of the lower mantle being rich to very rich, while the upper mantle is overall depleted in most of these elements. The surface (crust + atmosphere) is rich in C-H-O-N-S, but it actually constitutes the smallest reservoir on a planetary scale, at least for the Earth. Can this be applied elsewhere as a planetary rule? The processes that control the surficial vs. deep distribution of the C-H-O-N-S elements and how they change through time and from planet to planet is a rich and complex subject. To answer this question will require new geochemical data and the construction of internally consistent models allowing intra/extrapolation. These models should help us develop the evolutive pipeline, as illustrated in Fig. 6b. The community in this field is currently working hard on these topics.
As an indirect consequence of the fO2-dependant behaviour of the C-H-O-N-S elements, the composition and the speciation of the outgassed magmatic phases changes significantly in the P-fO2 parameter space. Figure 22 provides a schematic drawing of the composition of the equilibrium magmatic gas mixture. It demonstrates how changes in the size of a planet may affect the P-fO2 conditions of outgassing. This figure illustrates the diversity of volcanic outgassing, ranging from dry CH4 worlds to sulphur-rich water worlds. Note that very reduced conditions should suppress the degassing of S and N, which become magmatophile (as shown in Fig. 21). Very oxidized magmatism, on the other hand, would produce an SO2-rich mixture, with the possibility of trace amounts of O2 being directly outgassed from Fe-poor, S-rich oxidized magma. In principle, this diversity can be related to physical parameters such as the planetary size.

Hand-drawing of the range of magmatic atmospheres that can be generated by the degassing of a planet at different redox states and atmospheric pressure. Two types of Magmatic Pipeline are found in planetary systems: the magma ocean and post magma ocean volcanic degassing (from plumes & arcs)
In addition to the chemical controls on mantle melting and outgassing, the style of convection, i.e. plate tectonics vs. stagnant lid, have a first order impact on the fluxes of C-H-O-N-S elements between the surface and deep geochemical reservoirs. Plate tectonics make global mantle convection much more efficient resulting in enhanced mantle melting by decompression, and therefore enhanced outgassing. This also results in thinning of the oceanic lithosphere, which limits the depth of hydrothermal C-H-O-N-S deposits. Finally, most of the C-H-O-N-S deposits can be reintroduced into the atmosphere via arc-volcanoes, which at present-day inject the most oxidizing magmatic volatiles in the atmosphere. Therefore, plate tectonics greatly increase the mass of C-H-O-N-S being outgassed in addition to sustaining the fluxes and the availability of these elements for atmospheric processes.
Finally, the topic of mantle degassing has been historically addressed using noble gases. These gases do indeed have a great affinity for the gas phase, and display no chemical reactivity, making them insensitive to fO2 conditions during magmatic processes. Future research should therefore consider a simultaneous treatment of noble gases and the C-H-O-N-S elements during the various differentiation stages. There is a further class of volatiles which have not been looked at in this review: the halogens (F, Cl, I, Br). Seawater on Earth is salty and evidence for a chlorine-rich surface on Mars has long been available (Keller et al. 2006), not to mention the recent discovery of salt water on Ceres (Raymond et al. 2020). Our knowledge of the behaviour of these elements in a solar system context (Filiberto and Treiman 2009) is poor in comparison to their role and abundance among the planetary suites.
