
Abstract
A series of novel imidazopyridine derivatives of indole has been synthesized. All the synthesized derivatives were evaluated for their antiproliferative activity against A-549, T-47D, Hep-G2 and MCF-7 human cancer cell lines. The results demonstrated that some of these derivatives exhibited moderate to excellent cytotoxic activities. Compounds 7a having a cyclohexyl ring substituted to the second amine of imidazopyridyl moiety and phenyl ring of the C-2 indole ring and 7f with a para-methylphenyl ring at the same position exhibited the highest activity against the A-594 cell line with IC50 of 11.48 μM and 10.66 μM, respectively. The results indicate that compounds 7a and 7f are more cytotoxic towards cancer cell lines compared with etoposide in vitro. In addition, compounds, 7d and 7j showed the most potent activity against Hep-G2, equal to etoposide as the standard drug. Also, most of the compounds were inactive against the T-47D and MCF-7 cell lines. The morphological analysis by the acridine orange/ethidium bromide double-staining test and flow cytometry analysis indicated that compounds 7a and 7f induced apoptosis in A-549 cells. Furthermore, in silico and in vitro results of the synthesized compounds showed good correlation with each other. Molecular docking results of the compounds of the 7a–k series with the cyclohexyl ring substituted to the second amine of the imidazopyridyl moiety compared with the 7l–t members with the t-butyl group at the same position confirmed the effect of the higher lipophilicity on hydrophobic interactions with the studied enzymes. Moreover, all the compounds showed higher affinity to tubulin than topoisomerase IIα enzyme.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is a complex pathological disorder described by a high proliferative index and the spread of abnormal cells from their site of origin [1]. Cancer is the leading origin of death in developed countries and the second leading cause of death in developing countries [2].
Anticancer drug discovery has been strongly focused on the development of drugs intended to act against a specific target with high potency and selectivity. Both topoisomerase II and microtubule are significant anticancer targets, and their respective inhibitors have been extensively used for cancer therapy [3,4,5]. Microtubules are key components of the cell structure which take part in a varied number of essential cellular functions. In mitosis, the cytoplasmic microtubule is disrupted and reformed as a spindle consisting of large numbers of short microtubules that surround each centrosome [6]. It should be mentioned that this process is significant for the suitable attachment and movement of chromosomes during numerous steps of the mitotic phase. Therefore, inhibition of microtubule formation leads to mitotic arrest and promotes vascular disruption which eventually leads to cell death [7].
Topoisomerases are important cellular enzymes essential for cell proliferation through solving topological problems in the process of DNA replication. Topoisomerase II produces the relaxation of DNA double helices by scissoring and religating the two strands. Because of the critical role of this enzyme for the cell proliferation process, topoisomerase has been one of the main targets in the anticancer drug development field [8]. Topoisomerase II inhibitors and microtubule inhibitors are often used in combination for cancer therapy. Many therapeutic treatments contain vincristine, vinblastine, paclitaxel or docetaxel in combination with doxorubicin or etoposide for the treatment of numerous hematological or solid tumors. These combinations have been reported not only to produce synergistic therapeutic effects but also to reduce nonhematologic toxicities [3]. Heterocyclic compounds are an exceedingly important class of compounds and they have attracted more attention for diverse biological studies [9,10,11].
Indole analogs comprise a group of the most extensively distributed nitrogen-containing heterocycles in nature having biological significance. The indole ring system is present in various marketed drugs. More often, indoles have been used as drugs for diseases related to the CNS, but in current years researchers have been trying to develop indole-based drugs for combating cancer [12,13,14,15,16]. Perchellet et al. synthesized a collection of novel 6,7-annulated-4-substituted indole compounds to discover a scaffold for anticancer activity. They showed that this class of compounds interacts with tubulin to reduce microtubule assembly [17]. A series of 3-amidoindole derivatives reported by Chen et al. have shown moderate inhibitory activity on tubulin polymerization [18]. In addition, a group of researchers have designed some new 2-phenylindole derivatives as tubulin polymerization inhibitors with low micromolar IC50 values which inhibited colchicine binding with a mean value > 70% [19]. Compounds A and B as the most potent derivatives of 3-amidoindole and 2-phenylindole scaffolds, respectively, are illustrated in Fig. 1.

Structures of indole-based and imidazopyridine-based anticancer agents (a–e) and the general structure of the designed compounds
Imidazopyridine is one of the most important fused heterocyclic systems and is known to display a wide range of inhibitory activities for a diverse number of biological targets [20]. This scaffold is also found in some marketed drugs, such as zolpidem and zolimidine [21]. Over the past few years, considerable interest has been devoted to the synthesis and evaluation of the anticancer activity of imidazopyridines [22]. Compounds C (ethyl 6-(5-(phenyl sulfonamide) pyridine-3-yl) imidazo[1,2-a]pyridine-3-carboxylate) [23] and D ((E)-6-chloro-3-((3-(4-fluorophenyl)imidazo [1,5-a]pyridin-1-yl)methylene)indolin-2-one) [24] with promising anticancer activity have been introduced in such research (Fig. 1). Several derivatives of bicyclic N-fused aminoimidazoles have also been reported by Baviskar et al. They introduced compound E (Fig. 1) as a potent topoisomerase IIα catalytic inhibitor which acts via blocking the ATPase binding site of the enzyme without interacting DNA [25].
In the present research, indole and imidazopyridine scaffolds, as effective antiproliferative agents, were subjected to some structural modifications based on a hybridization approach [26]. Hybrid molecules take advantage of affecting different biomolecules, thus they are called multi-target compounds. Three structural motives were considered in the design of these hybrid molecules. First, the indole moiety for the inhibition of tubulin polymerization, and second, the imidazopyridine structure for its inhibitory activity on the ATPase domain of topoisomerase IIα. The third element which was implemented in the designed structures was a substituted phenyl ring that was proved to be efficient in the anticancer activity of compound B. The designed compounds were prepared in the laboratory then evaluated for their in vitro cytotoxic activities. A molecular docking approach against the desired targets was also exploited to confirm the effectiveness of the designed compounds in silico. The general structure of the designed compounds is provided in Fig. 1.
Experimental
Chemistry
All reagents and solvents used in this study are commercially available (from Merck chemical) and were used without further purification. Melting points were determined on a Kofler hot-stage apparatus (Reichert, Vienna, Austria) and uncorrected. Precoated Merck Silica gel 250 µm, F254 TLC aluminium sheets were utilized for thin-layer chromatographic analysis and spots were visualized under UV light at 254 nm. The IR spectra were taken using a Nicolet Magna FT-IR 550 spectrophotometer (potassium bromide disks) and only major peaks are reported in cm−1.
All NMR spectra were recorded on Bruker 500 MHz NMR instruments. Chemical shifts were reported in parts per million (ppm, δ), down-filed from tetramethylsilane coupling constant (J) values presented in Hz with spin multicities given as s (singlet), d (double), t (triplet) and m (multiplet). Purification of the compounds was carried out by silica gel column chromatography (230–400 mesh size) with the indicated eluent. Elemental analyses were carried out by a CHN-Rapid Heraeus elemental analyzer. The results of elemental analyses ©, H, N) were within ± 0.4% of the calculated values. For the ease of NMR assignment, all atoms of the scaffold are numbered by simple, prime and double-prime symbols. It should be clarified that this numbering system is different from the IUPAC numbering method. The numbered scaffold is provided in Scheme 1.

Synthesis of 2-(2-phenyl-1H-indol-3-yl)-imidazo[1,2-a]pyridin-3-amine derivatives (7a–t)
General procedure for the synthesis of 2-aryl-1H-indoles (3a–f)
Appropriate amounts of substituted acetophenone 1 (1 mmol) and phenyl hydrazine 2 (1 mmol) were mixed in ethanol (20 mL), and a few drops of glacial acetic acid were added. The solution was heated under reflux at 80 °C for 1–2 h. The solvent was evaporated in vacuo to give a solid that was added to polyphosphoric acid (30 mL), and the mixture was slowly heated to 120 °C and kept at this temperature for a few hours until the reaction was complete (TLC monitoring). The mixture was allowed to cool and then poured into cold water (50 mL). The acidic solution was neutralized by the slow addition of NaOH (1 M), and the solid precipitate of the crude product was collected. Purification by column chromatography (hexane/ethyl acetate) gave the substituted 2-aryl indoles 3a–t. Indoles 3a–t were all prepared by a recently reported procedure [27].
General procedure for synthesis of 2-arylindole-3-carbaldehydes (4a–f)
Under nitrogen gas, phosphorous oxychloride (10 mmol) was added dropwise to dry dimethylformamide (DMF) (10 mmol) while cooling in an ice bath, and the reaction mixture was stirred for 1 h. A solution of compound 3 (1 mmol) in DMF (50 ml) was added dropwise to the mixture with continuous stirring, which was then heated to 70 °C. The mixture was poured onto ice cold water (200 mL), naturalized with 40% NaOH, and extracted with chloroform. The chloroform extract was washed with water and dried over Na2SO4. The solvent was removed under vacuum. The residue was crystalized from an ethanol/water mixed solvent system [27].
