
Abstract
A magnetic ionic liquid supported on γ-Fe2O3 nanocatalyst was synthesized successfully and characterized by Fourier transform infrared spectroscopy, vibrating sample magnetometry, thermogravimetric analysis, differential scanning calorimetry, X-ray diffraction and scanning electron microscopy. The resulting nano-Fe3O4-supported, ionic liquid was an efficient catalyst for preparation of a series of tetracyclic quinazoline compounds by three components reaction of a mixture of isatoic anhydride and amine with ninhydrin in PEG and the desired products were obtained in good to excellent yields. High efficiency, waste-free, mild reaction conditions, effortless magnetic recovery and reusability up to four continuous cycles are the noteworthy features of the currently employed heterogeneous catalytic system.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
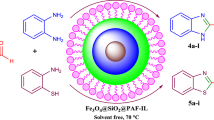
One of the 12 principles of green chemistry is the design of chemical processes that reduce or eliminate chemical waste and cost of production [1, 2]. This can be achieved by developing alternative and sustainable technologies that are non-toxic to living things and the environment [3]. The ionic liquids (ILs) method can be used to achieve this aim, because of their unique combination of thermal and chemical stability, low vapour pressure, low melting point, high conductivity, ability to dissolve organic and inorganic solutes and catalytic properties [4, 5]. Moreover, the relationship between the structure of ionic liquids and their specific performance further extends the scope of application of ILs. Use of heterogeneous catalysts in organic syntheses has gained more significant attention than that of homogeneous ones due to their excellent stability, recovery and reusability of the catalyst. On the basis of this awareness, ILs can modify different support materials, such as mesoporous silica [6], zeolites [7], polystyrene [8] and magnetic nanoparticles [9,10,11,12,13,14,15], and in this way the unique physicochemical properties of ILs can be transferred to substrates. The lack of mass transfer restrictions increase the number of accessible active sites of the catalyst, which is desirable from an economic and toxicological point of view [16]. Among these supports, magnetic nanoparticles (MNPs) have attracted more attention, because they are readily dispersed and provide a higher density of active sites available for catalysis. Moreover, magnetic nanoparticles are widely applied in drug delivery, medical diagnosis, environmental science, material science and catalysis [17, 18]. Knowing the importance of supported ionic liquids in catalysis and in continuation of our previous nanostudies herein, we represent an environmentally friendly approach for the preparation of magnetically ionic liquid supported on a γ-Fe2O3 nanocatalyst with high magnetic sensitivity and its application in multicomponent reactions (Scheme 1). Among fused heterocyclic compounds, tetracyclic quinazoline has recently attracted much attention due to its wide range of biological and pharmacological activities [19]. Furthermore, the quinazoline skeleton has been widely used as building blocks in various natural products [20, 21]. Consequently, several methods have been reported for promoting the preparation of tetracyclic quinazoline: (1) via the condensation reaction of isatoic anhydride, amine, and ninhydrin in the presence of HCl (2) utilizing the palladium-catalyzed carbonylation of commercially available 2-bromobenzyl amine and 2-bromoaniline as the starting materials (3) through radical approach for example using N-acylcyanamides as radical partners in cyclization cascade processes in the presence of AIBN/n-Bu3SnH. Although these methods are suitable for certain synthetic conditions, but there are still some drawbacks such as, low yield of products, toxic and corrosive. Also the used catalysts and solvents are not acceptable in the context of green synthesis. Thus, the development of a new, efficient and green approach for the synthesis of tetracyclic quinazoline compounds is highly desirable. Herein, we report the preparation of a novel heterogeneous catalyst, which proved to be highly efficient for the synthesis of tetracyclic quinazoline in PEG.

Schematic synthesis of nano-Fe3O4-supported, ionic liquid
Results and discussion
Scheme 1 illustrated the pathway of synthesis of nano-Fe3O4-supported, ionic liquid as a new magnetic, recyclable heterogeneous catalyst for synthesis of a series of tetracyclic quinazoline compounds.