References
A. Aiuppa, F. Gaillard, Volcanic gases, in Encyclopedia of Geochemistry (2016). https://doi.org/10.1007/978-3-319-39193-9_239-1
A. Aiuppa, D.R. Baker, J.D. Webster, Chem. Geol. 263, 1–18 (2009)
F. Albarède, Nature 461, 1227–1233 (2009)
F. Albarède et al., Meteorit. Planet. Sci. 50, 568–577 (2015). https://doi.org/10.1111/maps.12331
C.M.O.D. Alexander et al., Science 337, 721–723 (2012)
C.M.O.D. Alexander et al., Space Sci. Rev. 214, 1–47 (2018)
J.C. Alt, in Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions, ed. by S.E. Humphris, R.E. Zierenberg, L.S. Mullineaux, R.E. Thomson. Geophys. Monogr. Ser., vol. 91 (Am. Geophys Union, Washington, DC, 1995), pp. 85–114
J.C. Alt et al., Earth Planet. Sci. Lett. 327, 50–60 (2012)
D. Andrault et al., Geochem. Perspect. Lett. 6, 5–10 (2018)
K. Armstrong, Science 365, 903–906 (2019). https://doi.org/10.1126/science.aax8376
L.S. Armstrong et al., Geochim. Cosmochim. Acta 171, 283–302 (2015)
Y. Asahara et al., Phys. Earth Planet. Inter. 143–144, 421–432 (2004)
C. Aubaud et al., Earth Planet. Sci. Lett. 235(3–4), 511–527 (2005)
S. Aulbach, V. Stagno, Geology 44, 751–754 (2016). https://doi.org/10.1130/G38070.1
S. Aulbach, M. Massuyeau, F. Gaillard, Lithos 268–271, 364–382 (2017)
C. Avril et al., Meteorit. Planet. Sci. 48, 1415–1426 (2013)
D. Baker, R. Morretti, Rev. Mineral. Geochem. 73(1), 167–213 (2011). https://doi.org/10.2138/rmg.2011.73.7
M.D. Ballmer et al., Nat. Geosci. 10(3), 236 (2017)
J.A. Barrat et al., Geochim. Cosmochim. Acta 75, 3839–3852 (2011)
J.A. Barrat et al., Earth Planet. Sci. Lett. 478, 143–149 (2017)
T.J. Barrett et al., Meteorit. Planet. Sci. 51, 1110–1124 (2016)
H. Behrens, F. Gaillard, Elements 2, 275–280 (2006)
R.A. Berner, The Phanerozoic Carbon Cycle: CO2 and O2 (Oxford University Press, London, 2003)
A.J. Berry et al., Nature 455, 960–963 (2008). https://doi.org/10.1038/nature07377
A.J. Berry et al., Earth Planet. Sci. Lett. 483, 114–123 (2018)
J.P. Bibring et al., Science 312, 400–404 (2006)
J. Blichert-Toft et al., Lithos 37, 97–111 (1996)
J. Blundy et al., Nature 349, 321–324 (1991). https://doi.org/10.1038/349321a0
A. Boujibar et al., Earth Planet. Sci. Lett. 391, 42–54 (2014)
P.V. Brady, S.R. Gíslason, Geochim. Cosmochim. Acta 61, 965–973 (1997)
D. Breuer, in Solar System. Landolt-Börnstein, New Series VI/4B (Springer, Berlin, 2009), pp. 323–344
M. Brounce et al., Proc. Natl. Acad. Sci. USA 114, 8997–9002 (2017)
A. Burgisser et al., Comput. Geosci. 79, 11–14 (2015)
M. Burton et al., in Carbon in Earth. Reviews in Mineralogy and Geochemistry, ed. by R.M. Hazen, A.P. Jones, J.A. Baross (Mineralogical Society of America, Chantilly, 2013), pp. 323–354
V. Busigny et al., Earth Planet. Sci. Lett. 215, 27–42 (2003)
R.A. Cabral et al., Geochem. Geophys. Geosyst. 15, 4445–4467 (2014). https://doi.org/10.1002/2014GC005473
D. Canil, Nature 389, 842–845 (1997)
D. Canil, Earth Planet. Sci. Lett. 195, 75–90 (2002)
C. Cartier et al., Nat. Geosci. 7, 573–576 (2014). https://doi.org/10.1038/ngeo2195
P. Cartigny, F. Pineau, C. Aubaud, M. Javoy, Earth Planet. Sci. Lett. 265, 672–685 (2008)
D.C. Catling, J.F. Kasting, (Cambridge University Press, Cambridge, 2017)
D.C. Catling et al., Astrobiology 5, 415–438 (2005). https://doi.org/10.1089/ast.2005.5.415
N. Chabot, C.B. Agee, Geochim. Cosmochim. Acta 67, 2077–2091 (2003)
B. Charlier, O. Namur, Elements 15, 9–14 (2019)
H. Chi et al., Geochim. Cosmochim. Acta 139, 447–471 (2014)
D.M. Christie et al., Earth Planet. Sci. Lett. 79, 397–411 (1986)