2-Phenyl-1H-indole-3-carbaldehyde (4a)
Cream powder; Mp: 249–250 °C; Yield: 89%; IR (KBr, cm−1): 3144 (NH), 2858 (H–CO), 1625 (C=O), 1455 (C=C). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 7.24 (t, J = 7.9 Hz, 1H, H6), 7.29 (t, J = 7.9 Hz, 1H, H7), 7.51 (d, J = 7.9 Hz, 1H, H8), 7.56–7.62 (m, 3H, H3′,5′,4′), 7.78 (d, J = 9.3 Hz, 2H, H2′,6′), 8.21 (d, J = 7.9 Hz, 1H, H5), 9.96 (s, 1H, CHO), 12.40 (s, 1H, NH-indole). 13C NMR (125 MHz, DMSO-d6) (δ, ppm): 112.1, 113.5, 121.2, 122.5, 123.7, 125.8, 128.9, 129.2, 129.8, 130.1, 135.9, 149.1, 185.5. Anal. Calcd. for C15H11NO: C, 81.43; H, 5.01; N, 6.33. Found: C, 81.53; H, 4.85; N, 6.45.
2-(4-Hydroxyphenyl)-1H-indole-3-carbaldehyde (4b)
Cream powder; Mp: 225–227 °C; Yield: 86%; IR (KBr, cm−1): 3151 (OH), 3074 (NH), 2862 (H–CO), 1619 (C=O), 1455 (C=C), 1177 (C–O) 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 6.97 (d, J = 7.6 Hz, 2H, H3′,5′), 7.19–7.26 (m, 2H, H6,7), 7.45 (d, J = 7.5 Hz, 1H, H8), 7.60 (d, J = 7.6 Hz, 2H, H2′,6′), 8.17 (d, J = 7.5 Hz, 1H, H5), 9.92 (s, 1H, CHO), 10.04 (s, 1H, OH), 12.20 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 111.6, 112.7, 115.8, 120.4, 120.9, 122.2, 123.3, 125.9, 131.2, 135.8, 149.8, 159.1, 185.5. Anal. Calcd. for C15H11NO2: C, 75.94; H, 4.67; N, 5.93. Found: C, 76.04; H, 4.71; N, 5.72.
2-(4-Methoxyphenyl)-1H-indole-3-carbaldehyde (4c)
Cream powder, Mp: 207–209 °C; Yield: 90%. IR (KBr, cm−1): 3147 (NH), 2837 (H–CO), 1621 (C=O), 1454 (C=C), 1179 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 3.87 (s, 3H, OCH3), 7.16 (d, J = 8.7 Hz, 2H, H3′,5′), 7.20–7.28 (m, 2H, H6,7), 7.47 (d, J = 7.6 Hz, 1H, H8), 7.72 (d, J = 8.7 Hz, 2H, H2′,6′), 8.18 (d, J = 7.6 Hz, 1H, H5), 9.94 (s, 1H, CHO), 12.28 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 55.8, 111.8, 113.2, 114.7, 120.9, 122.0, 122.4, 123.7, 125.9, 131.4, 135.9, 149.3, 160.6, 185.4. Anal. Calcd. for C16H13NO2: C, 76.48; H, 5.21; N, 5.57. Found: C, 76.60; H, 5.29; N, 5.48.
2-(4-Chlorophenyl)-1H-indole-3-carbaldehyde (4d)
Cream powder, Mp > 250 °C; Yield: 88%. IR (KBr, cm−1): 3168 (NH), 2866 (H–CO), 1625 (C=O), 1450 (C=C). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 7.25 (t, J = 7.6 Hz, 1H, H6), 7.30 (t, J = 7.6 Hz, 1H, H7), 7.51 (d, J = 7.6 Hz, 1H, H8), 7.74 (d, J = 8.1 Hz, 2H, H3′,5′), 7.80 (d, J = 8.1 Hz, 2H, H2′,6′), 8.21 (d, J = 7.6 Hz, 1H, H5), 9.95 (s, 1H, CHO), 12.46 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 112.1, 113.6, 121.1, 122.6, 123.5, 123.9, 125.8, 129.0, 131.8, 132.0, 135.9, 147.4, 185.4. Anal. Calcd. for C15H10ClNO: C, 70.46; H, 3.94; N, 5.48. Found: C, 70.69; H, 3.85; N, 5.51.
2-(4-Bromophenyl)-1H-indole-3-carbaldehyde (4e)
Cream powder, Mp > 250 °C; Yield: 92%. IR (KBr, cm−1): 3167 (NH), 2840 (H–CO), 1623 (C=O), 1452 (C=C). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 7.25 (t, J = 7.6 Hz, 1H, H6), 7.30 (t, J = 7.6 Hz, 1H, H7), 7.51 (d, J = 7.6 Hz, 1H, H8), 7.74 (d, J = 8.3 Hz, 2H, H3′,5′), 7.80 (d, J = 8.3 Hz, 2H, H2′,6′), 8.20 (d, J = 7.6 Hz, 1H, H5), 9.95 (s, 1H, CHO), 12.47 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 112.1, 113.6, 121.1, 122.6, 123.5, 123.9, 125.5, 128.8, 131.9, 132.0, 136.2, 147.4, 185.2. Anal. Calcd. for C15H10BrNO: C, 60.02; H, 3.36; N, 4.67. Found: C, 60.21; H, 3.48; N, 4.60.
2-p-Tolyl-1H-indole-3-carbaldehyde (4f)
Cream powder, Mp: 239–241 °C, Yield: 88%. IR (KBr, cm−1): 3213 (NH), 2863 (H–CO), 1625 (C=O), 1452 (C=C). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 2.41 (s, 3H, CH3), 7.23 (t, J = 7.5 Hz, 1H, H6), 7.28 (t, J = 7.5 Hz, 1H, H7), 7.41 (d, J = 7.9 Hz, 2H, H3′,5′), 7.49 (d, J = 7.5 Hz, 1H, H8), 7.67 (d, J = 7.9 Hz, 2H, H2′,6′), 8.19 (d, J = 7.5 Hz, 1H, H5), 9.95 (s, 1H, CHO), 12.34 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 21.0, 112.0, 113.3, 121.1, 122.4, 123.6, 125.8, 126.9, 129.6, 129.9, 135.9, 139.7, 149.1, 184.9. Anal. Calcd. for C16H13NO: C, 81.68; H, 5.57; N, 5.97. Found: C, 81.78; H, 5.48; N, 5.79.
General procedure for synthesis of 2-(2-phenyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7a–t)
A mixture of 2-arylindole-3-carbaldehydes (4a–f) (1 mmol), diverse pyridine 2-amines (5) (1 mmol), varied isocyanides (6) (1.2 mmol) and ammonium chloride (1 mmol) were suspended in 15 mL of toluene. The reaction mixture was heated and stirred under reflux until the end of the reaction. After the completion of the reaction, the mixture was cooled to room temperature and then concentrated under vacuum and purified by ethyl acetate/n-hexane [30].
N-cyclohexyl-2-(2-phenyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7a)
White powder, Mp > 250 °C; Yield: 75%, IR (KBr, cm−1): 3359, 3310 (N–H stretching), 3058 (aromatic C–H stretching), 2926 (aliphatic C–H stretching), 1628 (C=N), 1582 (C=C), 1452 (CH2 bending), 1230 (C–N); 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.74–0.78 (m, 4H, cyclohexyl), 0.89–0.91 (m, 1H, cyclohexyl), 1.29–1.31 (m, 5H, cyclohexyl), 1.98 (m, 1H, NH), 3.08–3.16 (m, 1H, CH), 6.86 (t, J = 7.3 Hz, 1H, H6′′), 7.01 (t, J = 7.3 Hz, 1H, H5′′), 7.14 (t, J = 7.6 Hz, 2H, H6,7), 7.30–7.32 (m, 1H, H4′), 7.40 (t, J = 7.3 Hz, 2H, H3′,5′), 7.44 (d, J = 7.3 Hz, 1H, H8), 7.50 (d, J = 7.3 Hz, 1H, H4′′), 7.55 (d, J = 7.3 Hz, 2H, H2′,6′), 7.62 (d, J = 7.6 Hz, 1H, H5), 8.19 (d, J = 7.3, 1H, H6′′), 11.53 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 23.8, 23.3, 32.7, 54.52, 106.7, 111.0, 111.2, 116.4, 116.5, 119.2, 120.2, 121.8, 122.5, 122.7, 122.9, 125.8, 127.1, 127.5, 128.8, 129.1, 133.2, 136.0, 140.4. MS (ESI) m/z: 407 (MH+). Anal. Calcd for C27H26N4: C, 79.77; H, 6.45; N, 13.78; Found C, 79.91; H, 6.35; N, 13.90.
4-(3-(3-(cyclohexylamino)H-imidazo[1,2-a]pyridin-2-yl)-1H-indol-2-yl)phenol (7b)
Pale yellow powder, Mp: 177–180 °C; Yield: 82%. IR (KBr, cm−1): 3405 (O–H), 3343 (N–H stretching), 3057 (aromatic C–H stretching), 2927 (aliphatic C–H stretching), 1634 (C=N), 1584 (C=C), 1450 (CH2 bending), 1238 (C–N), 1176 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.72–0.83 (m, 4H, cyclohexyl), 0.90–0.94 (m, 1H, cyclohexyl), 1.26–1.34 (m, 5H, cyclohexyl), 2.20 (m, 1H, NH), 2.95–2.96 (m, 1H, CH), 6.79 (d, J = 8.5 Hz, 2H, H3′,5′), 6.84 (t, J = 7.4 Hz, 1H, H6′′), 6.98 (t, J = 7.4 Hz, 1H, H5′′), 7.08–7.14 (m, 2H, H6,7), 7.35 (d, J = 8.5 Hz, 2H, H2′,6′), 7.39 (d, J = 7.9 Hz, 1H, H8), 7.49 (d, J = 7.4 Hz, 1H, H4′′), 7.64 (d, J = 7.9 Hz, 1H, H5), 8.16 (d, J = 7.4 Hz, 1H, H7′′), 9.67 (s, 1H, OH), 11.36 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 22.8, 24.4, 31.3, 55.6, 106.4, 110.7, 111.0, 114.8, 115.2, 117.5, 121.2, 122.1, 124.5, 126.0, 126.8, 130.0, 130.6, 134.4, 135.5, 137.6, 140.9, 143.2, 156.5. MS (ESI) m/z: 423 (MH+). Anal. Calcd for C27H26N4O: C, 76.75; H, 6.20; N, 13.26; Found C, 76.51; H, 6.10; N, 13.37.
