
Abstract
Fe3O4 nanoparticles were coated with silica followed by surface modification using a silylating agent and piperidine to obtain piperidinium benzene-1,3-disulfonate salt (PBDS) silica-coated magnetic nanoparticles (SCMNPs). The obtained catalyst was then analyzed using various characterization techniques, including thermogravimetric analysis (TGA), scanning electron microscopy (SEM) and transmission electron microscopy (TEM) imaging, and X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET), and energy-dispersive X-ray (EDX) analyses. The heterogeneous catalytic activity was investigated in one-pot pseudo-three-component synthesis of pyran derivatives, revealing efficient action and giving the products in high yield under green and sustainable conditions.
Graphical Abstract
A novel efficient green reusable catalyst was obtained from silica-coated magnetic nanoparticles functionalized with piperidinium benzene-1,3-disulfonate for synthesis of 9-aryl-9H-2,4,5,7-tetramethyl-diuracilopyrans as well as 9-aryl-3,4,6,7-tetrahydro-2H-pyran-1,8(5H,9H)-diones.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pyran is a known structural framework found in the skeleton of many natural products such as (−)-hennoxazole A, an antiviral agent, annularins, and elijopyrone [1–4]. Additionally, compounds based on pyran are pharmacologically active, showing effects such as antituberculosis [5], antitumor [6, 7], anti-human immunodeficiency virus (HIV) [8], antifungal [9], calcium channel antagonist [10], and antimicrobial activities [11], as well as having clinical applications [12, 13]. Recently, a novel pyran-based phosphodiesterase 9A (PDE9A) inhibitor showed good activity experimentally against neurodegenerative diseases such as Down’s syndrome, Parkinson’s disease, Alzheimer’s disease, and schizophrenia [14]. In addition to the biological activities of pyran compounds, they are good precursors for preparation of organic compounds including polyazanaphthalenes, chromene derivatives, lactones, pyridine-2-ones, imidoesters, and pyrimidines [15–18].
Considering the importance of pyran derivatives, 4H-pyrans are prepared through several synthetic procedures, using different catalysts such as magnesium oxide [19], silica-bonded 1,4-diazabicyclo[2.2.2]octane (DABCO) [20], and SnCl2-loaded silica nanoparticles [21]. Green chemistry principles suggest the use of green solvents and green catalysts to reduce waste treatment and enhance the economics of chemical processes, associated with reduced environmental problems [22].
It is noteworthy that some methods published in literature to prepare 4H-pyrans suffer from serious limitations such as complex synthetic pathways, long reaction time, hard workup, and catalyst nonreusability. Therefore, according to environmentally benign design, research has focused on inexpensive and efficient processes to prepare green catalysts which can be separated easily.
Despite the remarkable efficiency of homogeneous catalysts, their separation for reuse in subsequent reactions is extremely difficult. Thus, production and development of new efficient heterogeneous catalysts is reasonably necessary, based on immobilization of homogeneous as well as organocatalysts on substrates, being of great interest to chemists. One of the most applicable such substrates is magnetic nanoparticles (MNPs), which can be separated from a mixture using an external magnet and reused several times [23–26]. Use of MNPs, e.g., nanoparticles of iron oxides (IOs), as a substrate for heterogeneous catalysts has attracted the attention of researchers [27–32]. However, hydrophobic surface and instability in strongly acidic solutions are some of the disadvantages of MNPs, limiting their applications [33]. Functionalization of the surface of MNPs can help to overcome such limitations [34–36].
Correspondingly, ionic liquids (ILs), as solvents or catalysts, have gained wide recognition due to their unique properties of reusability, environmental compatibility, and easy isolation from the reaction mixture [36–43]. Immobilizing functional ILs on a surface can decrease many of their negative properties, such as high viscosity. Hence, ILs are good candidates for functionalizing the surface of MNPs, since MNPs and ILs complement each other, addressing the aforementioned drawbacks [44–47].
In this work, we synthesized several new pyran derivatives using a new IL-functionalized MNP catalyst through a pseudo-three-component condensation reaction of aryl aldehydes and N,N-dimethylbarbituric acid and/or cyclohexane-1,3-diones. We report herein synthesis of this important class of heterocycles from cheap and easily available starting materials using a recyclable and efficient catalyst under green conditions.
Experimental
Materials and methods
All commercially available chemicals were obtained from Merck and Fluka and used without further purification. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker Advance 400 and 100 MHz FT NMR spectrometers, respectively (undertaken at University of Mazandaran, Iran). Infrared (IR) spectroscopy was performed on a PerkinElmer GX Fourier-transform infrared (FT-IR) spectrometer. Mass spectra were recorded on a 5973 Network mass-selective-detector mass spectrometer (undertaken at University of Tehran, Iran). Elemental analyses (CHN) were performed using an Elemental Combustion System 4010 (undertaken at University of Tehran, Iran). Thermogravimetric analysis (TGA) was performed on a Pyris Diamond. Energy-dispersive X-ray analysis of the prepared catalyst was performed on a FESEM-SIGM (Germany) instrument. Scanning electron microscopy (SEM) was performed on an EM3200 instrument operated at accelerating voltage of 30 kV. Transmission electron microscopy (TEM) was performed on a ZEISS EM3200 instrument. Brunauer–Emmett–Teller (BET) analysis was performed on a BELSORP instrument.
Preparation of magnetic nanoparticles (MNPs)
Magnetite phase was synthesized by adding FeCl3 (3 mL, 2 M dissolved in 2 M HCl) to 10.33 mL double-distilled water, followed by dropwise addition of Na2SO3 (2 mL, 1 M) for 3 min under magnetic stirring. The solution color changed from red to light yellow, then 80 mL NH3·H2O solution (0.85 M) was added under severe stirring. After 30 min, the obtained MNPs were separated using a magnet, then washed with distilled water to reach pH <7.5.
Preparation of silica-coated magnetic nanoparticles (SCMNPs)
MNPs@SiO2 were synthesized through a modified Stöber method [48]. MNPs (4.3 mmol, 1 g) were dispersed for 30 min in a mixture of deionized water (80 mL), ethanol (20 mL), and ammonia solution (4.0 mL, 25 % in water) using ultrasound treatment. Then, tetraethylorthosilicate (11 mmol, 2.0 mL) was added to the mixture and stirred for 3 h at room temperature. The resultant product was carefully washed with deionized water and then washed twice with ethanol and dried at 60 °C under vacuum condition (Scheme 1).