Characterization
The synthesized catalyst was characterized on the basis of NMR spectroscopy, Fourier transform infrared (FT-IR), thermogravimetric analysis, differential scanning calorimetry (TG/DSC), X-ray diffraction (XRD), vibrating sample magnetometry (VSM), and scanning electron microscopy (SEM). In the IR spectrum of Fe3O4/SiO2-Propyl-Cl (Fig. 1a) a broad band at 3400 cm−1 that can be attributed to the stretching vibration frequency of the O–H band of Fe3O4 nanoparticles, while the two characteristic absorption bands at 579 and 464 cm−1 correspond to the Fe–O stretching modes of the Fe3O4 lattice. Also, the peak at 1091 cm−1 is due to the Si–O–Si stretching vibrations. Moreover, the bands at 2926 and 2963 cm−1 are attributed to the stretching modes of C–H (Fig. 1b). In comparison to the IR spectra of Pip-His and Fe3O4/SiO2-Propyl-Pip-His (Fig. 1b, c), it is obvious that a band at 1470 cm−1 appears at the same regions in two spectra; (Fig. 1c) characteristic stretching of the C=C band can be assigned to the existence of Pip-His around the magnetic cores. The presence of a strong broad stretching band in the 3397 cm−1 region (Fig. 1d) correspons to the O–H band of sulfonic acid (Fig. 1d).

(a) FT-IR spectra of Fe3O4/SiO2-Propyl-Cl, (b) Pip-His, (c) Fe3O4/SiO2-Propyl-His, (d) Fe3O4/SiO2-Propyl-Pip-His-SO3H)

XRD pattern of magnetic ionic liquid
The XRD pattern of Fe3O4/SiO2-Propyl-Pip-His shows the typical peaks at 30.07°, 35.52°, 43.12°, 53.36°, 57.19°, 62.79°, respectively which is consistent that the standard Fe3O4 XRD spectrum (Fig. 2) [22]. The stability of sulfamic acid-functionalized Fe3O4 was investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), indicating the formation of bonds between MNPs and the catalyst. Sulfamic acid-functionalized Fe3O4 shows three-step weight loss behaviour. (1) Mass loss below 150 °C was attributed to the loss of adsorbed solvent or trapped water from the catalyst. (2) Around 300 °C, a large weight reduction occurred, which can be mainly ascribed to the decomposition of SO3H groups (Fig. 3). (3) Above 700 °C, the occurrence of further mass losses at higher temperature resulted from the decomposition of the silica shell. The DSC thermogram of sulfamic acid-functionalized Fe3O4 showed an exothermic peak at 98 °C, which is due to dehydration. Also, the DSC thermogram of nanoparticles showed an endothermic peak at 200–600 °C as a result of oxidation–reduction reactions. Moreover, it showed an exothermic peak at 680 °C, probably due to the decomposition of trialkoxysilane moiety (Fig. 4).

TGA and DSC thermograms of magnetic ionic liquid

Magnetization curve of magnetic ionic liquid
The magnetization curves of Fe3O4/SiO2-Propyl-His and Fe2O3 were measured at room temperature using a vibrating sample magnetometer (VSM). As seen, the saturation magnetization of the sample decreases in comparison with Fe3O4 [23] due to functionalization of magnetic nanoparticles. The morphology of the nanocomposite was observed on scanning electron microscopy. Figure 5 displays the SEM micrograph of Fe3O4/SiO2-Propyl-Pip-His-SO3H. As can be seen, the nanocomposite has spherical morphology.

SEM images of magnetic ionic liquid
Catalytic activity of Fe3O4/SiO2-Propyl-His was next evaluated in synthesis of a series of tetracyclic quinazoline compounds.