N-cyclohexyl-2-(2-(4-methoxyphenyl)-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7c)
Cream powder, Mp > 250 °C; Yield: 79%. IR (KBr, cm−1): 3409 (N–H stretching), 3063 (aromatic C–H stretching), 2929 (aliphatic C–H stretching),1653 (C=N), 1611 (C=C), 1454 (CH2 bending), 1250 (C–N), 1178 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.78–0.79 (m, 4H, cyclohexyl), 0.90–0.92 (m, 1H, cyclohexyl), 1.27–1.33 (m, 5H, cyclohexyl), 1.98 (m, 1H, NH), 3.19–3.20 (m, 1H, CH), 6.86 (t, J = 7.4 Hz, 1H, H6′′), 6.97–7.00 (m, 3H, H3′,5′, 5′′), 7.10–7.16 (m, 2H, H6,7), 7.41 (d, J = 7.9 Hz, 1H, H8), 7.48–7.50 (m, 3H, H4′′,2′,6′), 7.56 (d, J = 7.9 Hz, 1H, H5), 8.19 (d, J = 7.4 Hz, 1H, H7′′), 11.42 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 23.8, 25.2, 32.6, 54.5, 55.3, 110.8, 114.4, 116.6, 118.2, 119.6, 121.8, 123.1, 125.6, 126.5, 128.5, 129.5, 129.6, 131.8, 132.7, 133.4, 134.6, 135.1, 140.0, 158.8. MS (ESI) m/z: 437 (MH+). Anal. Calcd for C28H28N4O: C, 77.04; H, 6.46; N, 12.83; Found C, 77.15; H, 6.24; N, 12.71.
2-(2-(4-chlorophenyl)-1H-indol-3-yl)-N-cyclohexylH-imidazo[1,2-a]pyridin-3-amine (7d)
Dark cream powder, Mp > 250 °C; Yield: 78%, IR (KBr, cm−1): 3448 (N–H stretching), 3059 (aromatic C–H stretching), 2928 (aliphatic C–H stretching), 1628 (C=N), 1581 (C=C), 1451 (CH2 bending), 1230 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.73–0.81 (m, 4H, cyclohexyl), 0.89–0.91 (m, 1H, cyclohexyl), 1.31–1.33 (m, 5H, cyclohexyl), 1.99 (broad, 1H, NH), 3.48–3.50 (m, 1H, CH), 6.87 (t, J = 6.8 Hz, 1H, H6′′), 7.00 (t, J = 6.8 Hz, 1H, H5′′), 7.12–7.17 (m, 2H, H6,7), 7.44 (d, J = 8.0 Hz, 2H, H3′,5′), 7.49–7.54 (m, 3H, H5,8, 4′′), 7.57 (d, J = 8.0 Hz, 2H, H2′,6′), 8.22 (d, J = 6.8 Hz, 1H, H7′′), 11.60 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 23.9, 25.3, 32.8, 54.4, 107.3, 111.0, 116.5, 116.6, 119.4, 120.0, 120.1, 120.6, 122.1, 122.5, 122.8, 123.0, 126.2, 128.7, 129.2, 132.0, 133.8, 136.1, 140.4. MS (ESI) m/z: 441 (MH+). Anal. Calcd for C27H25ClN4: C, 73.54; H, 5.71; N, 12.71; Found C, 73.41; H, 5.90; N, 12.89.
2-(2-(4-bromophenyl)-1H-indol-3-yl)-N-cyclohexylH-imidazo[1,2-a]pyridin-3-amine (7e)
Cream to yellow powder, Mp = 242–244 °C; Yield: 76%, IR (KBr, cm−1): 3449 (N–H stretching), 3061 (aromatic C–H stretching), 2928 (aliphatic C–H stretching),1629 (C=N), 1581 (C=C), 1451 (CH2 bending), 1229 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.64–0.66 (m, 4H, cyclohexyl), 0.90–1.00 (m, 1H, cyclohexyl), 1.27–1.38 (m, 5H, cyclohexyl), 2.06 (broad, 1H, NH), 3.13–3.17 (m, 1H, CH), 7.10 (t, J = 6.8 Hz, 1H, H6′′), 7.24 (t, J = 6.8 Hz, 1H, H5′′), 7.47–7.55 (m, 5H, H5,6,7,8,4′′), 7.62 (d, J = 7.9 Hz, 2H, H3′,5′), 7.82–7.86 (m, 2H, H2′,6′), 8.77 (d, J = 6.8 Hz, 1H, H7′′), 12.28 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 24.0, 24.8, 32.7, 53.7, 111.8, 111.9, 119.0, 1119.1, 120.4, 121.6, 122.9, 123.0, 124.9, 125.0, 128.2, 128.6, 129.2, 130.5, 131.2, 131.9, 135.9, 136.1, 136.4. MS (ESI) m/z: 485 (MH+). Anal. Calcd for C27H25BrN4: C, 66.81; H, 5.19; N, 11.54 Found C, 66.61; H, 5.05; N, 11.64.
N-cyclohexyl-2-(2-p-tolyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7f)
White powder, Mp > 250 °C; Yield: 72%, IR (KBr, cm−1): 3351, 3131 (N–H stretching), 3063 (aromatic C–H stretching), 2927 (aliphatic C–H stretching),1624 (C=N), 1583 (C=C), 1453 (CH2 bending), 1225 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.76–0.78 (m, 4H, cyclohexyl), 0.90–0.91 (m, 1H, cyclohexyl), 1.26–1.31 (m, 5H, cyclohexyl), 1.99 (broad, 1H, NH), 2.30 (s, 3H, CH3), 3.07–3.08 (m, 1H, CH), 6.85 (t, J = 7.5 Hz, 1H, H6′′), 7.00 (t, J = 7.5 Hz, 1H, H5′′), 7.12–7.15 (m, 2H, H6,7), 7.21 (d, J = 8.0 Hz, 2H, H3′,5′), 7.41–7.44 (m, 3H, H2′,6′,8), 7.49 (d, J = 7.5 Hz, 1H, H4′′), 7.60 (d, J = 8.0 Hz, 1H, H5), 8.18 (d, J = 7.5 Hz, 1H, H7′′), 11.47 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 24.4, 25.2, 29.8, 32.6, 55.1, 114.21, 117.6, 119.4, 121,7, 122.5, 124.8, 125.5, 126.5, 127.0, 127.5, 128.3, 128.6, 129.3, 130.9, 135.1, 135.9, 137.0, 139.0, 141.2. MS (ESI) m/z: 421 (MH+). Anal. Calcd for C28H28N4: C, 79.97; H, 6.71; N, 13.32 Found C, 80.10; H, 6.90; N, 13.15.
4-(3-(3-(cyclohexylamino)-5-methylH-imidazo[1,2-a]pyridin-2-yl)-1H-indol-2-yl)phenol (7g)
Cream powder, Mp: 228–230 °C; Yield: 75%, IR (KBr, cm−1): 3349 (O–H), 3338 (N–H stretching), 3059 (aromatic C–H stretching), 2926 (aliphatic C–H stretching),1612 (C=N), 1575 (C=C), 1450 (CH2 bending), 1228 (C–N), 1178 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.61–0.79 (m, 6H, cyclohexyl), 1.24–1.25 (m, 4H, cyclohexyl), 1.98 (s, 1H, NH), 2.60–2.65 (m, 1H, CH), 2.83 (s, 3H, CH3), 6.51 (d, J = 7.0 Hz, 1H, H6′′), 6.80 (d, J = 8.0 Hz, 2H, H3′,5′), 6.97–7.03 (m, 2H, H6,7), 7.09 (t, J = 7.0 Hz, 1H, H5′′), 7.33 (d, J = 8.4 Hz, 1H, H8), 7.38–7.41 (m, 3H, H2′,6′,4′′), 7.63 (d, J = 8.4 Hz, 1H, H5), 9.67 (s, 1H, OH), 11.32 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 19.4, 23.8, 25.2, 32.1, 56.9, 112.5, 114.8, 114.9, 115.7, 118.9, 119.7, 121.2, 122.9, 124.0, 126.9, 128.4, 128.5, 129.1, 134.5, 135.5, 135.9, 136.1, 142.3, 157.1. MS (ESI) m/z: 437 (MH+). Anal. Calcd for C28H28N4O: C, 77.04; H, 6.46; N, 12.83; Found C, 76.85; H, 6.65; N, 12.97.
N-cyclohexyl-2-(2-(4-methoxyphenyl)-1H-indol-3-yl)-5-methylH-imidazo[1,2-a]pyridin-3-amine (7h)
Light cream powder, Mp: 203–205 °C; Yield: 69%, IR (KBr, cm−1): 3343, 3130 (N–H stretching), 3057 (aromatic C–H stretching), 2926 (aliphatic C–H stretching), 1606 (C=N), 1543 (C=C), 1452 (CH2 bending), 1242 (C–N), 1179 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.63–0.85 (m, 6H, cyclohexyl), 1.24–1.26 (m, 4H, cyclohexyl), 1.99 (s, 1H, NH), 2.78–2.79 (m, 1H, CH), 2.85 (s, 3H, CH3), 3.76 (s, 3H, CH3), 6.53 (d, J = 7.3 Hz, 1H, H6′′), 6.97–7.04 (m, 3H, H6,7,3′,5′), 7.12 (t, J = 7.3 Hz, 1H, H5′′), 7.34 (d, J = 9.0 Hz, 1H, H8), 7.41 (d, J = 7.3 Hz, 1H, H4′′), 7.53–7.58 (m, 4H, H5,2′,6′), 11.42 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 19.6, 23.5, 25.2, 32.1, 55.0, 56.41, 110.9, 112.5, 114.2, 114.9, 119.2, 119.7, 121.3, 123.0, 125.3, 127.2, 128.2, 128.5, 129.1, 134.9, 136.1, 140.3, 142.3, 146.3, 158.8. MS (ESI) m/z: 451 (MH+). Anal. Calcd for C29H30N4O: C, 77.30; H, 6.71; N, 12.43; Found C, 77.57; H, 6.51; N, 12.53.