R.N. Clayton, T.K. Mayeda, Geochim. Cosmochim. Acta 63, 2089–2104 (1999)
R.N. Clayton et al., J. Geophys. Res. 89, C245–C249 (1984)
L.I. Cleeves et al., Science 345, 1590–1593 (2014)
V. Clesi et al., Sci. Adv. 4, e1701876 (2018)
C.S. Cockell et al., Astrobiology 16(1), 89–117 (2016). https://doi.org/10.1089/ast.2015.1295
E. Cottrell, K.A. Kelley, Earth Planet. Sci. Lett. 305, 270–278 (2011)
E. Cottrell, K.A. Kelley, Science 340, 1314–1317 (2013)
C. Dalou et al., Geochim. Cosmochim. Acta 265, 32–47 (2019a)
C. Dalou et al., Proc. Natl. Acad. Sci. 116(29), 14485–14494 (2019b)
L.V. Danyushevsky et al., J. Petrol. 41(8), 1329–1364 (2000)
R. Dasgupta, Rev. Mineral. Geochem. 75, 183–229 (2013)
R. Dasgupta, D.S. Grewal, in Deep Carbon: Past to Present (Cambridge University Press, Cambridge, 2019)
R. Dasgupta et al., Geochim. Cosmochim. Acta 102, 191–212 (2013)
N. Dauphas, Nature 541(15), 521–524 (2017)
N. Dauphas, A. Morbidelli, in Treatise on Geochemistry 2nd edn. (2014), pp. 1–35. https://doi.org/10.1016/B978-0-08-095975-7.01301-2
G.F. Davies, Geology 20, 963–966 (1992)
J.M. De Moor et al., Earth Planet. Sci. Lett. 361, 379–390 (2014). https://doi.org/10.1016/j.epsl.2012.11.006
M. Debure et al., Sci. Rep. 9, 11720 (2019). https://doi.org/10.1038/s41598-019-48138-9
V. Dehant et al., Space Sci. Rev. (2019). https://doi.org/10.1007/s11214-019-0608-8
J.W. Delano, Orig. Life Evol. Biosph. 31, 311–341 (2001)
S. Ding et al., Earth Planet. Sci. Lett. 409, 157–167 (2015)
K.E. Donnelly et al., Earth Planet. Sci. Lett. 226, 347–366 (2004)
C. Dorn et al., Outgassing on stagnant-lid super-Earths. Astron. Astrophys. 614, A18 (2018)
M.J. Drake, Meteorit. Planet. Sci. 36, 501–513 (2001)
M.S. Duncan et al., Earth Planet. Sci. Lett. 466, 115–128 (2017). https://doi.org/10.1016/j.epsl.2017.03.008
M. Edmonds, T.M. Gerlach, Geology 35, 751–754 (2007)
C.S. Edwards, B.L. Ehlmann, Geology 43, 863–866 (2015). https://doi.org/10.1130/G36983.1
P.H. Edwards et al., Meteorit. Planet. Sci. 52, 2391–2410 (2017). https://doi.org/10.1111/maps.12953
J. Eguchi et al., Nat. Geosci. 13, 71–76 (2020). https://doi.org/10.1038/s41561-019-0492-6
B.L. Ehlmann et al., Geophys. Res. Lett. 37, L06201 (2010). https://doi.org/10.1029/2010GL042596
L.T. Elkins-Tanton, Earth Planet. Sci. Lett. 271(1–4), 181–191 (2008)
L.T. Elkins-Tanton, Annu. Rev. Earth Planet. Sci. 40, 113–139 (2012). https://doi.org/10.1146/annurev-earth-042711-105503
L.T. Elkins-Tanton, T.L. Grove, Earth Planet. Sci. Lett. 307, 173–179 (2011)
T. Encrenaz et al., Astron. Astrophys. 623, A70 (2019)
B. Fegley Jr., K. Lodders, N.S. Jacobson, Geochemistry (2020). https://doi.org/10.1016/j.chemer.2019.125594
B. Fegley, L.K. Schaefer, Treatise on Geochemistry, vol. 3 (2012). Chapter 13
J. Filiberto, A.H. Treiman, Geology 37, 1087–1090 (2009)
J. Filiberto et al., Meteorit. Planet. Sci. (2016). https://doi.org/10.1111/maps.12680
J. Filiberto et al., in Volatiles in the Martian Crust, ed. by J. Filiberto, S.P. Schwenser (Elsevier, Amsterdam, 2019), pp. 13–34. Chapter 2
T.P. Fischer et al., Science 297, 1154–1157 (2002)
T.R. Fischer et al., Proc. Natl. Acad. Sci. 117(16), 8743–8749 (2020)
M. Foerster et al., Partitioning of nitrogen during melting and recycling in subduction zones and the evolution of atmospheric nitrogen. Chem. Geol. 525, 334–342 (2019). https://doi.org/10.1016/j.chemgeo.2019.07.042
B.J. Foley, Astrophys. J. 812, 36 (2015)
B.R. Frost, Introduction to oxygen fugacity and its petrologic importance, in Oxide Minerals: Petrologic and Magnetic Significance, ed. by D.H. Lindsley. Rev. Mineral., vol. 25 (Mineral. Soc. Am., Washington, DC, 1991), pp. 1–9. 508 pp.