Isatoic anhydride with a benzyl amine and ninhydrin was selected as model reaction and the effect of various parameters such as amount of catalyst, nature of the solvents, and temperature were studied (Table 1). In order to reduce the formation of by-products, the isatoic anhydride and benzyl amine were mixed and stirred at 80 °C. Then, ninhydrin was added to this mixture. First, the effect of solvents was investigated and it was observed that the desired product was obtained in the PEG or DMSO solvent (Table 1, entries 1 and 5), while reaction in solvents such as toluene and water was ineffective. The effect of temperature was also investigated, and the highest yield was obtained at 80 °C. When the reaction was conducted at 100 and 130 °C, a low-yield was seen. The control experiment confirmed that the reaction did not occur in the absence of the catalyst (Table 1, entry 10). The influence of the amount of catalyst on the yield of the product was evaluated. It was observed that 30 mg of catalyst was the optimum. When the reaction conducted under the same conditions in the presence of Fe3O4, it was observed that the desired product was not obtained. We then investigated the substrate scope of this synthesis by subjecting the series of aliphatic and aromatic amines to the reactions with isatoic anhydride and, ninhydrin, the results are shown in Table 2. From these results, we observed that all of the reactions produced the corresponding tetracyclic quinazoline compounds in excellent yields (60–92%). A synthetic route is outlined in Scheme 2, Based on previous reports [24] carbonyl group of isatoic anhydride gives nucleophilic addition reaction, leading to ring opening followed by decarboxylation to compound B. Subsequently, nucleophilic attack by amines to the keto group of ninhydrin forms imines intermediate C; cyclization of this product leads to intermediates D; then nucleophilic attack of amine on the keto group provides a aziridine intermediate E, Finally, desired product is formed by rearrangement intermediate E.


Proposed mechanism for the synthesis of tetracyclic quinazoline compounds catalyzed by magnetic ionic liquid
The possibility of recovering and recycling the catalyst is an important issue from economic and environmental points of view. After performing the three-component reaction of a mixture of isatoic anhydride and amine with ninhydrin in PEG under the optimized conditions, EtOAc was added to the reaction mixture. The catalyst was separated by an external magnet from the reaction mixture (Fig. 6), washed three times with acetone and then with doubly distilled water several times. Then the recovered catalyst was used in the next run. This catalyst was recycled and reused at least four times without significant loss of its catalytic activity (Fig. 6).
In conclusion, we have developed a facile route to synthesis nano-Fe3O4–supported, ionic liquid and in addition, the excellent performance in synthesis of tetracyclic quinazoline compounds using isatoic anhydride, a benzyl amine and ninhydrin were investigated. According to the obtained results, the catalyst is stable and can be easily separated by magnetic decantation and reused many times with less deterioration in catalytic activity (Fig. 6).

Catalyst recycling study for the synthesis of quinazoline tetracyclic compounds
Experimental
Materials
All reagents and solvents used in this work were purchased from Sigma-Aldrich, Fluka or Merck Chemical Companies and used without further purification. NMR spectra were recorded on a Bruker AMX 300 MHz spectrometer and Bruker Avance III 400 MHz also 1H NMR, 13C NMR were recorded on either a Varian Mercury Plus operating at 400 MHz (1 H) or 100.6 MHz (13C), chemical shifts are given in ppm (d) relative to a TMS internal standard, and the coupling constants J reported in Hz. Melting points were measured with an Electrothermal 9100 apparatus. VSM measurements were performed using a vibrating sample magnetometer (VSM) MDKFD. The nanostructures were characterized using a Holland Philips X’pert X-ray powder diffractometer (XRD) (CoKa, radiation = 0.154056 nm). IR spectra were obtained as KBr pellets on a VRTEX 70 model BRUKER FTIR spectrophotometer. Thermogravimetric analysis (TGA) curves were recorded using STA NETZSCH 499 F3 Germany. The particle morphology was examined by SEM using a FESEM-TESCAN. Elemental analysis was examined by CHNSO COSTECH England.