2-(2-(4-chlorophenyl)-1H-indol-3-yl)-N-cyclohexyl-5-methylH-imidazo[1,2-a]pyridin-3-amine (7i)
Light green, Mp: 250 °C; Yield: 79%, IR (KBr, cm−1): 3143 (N–H stretching), 3044 (aromatic C–H stretching), 2930 (aliphatic C–H stretching),1653 (C=N), 1543 (C=C), 1446 (CH2 bending), 1262 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.68–0.74 (m, 4H, cyclohexyl), 0.85–0.87 (m, 1H, cyclohexyl), 1.24–1.25 (m, 5H, cyclohexyl), 1.99 (s, 1H, NH), 2.91 (s, 3H, CH3), 3.05–3.06 (m, 1H, CH), 6.56 (d, J = 6.8 Hz, 1H, H6′′), 7.06–7.12 (m, 2H, H6,7), 7.20 (t, J = 6.8 Hz, 1H, H5′′), 7.35 (d, J = 9.1 Hz, 1H, H8), 7.47–7.51 (m, 3H, H4′′,3′,5′), 7.57 (d, J = 9.1 Hz, H5), 7.64 (d, J = 8.5 Hz, 2H, H2′,6′), 11.63 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 19.8, 23.3, 25.05, 32.1, 55.1, 109.9, 112.0, 114.8, 116.2, 117.7, 119.4, 120.4, 122.2, 122.9, 124.6, 128.0, 129.0, 129.2, 130.1, 133, 136.0, 138.3, 139.8, 141.1. MS (ESI) m/z: 455 (MH+). Anal. Calcd for C28H27ClN4: C, 73.91; H, 5.98; N, 12.31 Found C, 73.65; H, 5.80; N, 12.54.
2-(2-(4-bromophenyl)-1H-indol-3-yl)-N-cyclohexyl-5-methylH-imidazo[1,2-a]pyridin-3-amine (7j)
Yellow powder, Mp: 230–233 °C; Yield: 81%, IR (KBr, cm−1): 3146 (N–H stretching), 3059 (aromatic C–H stretching), 2922 (aliphatic C–H stretching), 1651 (C=N), 1537 (C=C), 1450 (CH2 bending), 1230 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.66–0.74 (m, 5H, cyclohexyl), 1.24–1.25 (m, 5H, cyclohexyl), 1.98 (s, 1H, NH), 2.88 (s, 3H, CH3), 3.03–3.04 (m, 1H, CH), 6.54 (d, J = 7.0 Hz, 1H, H6′′), 7.00–7.05 (m, 2H, H6,7), 7.16 (t, J = 7.0 Hz, 1H, H5′′), 7.33 (d, J = 8.4 Hz, 1H, H8), 7.43–7.47 (m, 3H, H4′′,3′,5′), 7.55 (d, J = 8.4 Hz, H5), 7.64 (d, J = 8.4 Hz, 2H, H2′,6′), 11.60 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 20.1, 23.2, 24.7, 31.9, 54.9, 109.1, 110.7, 112.8, 116.4, 117.2, 119.4, 120.4, 121.5, 122.1, 124.7, 125.9, 127.6, 128.9, 129.6, 131.6, 133.3, 136.8, 137.6, 140.8. MS (ESI) m/z: 499 (MH+). Anal. Calcd for C30H31BrN4: C, 68.31; H, 5.92; N, 10.62 Found C, 68.15; H, 6.10; N, 10.41.
N-cyclohexyl-5-methyl-2-(2-p-tolyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7k)
White powder, Mp > 250 °C; Yield 71%, IR (KBr, cm−1): 3350, 3131 (N–H stretching), 3027 (aromatic C–H stretching), 2927 (aliphatic C–H stretching), 1453 ((CH2 bending), 1642 (C=N), 1583 (C=C), 1227 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.64 (m, 6H, cyclohexyl), 1.24–1.25 (m, 4H, cyclohexyl), 1.99 (s, 1H, NH), 2.31 (s, 3H, CH3), 2.79 (broad, 1H, CH), 2.86 (s, 3H, CH3), 6.54 (d, J = 6.4 Hz, 1H, H6′′), 7.00–7.06 (m, 2H, H6,7), 7.13 (t, J = 6.4 Hz, 1H, H5′′), 7.22 (d, J = 6.0, 2H, H3′,5′), 7.33 (d, J = 7.5 Hz, 1H, H8), 7.43 (d, J = 6.4 Hz, H4′′), 7.50 (d, J = 6.0 Hz, 2H, H2′,6′), 7.60 (d, J = 7.5 Hz, H5), 11.67 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 22.9, 23.5, 25.1, 30.4, 32.2, 56.0, 114.6, 115.2, 119.2, 120.4, 120.8, 122.4, 123.2, 124.0, 125.6, 126.5, 127.4, 128.2, 131.2, 133.6, 134.6, 136.0, 137.8, 138.3, 142.1. MS (ESI) m/z: 435 (MH+). Anal. Calcd for C29H30N4 C, 80.15; H, 6.96; N, 12.89 Found C, 80.35; H, 7.25; N, 12.63.
N-tert-butyl-2-(2-phenyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7l)
Creamish White powder, Mp > 250 °C; Yield 79%, IR (KBr, cm−1): 3432, 3339 (N–H stretching), 3061 (aromatic C–H stretching), 2963 (aliphatic C–H stretching), 1629 (C=N), 1584 (C=C), 1454 (CH2 bending), 1216 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.64 (s, 9H, t-butyl), 1.98 (s, 1H, NH), 6.84 (d, J = 6.7 Hz, 1H, H6′′), 7.03 (t, J = 6.7 Hz, 1H, H5′′), 7.13–7.19 (m, 2H, H6,7), 7.35 (t, J = 6.7 Hz, 1H, H8), 7.42–7.45 (m, 3H, H3′,5′,8), 7.50–7.53 (m, 3H, H2′,6′,4′′), 7.77 (d, J = 7.6 Hz, 1H, H5), 8.28 (d, J = 6.7 Hz, 1H, H7′′), 11.52 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 29.0, 55.08, 107.5, 110.8, 111.2, 116.4, 116.5, 119.3, 120.5, 123.2, 123.5, 123.7, 127.1, 127.2, 127.7, 128.8, 129.1, 133.4, 134.9, 136.4, 141.5. MS (ESI) m/z: 381 (MH+). Anal. Calcd for C25H24N4 C, 78.92; H, 6.36; N, 14.73 Found C, 79.14; H, 6.49; N, 14.69.
4-(3-(3-(tert-butylamino)H-imidazo[1,2-a]pyridin-2-yl)-1H-indol-2-yl)phenol (7m)
White powder, Mp < 250 °C; Yield 81%, IR (KBr, cm−1): 3449, 3339 (N–H stretching), 3060 (aromatic C–H stretching), 2923 (aliphatic C–H stretching), 1606 (C=N), 1544 (C=C), 1448 (CH2 bending), 1219 (C–N), 1180 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.64 (s, 9H, t-butyl), 1.98 (s, 1H, NH), 6.82–6.84 (m, 3H, H3′,5′, 6′′), 7.00 (t, J = 7.9 Hz, 1H, H5′′), 7.10 (t, J = 7.7 Hz, 1H, H6), 7.16 (t, J = 7.7 Hz, 1H, H7), 7.30 (d, 2H, H2′,6′), 7.38 (d, J = 7.7 Hz, 1H, H8), 7.51 (d, J = 7.9 Hz, 1H, H4′′), 7.76 (d, J = 7.7 Hz, 1H, H5), 8.27 (d, J = 7.9 Hz, 1H, H7′′), 11.34 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 28.9, 54.8, 106.3, 110.6, 115.7, 116.3, 116.4, 119.0, 120.1, 121.3, 123.0, 123.5, 124.2, 128.5, 128.7, 128.9, 135.3, 135.5, 136.1, 141.4, 157.2. MS (ESI) m/z: 397 (MH+). Anal. Calcd for C25H24N4O C, 75.73; H, 6.10; N, 14.13; Found C, 75.98; H, 5.97; N, 13.89.