D.J. Frost, C.A. McCammon, Annu. Rev. Earth Planet. Sci. 36, 389–420 (2008)
D.J. Frost et al., Nature 428, 409–412 (2004)
E. Füri, B. Marty, Nat. Geosci. 8, 515–522 (2015)
M. Gaborieau et al., Chem. Geol. 547, 119646 (2020)
F. Gaillard, B. Scaillet, Earth Planet. Sci. Lett. 403, 307–316 (2014)
F. Gaillard et al., Nature 478, 229 (2011)
F. Gaillard et al., Space Sci. Rev. 174, 251–300 (2013)
F. Gaillard et al., Chem. Geol. 418, 217–233 (2015)
F. Gaillard et al., in Deep Carbon: Past to Present, ed. by B. Orcutt, I. Daniel, R. Dasgupta (Cambridge University Press, Cambridge, 2019). https://doi.org/10.1017/9781108677950. Chapter 7
C. Ganino, N.T. Arndt, Geology 37(4), 323–326 (2009)
R. Greenwood et al., Nature 435, 916–918 (2005). https://doi.org/10.1038/nature03612
C. Greenwood et al., Sci. Adv. 4, eaao5928 (2018)
D.S. Grewal et al., Geochim. Cosmochim. Acta 251, 87–115 (2019a)
D.S. Grewal et al., Sci. Adv. 5, eaau3669 (2019b)
I. Halevy et al., Science 318, 1903 (2007). https://doi.org/10.1126/science.1147039
A.N. Halliday, Geochim. Cosmochim. Acta 105, 146–171 (2013)
L.J. Hallis et al., Science 350, 795–797 (2015)
T. Hammouda, S. Keshav, Chem. Geol. 418, 171–188 (2015)
J.J. Hanley, K.T. Koga, in The Role of Halogens in Terrestrial and Extraterrestrial Geochemical Processes, ed. by D.E. Harlov, L. Aranovich. Springer Geochemistry (2018). https://doi.org/10.1007/978-3-319-61667-4_2
S. Haruka, H. Kurokawa, H. Genda, Impact degassing and atmospheric erosion on Venus, Earth, and Mars during the late accretion. Icarus 317, 48–58 (2019)
E. Hauber et al., Geophys. Res. Lett. 38, 10 (2011)
E.H. Hauri et al., Science 333, 213–215 (2011)
E.H. Hauri et al., Earth Planet. Sci. Lett. 409, 252–264 (2015)
C. Hémond et al., Origin of MORB enrichment and relative trace element compatibilities along the Mid-Atlantic Ridge between \(10^{\circ}\) and \(24^{\circ}~\text{N}\). Geochem. Geophys. Geosyst. 7, Q12010 (2006). https://doi.org/10.1029/2006GC001317
C.D.K. Herd, Rev. Mineral. Geochem. 68, 527–553 (2008)
P.J. Heron, Geol. Soc. (Lond.) Spec. Publ. 470, 87–103 (2018). https://doi.org/10.1144/SP470-2018-97
C. Herzberg et al., Earth Planet. Sci. Lett. 292, 79–88 (2010)
S. Hier-Majumder, M.M. Hirschmann, Geochem. Geophys. Geosyst. 18, 3078–3092 (2017)
N. Hirano et al., Science 313, 1426–1428 (2006)
M.M. Hirschmann, Geochem. Geophys. Geosyst. 1, 1042 (2000). https://doi.org/10.1029/2000GC000070
M.M. Hirschmann, Science 325, 545–546 (2009). https://doi.org/10.1126/science.1176882
M.M. Hirschmann, Phys. Earth Planet. Inter. 179, 60–71 (2010)
M.M. Hirschmann, Am. Mineral. 101, 540–553 (2016)
M.M. Hirschmann, A.C. Withers, Earth Planet. Sci. Lett. 270, 147–155 (2008). https://doi.org/10.1016/j.epsl.2008.03.034
H.D. Holland, Geochim. Cosmochim. Acta 66, 3811–3826 (2002)
J.R. Holloway et al., Eur. J. Mineral. 4(1), 105–114 (1992)
D. Höning et al., Planet. Space Sci. 98, 5–13 (2014)
C. Houser, Earth Planet. Sci. Lett. 448, 94–101 (2016)
R. Hu et al., Nat. Commun. 6, 10003 (2015)
H. Hui et al., Nat. Geosci. 6, 177–180 (2013)
G. Iacono-Marziano et al., Geology 37, 319–322 (2009). https://doi.org/10.1130/G25446A
G. Iacono-Marziano et al., Earth Planet. Sci. Lett. 357–358, 319–326 (2012a)
G. Iacono-Marziano et al., Geochim. Cosmochim. Acta (2012b). https://doi.org/10.1016/j.gca.2012.08.035
G. Iacono-Marziano et al., Earth Planet. Sci. Lett. 357–358, 308–318 (2012c)
R. Iizuka-Oku et al., Nat. Commun. 8, 14096 (2017). https://doi.org/10.1038/ncomms14096