Catalyst preparation
Preparation of 1, 1-(piperazine-1, 4-diyl) diethanone
A mixture of piperazine (2 mmol), acetyl chloride (5 mmol) and potassium carbonate (2 mmol) was stirred in acetonitrile (5 mL), under reflux conditions at 80 °C for 24 h. After completion of the reaction, the mixture was cooled to room temperature, filtered, and the white powder was washed with ethanol. Mp: 143–145 [25], FT-IR (KBr): νmax/cm−1: 580.61, 765.33, 872.31, 959.45, 1006.50, 1068.87, 1252.77, 1321.35, 1362.36, 1444.52, 1551.32, 1621.48, 2420.73, 2595.62, 3017.17, 3204.63.
Preparation of Pip-His
A mixture of 1,1-(piperazine-1, 4-diyl) diethanone (2.0 mmol), histidine ethyl ester hydrochloride (4.0 mmol) and 2 mmol carbonate potassium in dry toluene was heated at 100 °C for 24 h. After completion of the reaction, the reaction mixture was cooled to room temperature, filtered and washed with ethanol. Finally, a white powder was obtained. 1H NMR (400 MHz, D2O) δ 1.09 (t, J = 7, 3H), 1.97 (s, 3H), 2.71 (s, 1H), 2.83–2.79 (m, 1H, CαH), 2.94–2.91 (m, 2H, CβH), 6.73–6.85 (m, 1H, ArH), 7.95–7.97 (m, 1H, ArH), FT-IR (KBr): νmax/cm−1: 697.87, 789.43, 864.13, 1034.38, 1415.43, 1658.49, 2880.43, 2928.39, 3415.78.
Preparation of Fe3O4
A mixture of FeCl3·6H2O (5.838 g) and FeCl2·4H2O (2.147 g) was dissolved in 100 mL of deionized water in a three-necked round-bottom flask (250 mL) at 80 °C under N2 atmosphere. Then, 10 mL of aqueous NH3 solution (32%) was added to the mixture over 30 min with vigorous mechanical stirring. The resulting black solid was washed with double-distilled water until neutrality, further washed twice with ethanol and dried at 80 °C in vacuum.
Preparation of Fe3O4@SiO2
Fe3O4 (0.50 g) was dispersed in a mixture of ethanol (50 mL), deionized water (5 mL) and TEOS (0.20 mL), followed by the addition of 2 mL of concentrated ammonia solution. This solution was stirred mechanically for 38 h at room temperature. Then the product, Fe3O4@SiO2, was separated by an external magnet and washed with deionized water and ethanol three times and dried at 80 °C.
Preparation of Fe3O4/SiO2-Propyl-Cl
The prepared Fe3O4/SiO2 (1 g) was sonicated in dry toluene (50 mL) for 30 min. (3-chloropropyl) trimethoxysilane (CPTMS) (5 mmol, 1 mL) was added to the dispersed Fe3O4/SiO2 in dry toluene and stirred for 24 h under reflux conditions, and the obtained chloropropyl-functionalized solid (Fe3O4/SiO2-Propyl-Cl) was washed with ethanol three times and dried at 80 °C.
Preparation of Fe3O4/SiO2-Propyl-His
Fe3O4/SiO2-Propyl-Cl (1 g) and KOH (2 mmol, 0.112 g) were added to the solution of Pip-His (2 g) in dry toluene (100 mL). The mixture was sonicated for 0.5 h, followed by stirring for 28 h under reflux conditions. The obtained solid was magnetically collected from the solution and washed with water/ethanol (20:10 mL) three times and dried at 80 °C.
Preparation of Fe3O4/SiO2-Propyl-Pip-His-SO3H)
To a mixture of piperazine-modified silica-coated Fe3O4 MNPs (Fe3O4/SiO2-Propyl-Pip) (0.5 g) in dry CH2Cl2 (3 mL), chlorosulfonic acid (ClSO3H, 1 mL) was added dropwise to a cooled ice-bath over a period of 30 min at room temperature then the mixture was filtered and washed with dried CH2Cl2 and dried at room temperature to afford the title compound.