N-tert-butyl-2-(2-(4-methoxyphenyl)-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7n)
Cream powder, Mp > 250 °C; Yield 75%, IR (KBr, cm−1): 3338 (N–H stretching), 3061 (aromatic C–H stretching), 2965 (aliphatic C–H stretching), 1615 (C=N), 1539 (C=C), 1454 (CH2 bending), 1248 (C–N), 1180 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.65 (s, 9H, t-butyl), 1.99 (s, 1H, NH), 3.78 (s, 3H, CH3), 6.84 (t, J = 7.3 Hz, 1H, H6′′), 7.01–7.03 (m, 3H, H3′,5′,5′′), 7.12 (t, J = 7.6 Hz, 1H, H6), 7.17 (t, J = 7.6 Hz, 1H, H7), 7.40 (d, J = 7.6 Hz, 1H, H8), 7.44 (d, J = 8.1 Hz, 2H, H2′,6′), 7.51 (d, J = 7.3 Hz, 1H, H4′′), 7.71 (d, J = 7.6 Hz, 1H, H5), 8.29 (d, J = 7.3 Hz, 1H, H7′′), 11.41 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 28.9, 55.2, 54.8, 106.7, 110.4, 111.0, 114.6, 116.3, 118.8, 119.9, 121.4, 122.8, 123.5, 123.7, 125.5,128.4, 128.6, 128.9, 135.0, 136.3, 141.5, 158.7. MS (ESI) m/z: 411 (MH+); Anal. Calcd for C26H26N4O C, 76.07; H, 6.38; N, 13.65; Found C, 76.23, 6.49, 13.51.
N-tert-butyl-2-(2-(4-chlorophenyl)-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7o)
Cream powder, Mp > 250 °C; Yield 78%, IR (KBr, cm−1): 3421, 3345 (N–H stretching), 3063 (aromatic C–H stretching), 2966 (aliphatic C–H stretching), 1641 (C=N), 1578 (C=C), 1452 (CH2 bending), 1216 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.67 (s, 9H, t-butyl), 1.98 (s, 1H, NH), 6.90 (t, J = 6.8 Hz, 1H, H6′′), 7.03 (t, J = 6.8 Hz, 1H, H5′′), 7.16 (t, J = 7.7 Hz, 1H, H6), 7.22 (t, J = 7.7 Hz, 1H, H7), 7.43 (d, J = 7.7 Hz, 1H, H8), 7.48 (d, J = 8.2 Hz, 2H, H3′,5′), 7.52–7.55 (m, 3H, H2′,6′, 4′′), 7.68 (d, J = 7.7 Hz, 1H, H5), 8.35 (d, J = 6.8 Hz, 1H, H7′′), 11.61 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 29.3, 54.8, 111.2, 113.4, 115.2, 116.2, 118.1, 119.5, 120.3, 122.2, 122.9, 123.9, 124.1, 128.6, 128.8, 132.0, 132.2, 133.8, 136.3, 137.2, 141.2. MS (ESI) m/z: 415 (MH+). Anal. Calcd for C25H23ClN4 C, 72.37; H, 5.59; N, 13.50, Found C, 72.10; H, 5.37; N, 14.13.
N-tert-butyl-2-(2-(4-bromophenyl)-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7p)
Cream powder, Mp > 250 °C; Yield 78%, IR (KBr, cm−1): 3424, 3342 (N–H stretching), 3060 (aromatic C–H stretching), 2965 (aliphatic C–H stretching), 1626 (C=N), 1577 (C=C), 1452 (CH2 bending), 1228 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.67 (s, 9H, t-butyl), 1.98 (s, 1H, NH), 6.86 (t, J = 6.8 Hz, 1H, H6′′), 7.03 (t, J = 6.8 Hz, 1H, H5′′), 7.14–7.19 (m, 2H, H6,7), 7.43 (d, J = 7.9 Hz, 1H, H8), 7.48–7.52 (m, 3H, H3′,5′,4′′), 7.62 (d, J = 7.9 Hz, 2H, H2′,6′), 7.68 (d, J = 7.9 Hz, 1H, H5), 8.33 (d, J = 6.8 Hz, 1H, H7′′), 11.59 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 29.2, 54.8, 108.0, 110.9, 111.2, 116.5, 119.4, 120.4, 120.7, 122.1, 124.0, 128.6, 129.0, 129.1, 131.8, 132.6, 133.4, 133.7, 136.1, 136.4, 141.5. MS (ESI) m/z: 459 (MH +); Anal. Calcd for C25H23BrN4 C, 65.36; H, 5.05; N, 12.20; Found C, 65.21; H, 5.25; N, 11.95.
4-(3-(3-(tert-butylamino)-5-methylH-imidazo[1,2-a]pyridin-2-yl)-1H-indol-2-yl)phenol (7q)
Creamish white powder, Mp > 250 °C; Yield 78%, IR (KBr, cm−1): 3421 (O–H), 3337 (N–H stretching), 3060 (aromatic C–H stretching), 2967 (aliphatic C–H stretching), 1627 (C=N), 1578 (C=C), 1455 (CH2 bending), 1226 (C–N), 1170 (C–O). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.66 (s, 9H, t-butyl), 2.23 (s, 1H, NH), 2.78 (s, 3H, CH3), 6.47 (d, J = 8.4 Hz, 1H, H6′′), 6.80 (t, J = 8.0 Hz, 1H, H3′,5′), 6.98–7.04 (m, 2H, H6,7), 7.09 (t, J = 8.4 Hz, 1H, H5′′), 7.35 (d, J = 7.1 Hz, 1H, H8), 7.38–7.41 (m, 3H, H2′,6′,4′′), 7.60 (d, J = 7.1 Hz, 1H, H5), 9.63 (s, 1H, OH), 11.63 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 21.53, 29.45, 53.59, 110.6, 112.4, 115.7, 120.7, 121.7, 122.9, 124.9, 125.6, 127.3, 128.8, 130.4, 131.2, 132.8, 134.1, 135.7, 137.9, 138.1, 142.7, 152.8. MS (ESI) m/z: 411 (MH +); Anal. Calcd for C26H26N4O C, 76.07; H, 6.38; N, 13.65; Found C, 76.29; H, 6.13; N, 13.47.
N-tert-butyl-2-(2-(4-chlorophenyl)-1H-indol-3-yl)-5-methylH-imidazo[1,2-a]pyridin-3-amine (7r)
Creamish white powder, Mp: 245–247 °C; Yield 71%, IR (KBr, cm−1): 3343 (N–H stretching), 3061 (aromatic C–H stretching), 2968 (aliphatic C–H stretching), 1628 (C=N), 1582 (C=C), 1453 (CH2 bending), 1233 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.58 (s, 9H, t-butyl), 1.98 (s, 1H, NH), 2.86 (s, 3H, CH3), 6.57 (d, J = 7.2 Hz, 1H, H6′′), 7.03–7.09 (m, 2H, H6,7), 7.16 (t, J = 7.2 Hz, 1H, H5′′), 7.37 (d, J = 8.8 Hz, 1H, H8), 7.43 (d, J = 7.2 Hz, 1H, H4′′), 7.56 (d, J = 8.0 Hz, 2H, H3′,5′), 7.62–7.65 (m, 3H, H2′,6′,5), 11.58 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 20.0, 28.9, 54.2, 108.7, 111.3, 113.3, 114.7, 115.0, 119.6, 120.2, 120.7, 122.2, 123.5, 128.9, 131.8, 132.3, 133.4, 133.6, 136.1, 136.2, 136.5, 142.8; MS (ESI) m/z: 429 (MH+). Anal. Calcd for C26H25N4 C, 72.80; H, 5.87; N, 13.06; Found C, 73.05; H, 6.0; N, 12.89.
N-tert-butyl-2-(2-(4-bromophenyl)-1H-indol-3-yl)-5-methylH-imidazo[1,2-a]pyridin-3-amine (7s)
Creamish white powder, Mp > 250 °C; Yield 71%, IR (KBr, cm−1): 3341 (N–H stretching), 3059 (aromatic C–H stretching), 2965 (aliphatic C–H stretching), 1625 (C=N), 1580 (C=C), 1456 (CH2 bending), 1234 (C–N). 1H NMR (500 MHz, DMSO-d6) (δ, ppm): 0.58 (s, 9H, t-butyl), 1.98 (s, 1H, NH), 2.86 (s, 3H, CH3), 6.57 (d, J = 7.4 Hz, 1H, H6′′), 7.03–7.09 (m, 2H, H6,7), 7.16 (t, J = 7.4 Hz, 1H, H5′′), 7.36 (d, J = 8.8 Hz, 1H, H8), 7.43 (d, J = 7.4 Hz, 1H, H4′′), 7.56 (d, J = 8.1 Hz, 2H, H3′,5′), 7.61–7.64 (m, 2H, H2′,6′,5), 11.57 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 20.3, 28.9, 54.3, 108.6, 113.2, 115.0, 115.1, 119.6, 120.3, 120.7, 121.1, 122.2, 122.3, 123.6, 126.1, 129.0, 131.7, 131.8, 132.2, 136.1, 136.6, 142.8. MS (ESI) m/z: 473 (MH+); Anal. Calcd for C26H25BrN4 C, 65.96; H, 5.32; N, 11.83; Found C, 65.79; H, 5.20; N, 12.15.
N-tert-butyl-5-methyl-2-(2-p-tolyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7t)
Creamish white powder, Mp > 250 °C; Yield 77%, IR (KBr, cm−1): 3338 (N–H stretching), 3026 (aromatic C–H stretching), 2965 (aliphatic C–H stretching), 1625 (C=N), 1577 (C=C), 1453 (CH2 bending), 1217 (C–N). 1H NMR (500 MHz, DMSO-d6) ( δ, ppm): 0.58 (s, 9H, t-butyl), 2.19 (s, 1H, NH), 2.32 (s, 3H, CH3), 2.82 (s, 3H, CH3), 6.56 (d, J = 7.1 Hz, 1H, H6′′), 7.02–7.09 (m, 2H, H6,7), 7.14 (t, J = 7.1 Hz, 1H, H5′′), 7.24 (d, J = 7.3 Hz, 2H, H3′,5′), 7.37 (d, J = 8.2 Hz, 1H, H8), 7.41 (d, J = 7.1 Hz, 1H, H4′′), 7.47 (d, J = 7.3 Hz, 2H, H2′,6′), 7.70 (d, J = 8.2 Hz, 1H, H5), 11.45 (s, 1H, NH-indole). 13C NMR (125 MHZ, DMSO-d6) (δ, ppm): 20.2, 20.7, 28.7, 54.4, 107.5, 111.0, 113.1, 115.0, 119.4, 120.1, 121.7, 123.3, 125.8, 126.8, 126.9, 128.9, 129.5, 130.4, 135.0, 136.4, 136.7, 137.1, 142.8. MS (ESI) m/z: 409 (MH+); Anal. Calcd for C27H28N4 C, 79.38; H, 6.91; N, 13.71; Found C, 79.21; H, 7.05; N, 13.64.