M. Javoy et al., Earth Planet. Sci. Lett. 293, 259–268 (2010)
S.R. Jurewicz et al., Earth Planet. Sci. Lett. 132, 183 (1995)
A.A. Kadik et al., Geochem. Int. 49, 429–438 (2011)
A.A. Kadik et al., Geochem. Int. 52, 707–725 (2014)
A.A. Kadik et al., Geochem. Int. 55, 151–162 (2017)
G.W. Kallemeyn et al., Geochim. Cosmochim. Acta 58, 2873–2888 (1994)
B.S. Kamber et al., Earth Planet. Sci. Lett. 240, 276–290 (2005)
S-I. Karato, Earth Planet. Sci. Lett. 301, 413–423 (2011)
J.F. Kasting, Icarus 74, 472–494 (1988). https://doi.org/10.1016/0019-1035(88)90116-9
J.F. Kasting et al., J. Geol. 101(2), 245–257 (1993a)
J.F. Kasting et al., Icarus 101(1), 108–128 (1993b)
R.F. Katz et al., Geochem. Geophys. Geosyst. 4, 9 (2003)
H. Kawakatsu, S. Watada, Science 316, 1468–1471 (2007)
K. Keil, in Asteroids III, ed. by W.F. Bottke, A. Cellino, P. Paolicchi, R.P. Binzel (Univ. of Arizona Press, Tucson, 2002), pp. 573–584
P.B. Kelemen, C.E. Manning, Proc. Natl. Acad. Sci. (2015). https://doi.org/10.1073/pnas.1507889112
B. Keller, B. Schoene, Nature 485, 490–493 (2012). https://doi.org/10.1038/nature11024
J.M. Keller et al., Equatorial and mid-latitude distribution of chlorine measured by Mars Odyssey GRS. J. Geophys. Res., Planets 111, E03S08 (2006)
K.A. Kelley, E. Cottrell, Science 325(5940), 605–607 (2009)
H. Keppler, G. Golabek, Geochem. Perspect. Lett. 11, 12–17 (2019). https://doi.org/10.7185/geochemlet.1918
D.M. Kerrick, Rev. Geophys. 39, 565–585 (2001)
E.S. Kite et al., Astrophys. J. 700(2), 1732 (2009)
E.S. Kite et al., Astrophys. J. Lett. 887, L33 (2019). https://doi.org/10.3847/2041-8213/ab59d9
J. Korenaga, Philos. Trans. R. Soc., Math. Phys. Eng. Sci. 376(2132), 20170408 (2018). https://doi.org/10.1098/rsta.2017.0408
J. Krissansen-Totton, D.C. Catling, Nat. Commun. 8, 15423 (2017)
T.S. Kruijer et al., Proc. Natl. Acad. Sci. USA 114, 6712–6716 (2017)
J. Labidi et al., Nature 580, 367–371 (2020). https://doi.org/10.1038/s41586-020-2173-4
H. Lammer et al., Space Sci. Rev. 139, 399–436 (2008). https://doi.org/10.1007/s11214-008-9413-5
H. Lammer et al., Origin and evolution of the atmospheres of early Venus, Earth and Mars. Astron. Astrophys. Rev. 26, 1–73 (2018)
H. Lammer et al., Constraining the early evolution of Venus and Earth through atmospheric Ar, Ne isotope and bulk K/U ratios. Icarus 339, 113551 (2020)
M. Laumonier et al., Earth Planet. Sci. Lett. 457, 173–180 (2017)
C. Le Guillou et al., Geochim. Cosmochim. Acta 420, 162–173 (2015)
T. Lebrun et al., J. Geophys. Res. E 118, 1155–1176 (2013)
C. Lécuyer, Y. Ricard, Earth Planet. Sci. Lett. 165(2), 197–211 (1999)
H. Lee et al., Nat. Geosci. 9, 145–149 (2016). https://doi.org/10.1038/ngeo2622
M. LeVoyer et al., Nat. Commun. 8, 14062 (2017)
L. Li, C.B. Agee, Geophys. Res. Lett. 28, 81–84 (2001)
Z.X.A. Li, C.T.A. Lee, Chem. Geol. 235, 161–185 (2006). https://doi.org/10.1016/j.chemgeo.2006.06.011
Y.R. Li et al., Earth Planet. Sci. Lett. 415, 54–66 (2015)
Y. Li et al., Nat. Geosci. 9, 781–785 (2016)
Y. Li et al., Nat. Geosci. 13, 453–458 (2020). https://doi.org/10.1038/s41561-020-0578-1
Y. Lin et al., Nat. Geosci. 10, 14–18 (2017). https://doi.org/10.1038/ngeo2845
M-A. Longpré et al., Earth Planet. Sci. Lett. 460, 268–280 (2016). https://doi.org/10.1016/j.epsl.2016.11.043
T. Lyons, B.C. Gill, Elements 6, 93–99 (2010). https://doi.org/10.2113/gselements.6.2.93
V. Malavergne et al., Earth Planet. Sci. Lett. 394, 186–197 (2014)
V. Malavergne et al., Icarus 321, 473–485 (2019)
E. Marcq et al., Nat. Geosci. 6(1), 25–28 (2013)