Synthesis of tetracyclic quinazoline compound derivatives general procedure
A mixture of isatoic anhydride (1.0 mmol), amine (1 mmol) and Fe3O4/SiO2-Propyl-Pip-SO3H (0.03 g) in PEG-400, was stirred at 80 °C. After the reaction was completed, ninhydrin (1 mmol) was added into the resulting mixture. Then the mixture was heated at 80 °C and the reaction was monitored by TLC (hexane/ethyl acetate, 4:1) for appropriate time. After completion, the mixture was cooled to room temperature and 20 mL of ethyl acetate was added to the organic phase and washed with water, dried over anhydrous Na2SO4, and filtered. The residue was purified by column chromatography (silica gel, hexane/ethyl acetate, 4:1) to afford the corresponding product.
NMR data
Compound 1 (Table 2)
Yellow solid; mp: 179–182 °C; 1H NMR (400 MHz, DMSO) δ 4.43 (s, 2H), 7.41 (t, J = 4.2, 2H), 7.55 (d, J = 8.4, 2H), 7.90–7.81 (m, 3H), 8.07 (s, 1H), 8.14 (t, J = 7.6, 2H). 13C NMR (100 MHz, CDCl3): δ = 34.1, 114.2, 117.6, 119.2, 120.0, 124.1, 126.5, 126.6, 127.1, 127.2, 133.1, 133.6, 134.7, 137.4, 137.6, 143.0, 178.0, 180.8.
Compound 2 (Table 2)
Brown solid; mp: 198–200 °C; 1H NMR (400 MHz, DMSO) δ 3.3 (s, 2H), 5.0 (s, 3H), 7.38 (d, J = 12, 1H), 7.41 (d, J = 10, 1H), 7.55 (d, J = 8.4, 1H), 7.68 (s, 1H), 7.88–7.81 (m, 4H), 8.07 (S, 1h), 8.14 (t, J = 14.8, 2H). 13C NMR (100 MHz, CDCl3): δ = 50.9, 114.2, 117.6, 119.2, 120.0, 123.6, 123.8, 124.6, 126.4, 126.5, 127.2, 128.6, 132.5, 133.1, 133.6, 134.6, 137.6, 145.0, 177.9, 180.8.
Compound 3 (Table 2)
Orange solid; mp: 210–213; 1H NMR (400 MHz, DMSO) δ 6.67 (d, J = 7.6, 1H), 6.83–6.82 (m, 1H), 6.94 (d, J = 8.4, 2H), 7.24–7.19 (m, 3H), 7.36–7.35 (m, 1H), 7.73 (d, J = 7.6), 7.98–7.96 (m, 1H), 8.04–8.02 (m, 4H). 13C NMR (100 MHz, CDCl3): δ = 75.1, 114.5, 119.0, 124.5, 128.1, 128.3, 129.2, 134.0, 137.1, 138.1, 139.2, 146.1, 163.3, 195.
Compound 4 (Table 2)
White solid; mp: 241–245 °C; 1H NMR (400 MHz, DMSO) δ 1.5 (t, J = 7.4 3H), 2.52 (q, J = 7.4 3H), 7.23–7.17 (m, 4H), 7.68–7.64 (m, 3H), 7.95 (d, J = 8, 3H). 13C NMR (100 MHz, CDCl3): δ = 16.1, 18.6, 112.5, 123.7, 126.3, 126.5, 126.8, 128.6, 129.1, 131.8, 132.8, 134.1, 134.3, 144.4, 167.5, 180.6. FT-IR (KBr) νmax/cm−1: 462, 527, 680, 726, 1019, 1091, 1161, 1408, 1435, 1477, 1652, 1714, 2849, 2926, 3023, 3062. Anal. Calcd for C24H18N2O3: C, 75.38; H, 4.74; N, 7.33; O, 12.55, Found: C, 74.0; H, 6.19; N, 7.60; O, 12.21%.