Biology
Anti-proliferative assay
The cytotoxic activity of the synthesized compounds 7a–7t was assessed against three different cancer cell lines [31]. Tumor cell lines including lung cancer cell line (A-549), liver cancer cell line (Hep-G2), breast cancer cell line (MCF-7) and epithelial substrain cell line (TD-47) were obtained from the National Cell Bank of Iran and grown in RPMI-1640 medium (Gibco, UK). Exponentially growing cells (1 × 104 cells/well) were seeded in 96-well with in RPMI1640 medium at 37 °C under 5% CO2 supplemented with 10% FBS, 1% l-glutamine, and penicillin–streptomycin, and incubated overnight. The cells were treated by different concentrations of test compounds and allowed to incubate for 48 h in a humidified atmosphere. All compounds were initially dissolved in DMSO, and the final concentration of DMSO was less than 1% in all the concentrations of the applied compounds. Etoposide and colchicine were used as positive controls. After 48 h of further incubation, the medium was replaced with MTT (1 mg/mL), followed by 4 h incubation. After the formation of blue formazan crystals, the culture medium was replaced with 100 µL of DMSO and the absorbance values were measured using a multi-well plate reader (Gen5; Epoch, BioTek, USA) at 492-nm wavelength. The IC50 values were compared with the control and expressed in mean ± SD from the dose–response logarithmic curves of at least three independent experiments.
Acridine orange/ethidium bromide double staining
Apoptosis in the treated cancer cells was determined morphologically after staining with acridine orange/ethidium bromide using fluorescence microscopy (Zeiss, Germany) [31]. A-549 cells grown in 12-well plates (5 × 105 cells/well) were treated with and without IC50 concentrations of compound 7a and 7f for 24 h. Then, the cells were washed three times with PBS. Finally, ethidium bromide/acridine orange (1:1, 100 mg/mL) solution was added to the cell suspension, and the nuclear morphology was evaluated by fluorescence microscopy. All the experiments were repeated three times.
Flow cytometry analysis of the apoptotic cells with Annexin V-FITC/PI staining
Flow cytometry analyses for 7a and 7f were performed to confirm apoptosis induced in comparison with the standard drug etoposide [31]. In brief, 5 × 105 cells/well of A-549 were treated with the IC50 doses of the most potent compounds, 7a and 7f. After 24 h, the cells were washed twice with cold PBS, collected by centrifugation and resuspended in 1× annexin V binding buffer [0.1 M Hepes/NaOH (pH 7.4), 1.4 M NaCl, 25 mM CaCl2]. Then, the cells were double-stained with 5 µL of annexin V-FITC and 5 µL of PI, and the cells were gently vortexed and incubated at room temperature for 15 min in the dark before flow cytometry. Finally, 400 μL of 1× annexin binding buffer was added into the suspension, and the cells were analyzed using a flow cytometer within 1 h.
Molecular docking study
Computer-simulated docking studies were accomplished by the AutoDock 4.2 software. The Lamarckian Genetic Algorithm of the AutoDock 4.2 program was used as the search algorithm. The Graphical User Interface program, AutoDock Tools 1.5.6 was used to prepare, run, and analyze the docking simulations. Molecular docking of compounds was performed with two crystal structures (PDB ID: 1SA0 and 1ZXM for tubulin and topoisomerase II, respectively) by the Auto-Dock Tools 1.5.6. All two-dimensional structures of the compounds were built using ChemDraw Ultra 10.0 (Cambridge Software), and then moved into the Hyperchem 8.0 software (Release 8.0 for Windows, Molecular Modeling System, HyperCube 2007). Molecules were subjected to energy minimization with MM + force field and then the PM3 semi-empirical technique. Then, the partial charges of the atoms were calculated by the Gasteiger–Marsili procedure implemented in the AutoDock Tools package [32]. The non-polar hydrogens of compounds were merged and then the crystal structures of protein were taken from the Protein Data bank (www.rcsb.org). All bound water and ligands were eliminated from the protein, and polar hydrogen atom were added to the protein as it was required for the electrostatics interactions, and then non-polar hydrogen atoms were merged. In all the dockings, a grid map with 60 grid points in the X, Y, and Z directions was built. Among the three different search algorithms offered by AutoDock 4.2, the Lamarckian genetic algorithm approach was applied. For all docking procedures, 100 independent runs with step sizes of 0.2 Å for translations and 5° for orientations and torsions were considered. For the Lamarckian genetic algorithm method, a maximum number of 25 × 105 energy evaluations, 27,000 maximum generations, a gene mutation rate of 0.02. and a cross-over rate of 0.8 were used. At the end of docking, the structures were ranked by energy. Ligand–receptor interactions were all visualized on the basis of the docking results using Discovery Studio Visualizer 4.0.
Results and discussion
Chemistry
The overall procedure for the synthesis of the designed compounds 7a–t is illustrated in Scheme 1 (above). As illustrated in this scheme, the reaction of acetophenone derivatives and phenyl hydrazine in the presence of acetic acid and polyphosphoric acid in refluxing EtOH afforded 2-aryl-1H-indoles 3a–f in good yield. Subsequently, treatment of compounds 3a–f with phosphorous oxychloride in DMF led to 2-aryl-1H-indole-3-carbaldehyde derivatives 4a–f [27]. Finally, compound 4, 2-aminopyridines and isocyanides were refluxed in toluene to generate imidazopyridines-linked 2-aryl-1H-indoles 7a–t in a good yield.
The structures of the title compounds 7a–t were determined by 1H and 13CNMR, IR and elemental analyses. As exemplified with the IR analysis of N-cyclohexyl-2-(2-p-tolyl-1H-indol-3-yl)H-imidazo[1,2-a]pyridin-3-amine (7f) displayed major adsorption bands at 1624 cm−1 and 1225 cm−1, which were due to the stretching vibrations of C=N and C–N bonds, respectively. Furthermore, two bands were observed at 3351 and 3131 cm−1 which were assigned to the ν(N–H) stretching of compound 7f.
The 1H NMR spectrum for compound 7f showed protons resonance at 6.85–8.18 ppm, which was due to the aromatic, pyridine and indole rings. In addition, two single resonances for the NH groups appeared at 1.99 and 11.47 ppm, which were due to the cyclohexyl and indole rings, respectively. Also, the signals related to the protons of the cyclohexyl group appeared at 0.9–0.91 ppm, 1.26–1.31 ppm, and a multiplet at 3.07–3.08 ppm for CH–N of the cyclohexyl ring. Finally, the single resonance at 2.3 ppm was related to the methyl group as R1.
Biological
Anti-proliferative activity
Derivatives of series 7a–t were synthesized and screened against A-549 (lung), Hep-G2 (liver), MCF-7 and T-47D (breast) cell lines (Table 1). As shown in Table 1, compound 7f, having the N1-cyclohexyl-imidazo[1,2-a]pyridin-3-amine moiety substituted on the C-3 position of the indole ring and the tolyl ring at the C-2 position, exhibited the highest inhibitory activity against A-549 with IC50 = 10.34 μM, which was more potent than etoposide (IC50 = 36.72 µM) and other synthesized derivatives. Compound 7d with the N1-cyclohexyl-imidazo[1,2-a]pyridin-3-amine moiety substituted on the C-3 position of the indole ring and the para chloro phenyl ring at the C-2 position, exhibited the highest inhibitory activity against Hep-G2 with IC50 = 35.79 μM compared with etoposide (IC50 = 37.13 µM) and colchicine (IC50 = 1.76 µM). However, compound 7j, having the N1-cyclohexyl-5-methyl-imidazo[1,2-a]pyridin-3-amine at the C-3 position of the indole ring and the para bromo phenyl ring at the C-2 position, had the highest inhibitory activity against T-47D and MCF-7 cell lines with IC50 = 69.35 μM and 49.69 μM, respectively. All compounds showed higher activity against the two A-549 and Hep-G2 cell lines than against other cell lines.

MCF-7 (breast cancer) cell line:
Compound 7j was found to be the most potent one against the MCF-7 cell line among the studied compounds. Others had weak cytotoxic activity against this cell line. Replacement of the cyclohexyl ring as R3 in the structure of 7g–k with the t-butyl group giving 7q–t led to a decrease of cytotoxic activity against the MCF-7 cell line (Table 1). The presence of the cyclohexyl group substituted on the amine group of the imidazopyridyl moiety and the more lipophilic halogen group at the para position of the phenyl ring in the structure of compound 7j have important roles in inhibitory activity against the MCF-7 cell line. However, all compounds showed weaker activity than colchicine and etoposide drugs against the MCF-7 cell line.