Y. Marrocchi, Earth Planet. Sci. Lett. 482, 23–32 (2018)
B. Marty, Earth Planet. Sci. Lett. 313–314, 56–66 (2012). https://doi.org/10.1016/j.epsl.2011.10.040
B. Marty et al., Earth Planet. Sci. Lett. 441, 91–102 (2016)
H. Massol et al., Space Sci. Rev. 205(1), 153–211 (2016)
F.M. McCubbin et al., Proc. Natl. Acad. Sci. 107, 11223–11228 (2010)
F.M. McCubbin et al., Geology 40, 683–686 (2012a)
F.M. McCubbin et al., Geophys. Res. Lett. 39, L09202 (2012b). https://doi.org/10.1029/2012GL051711
F.M. McCubbin et al., Am. Mineral. 100(8–9), 1668–1707 (2015)
W.F. McDonough, in The Mantle and Core, ed. by R.W. Carlson. Treatise on Geochemistry, vol. 2 (Elsevier-Pergamon, Oxford, 2003), pp. 547–568
W. McEwen et al., Icarus 135, 181–219 (1998)
D. McKenzie et al., Earth Planet. Sci. Lett. 233, 337–349 (2005)
H.Y. McSween et al., Meteorit. Planet. Sci. 48, 2090–2104 (2013)
N. Metrich, P.J. Wallace, Volatile abundances in basaltic magmas and their degassing paths tracked by melt inclusions. Rev. Mineral. Geochem. 69(1), 363–402 (2008). https://doi.org/10.2138/rmg.2008.69.10
N. Metrich et al., Earth Planet. Sci. Lett. 167, 1–14 (1999)
S. Mikhail, D.A. Sverjensky, Nat. Geosci. 7(11), 816 (2014)
W.G. Miller et al., Earth Planet. Sci. Lett. 523, 115699 (2019)
N.L. Mironov, M.V. Portnyagin, Petrology 26, 145–166 (2018). https://doi.org/10.1134/S0869591118020030
E. Mitchell et al., Geochem. Geophys. Geosyst. 11, Q02X11 (2010). https://doi.org/10.1029/2009GC002783
A. Morbidelli et al., Icarus 267, 368–376 (2016)
M. Moreira, Geochem. Perspect. 2, 244–395 (2013)
R. Moretti, P. Papale, Chem. Geol. 213, 265–280 (2004)
Y. Moussallam et al., Earth Planet. Sci. Lett. 393, 200–209 (2014). https://doi.org/10.1016/j.epsl.2014.02.055
B. Mysen, Nitrogen in the Earth: abundance and transport. Prog. Earth Planet. Sci. 6, 38 (2019). https://doi.org/10.1186/s40645-019-0286-x
O. Namur et al., Earth Planet. Sci. Lett. 448, 102–114 (2016)
J.A.M. Nanne et al., Earth Planet. Sci. Lett. 511, 44–54 (2019)
A. Nikolaou et al., Astrophys. J. 875, 11 (2019). https://doi.org/10.3847/1538-4357/ab08ed
L.R. Nittler et al., in Mercury: The View After MESSENGER, ed. by S.C. Solomon, L.R. Nittler, B.J. Anderson. Cambridge Planetary Science Series (2017). 58 pp.
L. Noack et al., Planet. Space Sci. 98, 14–29 (2014)
L. Noack et al., Int. J. Adv. Syst. Meas. 9(1/2), 66–76 (2016)
L. Noack et al., Volcanism and outgassing of stagnant-lid planets: implications for the habitable zone. Phys. Earth Planet. Inter. 269, 40–57 (2017)
T. Okuchi, Science 278, 1781–1784 (1997)
S. Okumura, N. Hirano, Geology 41, 1167–1170 (2013)
P.L. Olson, Z.D. Sharp, Phys. Earth Planet. Inter. 294, 106294 (2019)
S.L. Olson et al., Earth Planet. Sci. Lett. 506, 417–427 (2019)
H.St.C. O’Neill, M.I. Pownceby, Contrib. Mineral. Petrol. 114, 296–314 (1993)
B.N. Orcutt et al. (eds.), Deep Carbon: Past to Present (Cambridge University Press, Cambridge, 2019)
G. Ortenzi et al., Sci. Rep. 10, 10907 (2020). https://doi.org/10.1038/s41598-020-67751-7
K. Ozaki et al., Geobiology 17, 3–11 (2019)
R.I.T.A. Parai, S. Mukhopadhyay, Earth Planet. Sci. Lett. 317, 396–406 (2012)
L. Piani et al., Earth’s water may have been inherited from material similar to enstatite chondrite meteorites. Science 369, 1110–1113 (2020). https://doi.org/10.1126/science.aba1948
R.T. Pierrehumbert, Nat. Geosci. 6, 81–83 (2013). https://doi.org/10.1038/ngeo1711
T. Plank et al., Earth Planet. Sci. Lett. 364, 168–179 (2013)
A.C. Plesa et al., Geophys. Res. Lett. 45(22), 12,198–12,209 (2018)