Compound 5 (Table 2)
White solid; mp: 261–265 °C; 1H NMR (400 MHz, CDCl3) δ 1.3 (d, J = 8, 6H), 2.88 (m, 1H), 7.49 (d, J = 8.8, 2H), 7.79–7.75 (m, 2H), 7.83–7.81 (m, 3H), 8.0–7.98 (m, 3H), 8.2 (d, J = 7.6, 1H), 8.3 (t, J = 7.2, 1H). 13C NMR (100 MHz, CDCl3): δ = 10.2, 30.0, 114.7, 115.0, 123.3, 125.3, 128.3, 129.0, 134.9, 138.6, 152.2, 162.4, 200.2. FT-IR (KBr) νmax/cm−1: 540, 662, 737, 819, 901, 951, 1033, 1249, 1312, 1408, 1511, 1593, 1630, 1708, 2924, 3024. Anal. Calcd for C25H20N2O3: C, 75.74; H, 5.09; N, 7.07; O, 12.11, Found: C, 75.08; H, 6.19; N, 7.60; O, 11. 13%.
Compound 6 (Table 2)
Orange solid; mp: 151–155 °C; 1H NMR (400 MHz, CDCl3) δ 1.1–1.0 (m, 3H), 1.4–1.3 (m, 3H), 1.74–1.70 (m, 3H), 7.17 (d, J = 8, 2H), 7.29 (q, J = 6.8, 2H), 7.66–7.62 (m, 2H), 8.17 (d, J = 7.6, 2H). 13C NMR (100 MHz, CDCl3): δ = 14.1, 20.1, 29.9, 40.6, 114.2, 115.5, 122.9, 127.8, 135.4, 139.9, 150.6, 162.3, 192.3.
Compound 7 (Table 2)
Brown solid; mp: 195–199 °C; 1H NMR (400 MHz, CDCl3) δ 7.95–7.91 (m, 2H), 8.1–8.06 (m, 3H), 8.1 (d, J = 8 2H), 8.2 (d, J = 7.6, 2H).
Compound 8 (Table 2)
White solid; mp: 161–164 °C; 1H NMR (300 MHz, CDCl3) δ 1.75–1.59 (m, 10H), 2.51–2.41 (m, 1H), 7.04 (d, J = 8, 2H), 7.26–7.19 (m, 2H), 7.62–7.55 (m, 2H), 8.1. (d, J = 8, 2H). 13C NMR (75 MHz, CDCl3): δ = 25.9, 28.3, 28.9, 52.7, 114.1, 114.7, 122.3, 127.4, 134.7, 139.3, 150.2, 162.2, 184.7. FT-IR (KBr) νmax/cm−1:680, 725, 899, 973, 1019, 1075, 1185, 1302, 1374, 1429, 1475, 1609, 1655, 1726, 2858, 2927. Anal. Calcd for C22H20N2O3: C, 73.32; H, 5.59; N, 7.77; O, 13.32, Found: C, 72.14, H, 6.06, N, 9.60; O, 12.2%.
Compound 9 (Table 2)
White solid; mp: 175–180 °C; 1H NMR (400 MHz, DMSO) δ 1.31–1.20 (m, 14H), 3.43–3.34 (m, 1H), 7.21–7.14 (q, J = 8.5, 1H), 7.57 (t, J = 7.2, 1H), 7.69–7.62 (m, 1H), 7.84 (q, J = 4.9, 2H), 7.92 (d, J = 8, 1H), 8.17 (d, J = 8, 1H), 8.4 (s, 1H). 13C NMR (100 MHz, CDCl3): δ = 15.9, 26.1, 29.1, 40.3, 40.6, 115.2, 121.9, 122.8, 126.7, 127.4, 127.5, 134.6, 135.2, 146.6, 147.8, 159.8, 180.9. FT-IR (KBr) νmax/cm−1:463, 537, 676, 743, 821, 942, 1022, 1100, 1153, 1403, 1609, 1656, 1711, 2846, 2923, 3020, 3056. Anal. Calcd for C24H24N2O3: C, 7.21; H, 6.22; N, 7.21; O, 12.36, Found: C, 75.14, H, 6.26, N, 8; O, 10.6%.