A-549 (lung cancer) cell line:
Compounds 7a and 7f exhibited the highest cytotoxic activity (IC50 = 11.48 μM and 10.66 μM, respectively) against the A-549 cell line. Here again, compounds having the cyclohexyl group substituted on the amine group of the imidazopyridyl moiety showed higher activity against the A-549 cell line than compounds bearing the t-butyl amine group at the same position (Table 1). Among the studied compounds, those with the cyclohexyl ring and hydrogen substitution as R3 and R2, respectively, the cytotoxic activity order was 7f > 7a > 7e > 7d > 7c > 7b. This order can be explained by the lipophilicity of the groups substituted on the phenyl ring at C-2 of the indole ring. Moreover, between the compounds with the t-butyl group substituted on the imidazopyridyl moiety, a compound with the para bromo phenyl ring at C-2 of the indole ring, 7p showed the highest activity among the other compounds. According to the results in this series, the presence of lipophilic groups at the phenyl ring at the C-2 indole ring moiety had a positive effect. Compounds 7a–h (except for b) showed more potent cytotoxic activity compared with etoposide (IC50 = 10.66–36.57 μM vs. 39.18 μM). Furthermore, replacing hydrogen with methyl at the imidazopyridyl moiety decreased activity against the A-549 cell line.
Hep-G2 (liver cancer) cell line:
Here again, compounds having the cyclohexyl amine substituted on the imidazopyridyl moiety showed higher activity than compounds bearing the t-butyl amine group at the same position (Table 1). Compound 7d ®1 = Br and R3 = cyclohexyl) revealed the highest inhibitory activity against the Hep-G2 cell line (IC50 = 36.12 µM). The presence of a lipophilic group substituted on the phenyl ring at the C-2 position of the indole ring resulted in higher activity against the Hep-G2 cell line. Replacement of the chloro group (7d) in the para position of the phenyl ring with methoxy (7c) or hydroxyl (7b) led to a decrease in the cytotoxicity. Compounds 7q–t demonstrated no cytotoxic activity against this cell line (IC50 > 100). Interestingly, the presence of methyl at the R2 and tert-butyl at the R3 positions drastically decreased cytotoxicity against the Hep-G2 cell line.
T-47D (breast cancer) cell lines:
None of the synthesized compounds demonstrated cytotoxicity against the T-47D cell line (IC50 > 100), except 7g, 7h and 7j which showed IC50 = 92.92, 83.42 and 69.35 µM, respectively. Among these compounds, 7j revealed the most potent activity in this series against the T-47D cell line (Table 1). Based on structure–activity relationships of these compounds, the presence of a lipophilic group (e.g., Br) on the phenyl ring at the C-2 position of the indole ring had a toxic effect against the T-47D cell line. Compounds with the cyclohexyl ring as R3 and the methyl group as R2 showed higher toxicity against this cell line compared with the other compounds. On the whole, all the prepared compounds were less cytotoxic against T-47D compared with other cell lines.
To further assess the cytotoxic effects of the synthesized compounds, compounds 7a and 7f (the most potent compounds against A-549), were evaluated against the normal human cell lines HDF. The results revealed that these compounds were noncytotoxic at 100-μM concentrations to the studied normal cells.
Morphological analysis
The morphological assessment of the A-549 cells by the acridine orange/ethidium bromide double-staining method was used to reveal the potential of compounds 7a and 7f as apoptotic inducers [28]. Living cells have a normal green nucleus, but apoptotic cells show orange-stained nuclei with chromatin condensation or fragmentation. Accordingly, compounds 7a and 7f were investigated in comparison with etoposide for identification of apoptosis induced in A-549 cells.
Analysis of the acridine orange/ethidium bromide double-staining of the selected most potent compounds, 7a and 7f, in the A-549 cell line is shown in Fig. 2, in which the viable cells are observed to be green but the apoptotic cells with chromatin condensation or fragmentation show orange-stained nuclei. Morphological findings indicated that compounds 7a and 7f reduced cell viability and induced apoptosis in A-549 cells. The lung cancer cells treated with 7a and 7f showed higher apoptosis compared with etoposide as the reference drug.

Acridine orange/ethidium bromide double-staining of cancer cells with characteristic symptoms of apoptosis of A-549. a DMSO 1% as control, b–d cells treated with IC50 concentrations of etoposide, compounds 7a and 7f, respectively, for 24 h. White arrows indicate live cells, dashed arrows show apoptosis. The images of the cells were taken with a fluorescence microscope at magniciation of ×400
Flow cytometry analysis
The apoptosis induction caused by the prepared compounds was also confirmed by flow cytometry analysis. Annexin V-FITC/PI was used as a quantitative method for determining apoptosis. The results indicated that the prepared compounds could induce apoptosis in A-549 cancer cells. Flow cytometric analysis revealed that cells undergo apoptosis after treatment with the test compounds. As illustrated in Fig. 3, the apoptotic index of compound 7a and 7f was compared with the negative control and etoposide in the A-549 cells. Double-staining followed by flow cytometric analysis revealed the percentages of apoptotic cells as 39.3% and 33.9% resulting from the treatment with compounds 7a and 7f after 24 h incubation, respectively. In addition, the corresponding value obtained after the treatment with etoposide was 32.1% following 24 h incubation (Fig. 3).

Flow cytometric analysis of A-549 cells treated with the prepared compounds 7a and 7f. Cells were stained with annexin V-FITC/PI and quantitated by flow cytometry. Cells were treated with DMSO 1% (negative control) or with IC50 values of etoposide (positive control)
Molecular docking study
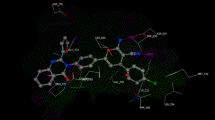
Molecular docking as an important method in structure-based computer-assisted drug design for predicting the main binding mode(s) of a ligand with a protein of known three-dimensional structure [29]. Molecular docking simulations and analysis of the binding modes of the designed compounds within tubulin and ATPase domain of topoisomerase IIα active sites were performed to rationalize the anticancer activity results. At first, the binding sites of colchicine and adenylyl-imidodiphosphate (ANP) were precisely characterized inside the active site of tubulin and the ATPase domain of topoisomerase IIα, respectively (Fig. 4). The formation of hydrogen bonds between the carbonyl group attached to the cycloheptyl ring of colchicine and NH of Val181, as well as the methoxy group and the SH group of Cys241, can be seen in Fig. 4a,. In addition, hydrophobic interactions of different parts of colchicine with Lys352, Val315, Ala180, Met259, Leu255, Leu248, Ala250, and Asn258 have been well established. Figure 4b also clearly represents the hydrogen bonding interaction of some parts of ANP with Gly166, Tyr165, Asn163, Ser149, Gln376, Asn150, Asn91, Arg162, and Asn120 residues. Hydrophobic interactions of ANP with Lys168, Ala167, Lys378, Gly161, Arg1 and Asp94 have also been fully recognized, as indicated in Fig. 4.

Binding sites of a colchicine and b ANP in the active site of tubulin and ATPase domain of topoIIα. Green dashed lines indicate hydrogen bonds
Docking studies revealed that the binding modes of the most active compounds are coherent with colchicine and ANP.
Binding mode of the prepared compounds
Molecular interactions established for the studied compounds inside the ATPase domain of topoisomerase IIα and tubulin obtained by molecular docking protocol are summarized in Tables 2 and 3, respectively.
Compounds 7c with methoxy at the para phenyl ring and cyclohexyl as R3, and 7j bearing bromo at the para phenyl ring again with cyclohexyl as R3 showed the highest free binding energy at topoisomerase IIα and tubulin active sites, respectively (Tables 2, 3). It seemed that the para methoxy- and para bromo-substituted phenyl might be responsible for additional hydrophobic interactions with the topoisomerase IIα and tubulin. In antiproliferative activity assay, compound 7c showed weak activity against T-47D and MCF-7 cell lines (Table 1), whereas 7j showed the highest activity against the Hep-G2, T-47D and MCF-7 cell lines among the compounds with R2 = CH3 and R3 = cyclohexyl. Moreover, this compound showed moderate activity against Athe -549 cell line in biological assays, as shown in Table 1.
Compound 7c was involved in hydrophobic interactions with the side chains of Ala92, Asn91, Thr159, Asp94, Ser149, Ile125, Gly164, Gly166, Ala167 and Thr147 residues of topoisomerase IIα. In addition, the carbonyl group of Asn95 formed a hydrogen bond with the hydrogen of NH attached to the imidazopyridine moiety. The amine group of Lys168 formed a hydrogen bond with the methoxy group of the phenyl moiety in 7c. This can be seen in Fig. 5 for compounds 7c and 7j inside both the active sites. Moreover, compound 7j interacts hydrophobically with Asn150, Asn91, Asn95, Thr215, Thr147, Asp94, Ser148, Arg98, Ser149 and Ile141 residues in topoisomerase IIα. However, this compound did not show any hydrogen bond with the active site residues, as can be seen in Fig. 5. Neither of the compounds 7c and 7j displayed any hydrogen bond in the tubulin protein. However, these two compounds interact with Leu255, Ala250, Asn249, Lys254, Leu248, Asn258, Met259, Ala316, Lys352, Thr353 and Ala317 residues inside the tubulin active site in a hydrophobic manner (Fig. 5).

Interactions of 7c and 7j in topoisomerase IIα (a, b) and in tubulin active sites (c, d)
Compound 7f revealed the highest activity among the first series against the A-549 cell line according to the antiproliferative activity assay (10.66 µM). Compound 7f did not show any hydrogen bonding with the amino acids at the tubulin active site, whereas the NH group of this compound exhibited a hydrogen bond with the carbonyl group of the Asn95 residue. Docking studies showed that the steric orientation of 7f was similar to colchicine and ANP inside their corresponding active sites. This can be seen in Fig. 6 by superimposing 7f with ANP and colchicine inside both the active sites.