J.P. Poirier, Phys. Earth Planet. Inter. 85, 319–337 (1994)
N. Potts et al., in 46th LPSC, Abstract #1372 (2015)
T.H. Prettyman et al., Icarus 259, 39–52 (2015)
G. Prouteau, B. Scaillet, J. Petrol. 54, 183–213 (2013). https://doi.org/10.1093/petrology/egs067
S.N. Raymond, A. Izidoro, Icarus 297, 134–148 (2017)
C.A. Raymond et al., Nat. Astron. 4, 741–747 (2020). https://doi.org/10.1038/s41550-020-1168-2
K. Righter et al., Meteorit. Planet. Sci. 43(10), 1709–1723 (2008)
K. Righter et al., Am. Mineral. 101, 1928–1942 (2016)
A. Rivoldini, T. Van Hoolst, O. Verhoeven, A. Mocquet, V. Dehant, Icarus 213, 451–472 (2011). https://doi.org/10.1016/j.icarus.2011.03.024
H. Rizo et al., Geochem. Perspect. Lett. 11, 6–11 (2019)
F. Robert, Space Sci. Rev. 106, 87–101 (2003)
A. Rohrbach, M.W. Schmidt, Nature 472, 209–212 (2011)
L. Rose-Weston et al., Geochim. Cosmochim. Acta 73, 4598–4616 (2009)
M. Roskosz et al., Geochim. Cosmochim. Acta 70, 2902–2918 (2006)
M. Roskosz et al., Geochim. Cosmochim. Acta 121, 15–28 (2013)
A.B. Rozel et al., Nature 545(7654), 332–335 (2017)
D.C. Rubie et al., Earth Planet. Sci. Lett. 301, 31–42 (2011)
D.C. Rubie et al., Icarus 248, 89–108 (2015)
A.E. Rubin, B.-G. Choi, Earth Moon Planets 105, 41–53 (2009). https://doi.org/10.1007/s11038-009-9316-9
A.E. Rubin, C. Ma, Meteoritic minerals and their origins. Chem. Erde 77, 325–385 (2017). https://doi.org/10.1016/j.chemer.2017.01.005
A.E. Saal et al., Nature 419, 451–455 (2002)
A.E. Saal et al., Nature 454, 192–195 (2008)
A.R. Sarafian et al., Meteorit. Planet. Sci. 48, 2135–2154 (2013)
A.R. Sarafian et al., Science 346, 623–626 (2014)
A.R. Sarafian et al., Philos. Trans. R. Soc., Math. Phys. Eng. Sci. 375, 20160209 (2017a)
A.R. Sarafian et al., Earth Planet. Sci. Lett. 459, 311–319 (2017b)
A.R. Sarafian et al., Geochim. Cosmochim. Acta 266, 568–581 (2019)
L. Schaefer, L. Elkins-Tanton, Philos. Trans. R. Soc., Math. Phys. Eng. Sci. 376(2132), 20180109 (2018)
L. Schaefer, B. Fegley, Icarus 186, 462–483 (2007)
H.E. Schlichting, S. Mukhopadhyay, Space Sci. Rev. 214, 34 (2018)
N. Schmerr, Science 380, 1480–1483 (2012)
M.W. Schmidt, S. Poli, in Treatise on Geochemistry, vol. 3, ed. by D.H. Heinrich, K.K. Turekian (Pergamon, Oxford, 2003), pp. 567–591
G. Schubert, D.T. Sandwell, Icarus 117, 173–196 (1995)
S. Seager, D. Drake, Annu. Rev. Astron. Astrophys. 48(1), 631–672 (2010). https://doi.org/10.1146/annurev-astro-081309-130837
A. Shahar et al., Science 352, 580–582 (2016)
W.C. Shanks III et al., Geochim. Cosmochim. Acta 45(11), 1977–1995 (1981)
Z.D. Sharp et al., Science 329, 1050–1053 (2010)
D. Sifré et al., Nature 509, 81–85 (2014)
N.H. Sleep, K. Zahnle, J. Geophys. Res., Planets 106(E1), 1373–1399 (2001)
M. Smit, K. Mezger, Nat. Geosci. 10, 788–792 (2017). https://doi.org/10.1038/ngeo3030
S.E. Smrekar et al., Science 328(5978), 605–608 (2010)
P.A. Sossi, B. Fegley, Rev. Mineral. Geochem. 84(1), 393–459 (2018). https://doi.org/10.2138/rmg.2018.84.11
I.M. Speelmanns et al., Geophys. Res. Lett. 45, 7434–7443 (2018)
J.R. Spencer et al., Science 288, 1208–1210 (2000)
T. Spohn et al., in Encyclopedia of the Solar System, ed. by T. Spohn, D. Breuer, T.V. Johnson (Elsevier, Amsterdam, 2014). https://doi.org/10.1016/C2010-0-67309-3
V. Stagno et al., Nature 493, 84–88 (2013)
B.D. Stanley et al., Geochim. Cosmochim. Acta 129, 54–76 (2014)
A. Steele et al., Am. Mineral. 97, 1256–1259 (2012)
D.A. Stolper, C.E. Bucholz, Proc. Natl. Acad. Sci. 116(18), 8746–8755 (2019). https://doi.org/10.1073/pnas.1821847116