References
E. García-Verdugo, B. Altava, M.I. Burguete, P. Lozano, S.V. Luis, Green Chem. 17, 2693 (2015)
R.D. Rogers, K.R. Seddon, Science 302, 792 (2003)
J.S. Wilkes, M.J. Zaworotko, Chem. Commun. 13, 965 (1992)
X. Liu, J. Ma, W. Zheng, Rev. Adv. Mater. Sci. 27, 43 (2011)
V. Campisciano, F. Giacalone, M. Gruttadauria, Chem. Rec. 17, 1 (2017)
M.R. Tchalala, J.K. El-Demellawi, E. Abou-Hamad, J.R.D. Retamal, P. Varadhan, J.H. He, S. Chaieb, Appl. Mater. Today 9, 10 (2017)
K. Sun, Y. Shi, W. Xu, N. Potter, Z. Li, J. Zhu, Chem. Eng. J. 313, 336 (2017)
G. Liu, P. Su, L. Zhou, Y. Yang, J. Sep. Sci. 40, 2603 (2017)
J. Lu, F. Ye, X. Huang, L. Wei, D. Yao, S. Li, H. Lai, J. Sep. Sci. 40, 1133 (2017)
E. Aliyari, M. Alvand, F. Shemirani, RSC Adv. 6, 64193 (2016)
X. Wang, G. Li, K.H. Row, J. Sep. Sci. 40, 3301 (2017)
A. Ghorbani-Choghamarani, M. Norouzi, J. Magn. Magn. Mater. 401, 832 (2016)
H. Abdolmohammad-Zadeh, S. Hassanlouei, M. Zamani-Kalajahi, RSC Adv. 7, 23293 (2017)
M.A. Zolfigol, R. Ayazi-Nasrabadi, S. Baghery, Appl. Organomet. Chem. 30, 273 (2016)
A. Rostami, B. Atashkar, H. Gholami, Chem. Commun. 37, 69 (2013)
R. Skoda-Földes, Molecules 19, 8840 (2014)
M. Afshari, M. Gorjizadeh, S. Nazari, M. Naseh, J. Magn. Magn. Mater. 363, 13 (2014)
A.S. Kumar, B. Thulasiram, S.B. Laxmi, V.S. Rawat, B. Sreedhar, Tetrahedron 70, 6059 (2014)
C. Shen, N.Y. Man, S. Stewart, X.F. Wu, Org. Biomol. Chem. 13, 4422 (2015)
A. Servais, M. Azzouz, D. Lopes, C. Courillon, M. Malacria, Angew. Chem. Int. Ed. 46, 576 (2007)
Y.Y. Han, H. Jiang, R. Wang, S. Yu, JOC 81, 7276 (2016)
A. Ghorbani-Choghamarani, G. Azadi, RSC Adv. 5, 9752 (2015)
S. Sobhani, Z. Pakdin-Parizi, Appl. Catal. A 479, 112 (2014)
V.N. Murthy, S.P. Nikumbh, S.P. Kumar, Y. Chiranjeevi, L.V. Rao, A. Raghunadh, Synlett 27, 2362 (2016)
T. Irikura, K. Masuzawa, K. Nishino, M. Kitagawa, H. Uchida, N. Ichinoseki, M. Ito, J. Med. Chem. 11, 801 (1968)
Acknowledgements
The authors would like to thank the research facilities of Ilam University, Ilam, Iran, for the financial support of this research project.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ghorbani-Choghamarani, A., Taherinia, Z. & Nikoorazm, M. Ionic liquid supported on magnetic nanoparticles as a novel reusable nanocatalyst for the efficient synthesis of tetracyclic quinazoline compounds. Res Chem Intermed 44, 6591–6604 (2018). https://doi.org/10.1007/s11164-018-3510-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-018-3510-1