Superimposition of 7f and colchicine inside the tubulin active site (a), superimposition of 7f and ANP inside the topoisomerase IIα active site (b)
Replacing the cyclohexyl ring of 7a–k with the t-butyl group of 7l–t (R3) revealed a decrease in the free binding energy at both active sites. For example, compound 7c with the cyclohexyl ring showed the highest free binding energy, while compound 7n bearing the t-butyl group showed the lowest free binding energy, as can be seen in Table 2. This can be explained by the higher hydrophobicity of compound 7c compared to 7n due to the greater lipophilicity of the cyclohexyl ring. The docking results also revealed that all the compounds except 7g and 7q formed cation–π stacking interactions with Lys168, Arg98 and Lys157 topoisomerase IIα amino acids (Fig. 7). None of the studied compounds participated in cation–π stacking interactions with tubulin amino acids.

Compound 7n inside the topoisomerase IIα enzyme binding site. Orientations of 7n phenyl ring, Arg98 guanidino and Lys168 amine groups in cation–π stacking interactions are demonstrated by conical labels
Molecular docking simulations showed that all compounds had more in silico affinity to tubulin compared to the topoisomerase IIα enzyme active site. This suggests that all the compounds might be stronger inhibitors of tubulin copmared with the topoisomerase IIα enzyme.
Conclusion
The present study describes the synthesis of a series of imidazopyridine derivatives of indole (7a–t). All the synthesized analogs were screened for antiproliferative activity against the A-594, Hep-G2, T-47D and MCF-7 cell lines using the MTT assay and with colchicine and etoposide as reference drugs. Structure–activity relationships analysis revealed that the presence of the cyclohexyl ring substituted to the second amine of the imidazopyridyl moiety, and that the phenyl ring (7a) and para-methylphenyl ring (7f) of the C-2 indole ring are appropriate for the cytotoxic activity against the A-594 cell line. In addition, compounds 7d and 7j with the para-chlorophenyl ring and the para-bromophenyl ring of the C-2 indole ring, respectively, showed the most potent activity against the Hep-G2 cell line. However, morphological analysis by the acridine orange/ethidium bromide double-staining test and flow cytometry analysis confirmed the induction of apoptosis in A-549 cells by compounds 7a and 7f. Molecular docking studies were performed to recognize the effects of substituents on the anticancer activity. Molecular docking studies established the binding modes of this series into tubulin and the ATPase domain of topoisomerase active sites, and also exposed the favorable interactions of the active molecules with the two enzyme residues. The higher ΔGbinding obtained for the compounds of the 7a–k series compared with the ΔGbinding of the 7l–t members confirmed the effect of the higher lipophilicity on hydrophobic interactions with the studied enzymes. Moreover, all the compounds showed a higher affinity to tubulin than to the topoisomerase IIα enzyme. Further investigations on these derivatives could lead to more potent compounds as promising candidates for the development of new anticancer chemotherapy.
References
H. Patel, N. Darji, J. Pillai, B. Patel, Int. J. Drug. Res. Tech. 2, 225 (2012)
S. Shaaban, A. Negm, E.E. Ibrahim, A.A. Elrazak, Oncol. Rev. 8, 246 (2014)
J.M. Yi, X.F. Zhang, X.J. Huan, S.S. Song, W. Wang, Q.T. Tian, Y.M. Sun, Y. Chen, J. Ding, Y.Q. Wang, C.H. Yang, Z.H. Miao, Oncotarget 6, 8960 (2015)
K.E. Hevener, A.T. Verstak, K.E. Lutat, D.L. Riggsbee, J.W. Mooney, Acta Pharm. Sin. B 8(6), 844 (2018)
D.S. Takur, Int. J. Pharm. Sci. Nanotechnol. 3(4), 1173 (2011)
N. Fani, A.K. Bordbar, Y. Ghayeb, S. Sepehri, J. Biomol. Struct. Dyn. 33(3), 471 (2015)
N. Fani, A.K. Bordbar, Y. Ghayeb, S. Sepehri, J. Biomol. Struct. Dyn. 33(10), 2285 (2015)
A. Basnet, P. Thapa, R. Karki, H. Choi, J.H. Choi, M. Yun, B.S. Jeong, Y. Jahng, Y. Na, W.J. Cho, Y. Kwon, C.S. Lee, E.S. Lee, Bioorg. Med. Chem. Lett. 20, 42 (2010)
S. Shaaban, F. Sasse, R. Diestel, B. Hinkelmann, Y. Muthukumar, R.P. Verma, C. Jacob, Eur. J. Med. Chem. 58, 192 (2012)
S. Shaaban, A. Negm, A.M. Ashmawy, D.M. Ahmed, L.A. Wessjohann, Eur. J. Med. Chem. 122, 55 (2016)
S. Shaaban, D. Vervandier-Fasseur, P. Andreoletti, A. Zarrouk, P. Richard, A. Negm, G. Manolikakes, C. Jacob, M. Cherkaoui-Malki, Bioorg. Chem. 80, 43 (2018)
P.C. Diao, K.H. Hong, Q. Li, M.J. Hu, Y.F. Ma, W.W. You, P.L. Zhao, Eur. J. Med. Chem. 134, 110 (2017)
D.R. Kerzare, P.B. Khedekar, J. Pharmsci. Biosci. Res. 6(1), 144 (2016)
S. Suzan, Curr. Org. Chem. 21(20), 2068 (2017)
A.M. Metwally, S. Shaaban, B.F. Abdel-Wahab, G.A. El-Hiti, Curr. Org. Chem. 13(14), 1475 (2009)
S.N. Baytas, N. Inceler, A. Yılmaz, A. Olgac, S. Menevse, E. Hamel, R. Bortolozzi, G. Viola, Bioorg. Med. Chem. 22, 3096 (2014)
J.P. Perchellet, E.M. Perchellet, C.R. Singh, Anticancer Res. 34(4), 1643 (2014)
P. Chen, Y.X. Zhuang, P.C. Diao, F. Yang, S.Y. Wu, L. Lv, W.W. You, P.L. Zhao, Eur. J. Med. Chem. 162, 525 (2019)
G.L. Regina, R. Bai, A. Coluccia, V. Famiglini, S. Pelliccia, S. Passacantilli, C. Mazzoccoli, V. Ruggieri, A. Verrico, A. Miele, L. Monti, M. Nalli, R. Alfonsi, L.D. Marcotullio, A. Gulino, B. Ricci, A. Soriani, A. Santoni, M. Caraglia, S. Porto, E.D. Pozzo, C. Martini, A. Brancale, L. Marinelli, E. Novellino, S. Vultaggio, M. Varasi, C. Mercurio, C. Bigogno, G.M. Dondio, E. Hamel, P. Lavia, R. Silvestri, J. Med. Chem. 58(15), 5789 (2015)
A.V. Subba Rao, M.V. Vishnu Vardhan, N.V. Subba Reddy, T. Srinivasa Reddy, S.P. Shaik, C. Bagul, A. Kamal, Bioorg. Chem. 69, 7 (2016)
T.S. Harrison, G.M. Keating, CNS Drugs 19, 65 (2005)
S. Shaaban, B.F. Abdel-Wahab, Mol Divers. 20(1), 233 (2016)
K. Okseon, J. Yujeong, H. Lee, S.S. Hong, S. Hong, J. Med. Chem. 54, 2455 (2011)
A. Kamal, V.S. Reddy, S. Karnewar, S.S. Chourasiya, A.B. Shaik, G.B. Kumar, C. Kishor, M.K. Reddy, M.P. Narasimha Rao, A. Nagabhushana, K.V. Ramakrishna, A. Addlagatta, S. Kotamraju, Chem. Med. Chem. 8(12), 2015 (2013)
A.T. Baviskar, C. Madaan, R. Preet, P. Mohapatra, V. Jain, A. Agarwal, S.K. Guchhait, C.N. Kundu, U.C. Banerjee, P.V. Bharatam, J. Med. Chem. 54, 5013 (2011)
K.T. Ashitha, C.T.F. Salfeena, J. Renjitha, V.P. Kumar, R. Parveen, B.S. Sasidhar, Curr. Bioact. Compd. 14(4), 445 (2018)
Z. Bakherad, M. Safavi, A. Fassihi, H. Sadeghi-Aliabadi, M. Bakherad, H. Rastegar, J.B. Ghasemi, S. Sepehri, L. Saghaie, M. Mahdavi, Res. Chem. Intermed. 45(5), 2827 (2019)
M. Safavi, N. Esmati, S.K. Ardestani, S. Emami, S. Ajdari, J. Davoodi, A. Shafiee, A. Foroumadi, Eur. J. Med. Chem. 58, 573 (2012)
S. Sepehri, S. Soleymani, R. Zabihollahi, M.R. Aghasadeghi, M. Sadat, L. Saghaie, A. Fassihi, Chem. Biodivers. 14(12), e1700295 (2017)
T. Akbarzadeh, S. Noushini, S. Taban, M. Mahdavi, M. Khoshneviszadeh, M. Saeedi, Mol. Divers. 19, 273 (2015)
M. Safavi, A. Ashtari, F. Khalili, S.S. Mirfazli, M. Saeedi, S. Ardestani, K.P. Ranjbar, R.M. Barazandeh Tehrani, B. Larijani, M. Mahdavi, Chem. Biol. Drug. Des. 92(1), 1373 (2018)
S. Sepehri, L. Saghaie, A. Fassihi, Mol. Inform. 36(3), 1 (2017)
Acknowledgements
This work was supported by grants from the Isfahan University of Medical Sciences and the Tehran University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bakherad, Z., Safavi, M., Sepehri, S. et al. Preparation of some novel imidazopyridine derivatives of indole as anticancer agents: one-pot multicomponent synthesis, biological evaluation and docking studies. Res Chem Intermed 45, 5261–5290 (2019). https://doi.org/10.1007/s11164-019-03915-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-019-03915-z