T-A. Suer et al., Earth Planet. Sci. Lett. 469, 84–97 (2017)
A. Suzuki et al., Science 269, 216–218 (1995). https://doi.org/10.1126/science.269.5221.216
R.B. Symonds et al., in Volatiles in Magmas, vol. 30, ed. by M.R. Carroll, J.R. Holloway (Mineralogical Society of America, Chantilly, 1994), pp. 1–60
M. Tang et al., Science 351(6271), 372–375 (2016). https://doi.org/10.1126/science.aad5513
S. Tappe et al., Earth Planet. Sci. Lett. 484, 1–14 (2018)
R.J. Thomas, D.A. Rothery, Volcanism on Mercury. Elements 15(1), 27–32 (2019)
D. Trail et al., Nature 480, 79–82 (2011)
J.M. Tucker, S. Mukhopadhyay, Evidence for multiple magma ocean outgassing and atmospheric loss episodes from mantle noble gases. Earth Planet. Sci. Lett. 393, 254–265 (2014)
J.M. Tucker et al., Earth Planet. Sci. Lett. 496, 108–119 (2018)
L.G. Vacher et al., Astrophys. J. Lett. 826, 1–6 (2016)
L.G. Vacher et al., Geochim. Cosmochim. Acta 281, 53–66 (2020). https://doi.org/10.1016/j.gca/2020.05.007
J. Van Hunen, J.F. Moyen, Annu. Rev. Earth Planet. Sci. 40, 195–219 (2012)
J. Wade, B.J. Wood, Earth Planet. Sci. Lett. 236, 78–95 (2005)
P. Wallace, J. Volcanol. Geotherm. Res. 140, 217–240 (2005)
M.E. Wallace, D.H. Green, Nature 335, 343–346 (1988)
P.H. Warren et al., Earth Planet. Sci. Lett. 311, 93–100 (2011)
J.T. Wasson, G.W. Kallemeyn, Philos. Trans. R. Soc. Lond. Ser. A 325, 535–544 (1988)
H.K. Wehrmann et al., Geochem. Geophys. Geosyst. 12, Q06003 (2011). https://doi.org/10.1029/2010GC003412
S.Z. Weider et al., Geophys. Res. Lett. 43, 3653–3661 (2016)
C. Werner et al., Carbon dioxide emissions from subaerial volcanic regions, in Deep Carbon: Past to Present, ed. by B. Orcutt, I. Daniel, R. Dasgupta (Cambridge University Press, Cambridge, 2019). https://doi.org/10.1017/9781108677950
D.T. Wetzel et al., Nat. Geosci. 8, 755–758 (2015)
D.A. Williams, R.R. Howell, in Io After Galileo. A New View of Jupiter’s Volcanic Moon, ed. by R.M. Lopes, J.R. Spencer. Springer Praxis Books Series, vol. 342 (2002), pp. 133–161. Chapter 7
B.J. Wood et al., Nature 467, E6–E7 (2010). https://doi.org/10.1038/nature09484
R.D. Wordsworth, Earth Planet. Sci. Lett. 447, 103–111 (2016)
R.D. Wordsworth et al., Icarus 222, 1–19 (2013). https://doi.org/10.1016/j.icarus.2012.09.036
X. Yang et al., Earth Planet. Sci. Lett. 393, 210–219 (2014)
K. Zahnle et al., Earth’s earliest atmospheres, in The Origin of Cellular Life, ed. by D. Deamer, J.W. Szostak (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 2010)
A.L. Zerkle, S. Mikhail, The geobiological nitrogen cycle: from microbes to the mantle. Geobiology 15, 343–352 (2017). https://doi.org/10.1111/gbi.12228
Y. Zhang, Q.Z. Yin, Proc. Natl. Acad. Sci. USA 109, 16579–16583 (2012)
M.Y. Zolotov, B. Fegley Jr., Icarus 132, 431–434 (1998)
M.Y. Zolotov, B. Fegley Jr., Icarus 141, 40–52 (1999)
M.Y. Zolotov et al., J. Geophys. Res., Planets 118, 138–146 (2013)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Understanding the Diversity of Planetary Atmospheres
Edited by François Forget, Oleg Korablev, Julia Venturini, Takeshi Imamura, Helmut Lammer and Michel Blanc
Rights and permissions
About this article

Cite this article
Gaillard, F., Bouhifd, M.A., Füri, E. et al. The Diverse Planetary Ingassing/Outgassing Paths Produced over Billions of Years of Magmatic Activity. Space Sci Rev 217, 22 (2021). https://doi.org/10.1007/s11214-021-00802-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11214-021-00802-1