Synthesis of PBDS–SCMNPs
Preparation of piperidine-modified SCMNPs
SCMNPs (1.0 g) were dispersed in dry toluene (50 mL) for 30 min, then reacted with 3-chloropropyltrimethoxysilane (CPTS, 2 mL) as silylating agent, which was added to the mixture dropwise. The mixture was heated under reflux condition for 24 h, and the obtained SCMNPs–Pr–Cl were filtered off, washed twice with toluene, and dried at 60 °C under vacuum condition. To prepare pi-SCMNPs, SCMNPs–Pr–Cl (1.0 g) were added to a mixture of dry toluene (50 mL) and piperidine (0.03 mmol, 3 mL). The reaction mixture was dispersed for about 30 min, then heated under reflux and stirring conditions for 72 h. The solid phase was filtered, washed with toluene and ethanol, and dried at 60 °C under reduced pressure (Scheme 1).
Synthesis of piperidinium benzene-1,3-disulfonate salt (PBDS) magnetic nanoparticles (MNPs) as catalyst
pi-SCMNPs (1.0 g) were suspended in dried acetonitrile (50 mL), and benzene-1,3-disulfonic acid (3 mL) was added to this suspension. The mixture was stirred for 24 h at ambient temperature, then filtered, washed twice with acetonitrile, and further washed with deionized water and dried at 60 °C under vacuum condition (Scheme 1).
General procedure for preparation of pyranodipyrimidines 3a–f and xanthene derivatives 5a–f
A mixture of either N,N-dimethylbarbituric acid 1 (0.312 g, 2 mmol) or 1,3-cyclohexanedione 4 (0.224 g, 2 mmol), aromatic aldehyde (1 mmol), and PBDS–SCMNPs (20 mg) as nanocatalyst was vigorously stirred in ethanol at 70 °C under reflux condition for appropriate times (Table 4). After reaction completion, monitored by thin-layer chromatography (TLC), the catalyst was separated using an external magnet. After that, the products were filtered and separated. Then, the obtained product was recrystallized in EtOH to obtain highly purified product. The PBDS–SCMNPs were washed for three to four times with ethanol and then dried at 50 °C for 3 h to recycle the catalyst for use in alternative reactions. In this study, PBDS–SCMNPs were used as an efficient catalyst for five times without significant loss of catalytic activity (Fig. 1).

Investigating the reusability of PBDS–SCMNPs as the catalyst in the model reaction
Results and discussion
Characterization of PBDS–SCMNPs
The synthesized PBDS–SCMNPs, as a novel and efficient catalyst, were characterized using FT-IR, thermogravimetric analysis (TGA), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD) analysis, energy-dispersive X-ray spectroscopy (EDX), vibrating-sample magnetometry (VSM), and Brunauer–Emmett–Teller (BET) specific surface area analysis.
FT-IR spectroscopy was used to compare the produced Fe3O4 MNPs, SCMNPs, and other core–shell surface-modified samples (Fig. 2). The FT-IR spectrum of the magnetic Fe3O4 MNPs showed a characteristic absorption peak of Fe–O bond at about 585 cm−1. The absorption peaks of the silica shell in the SCMNPs at around 1100 and 810 cm−1 can be ascribed to asymmetric and symmetric stretching vibrations of Si–O–Si bond in oxygen–silicon tetrahedral, respectively. Moreover, the absorption peaks of the SCMNPs–Pr–Cl core–shell MNPs at 2957 cm−1 are connected to stretching vibration of C–H groups. Additionally, the absorption band at 3408 cm−1 is related to stretching vibration of N–H on the piperidinium moiety, and the peak appearing at 1645 cm−1 is attributed to stretching vibration of C=C bonds of benzene-1,3-disulfonate. The two peaks observed at 1220 and 1105 cm−1 are related to vibrational modes of sulfonate groups.

IR spectra of MNPs (a), SCMNPs (b), SCMNPs–Pr–Cl (c), pi-SCMNPs (d), and PBDS–SCMNPs (e)
TGA was applied to investigate the possible thermal stability/degradation and to determine the content of organic groups loaded on the surface of the SCMNPs. The first signal in both TGA and derivative thermogravimetric analysis (DTG) at around 100 °C is due to loss of water adsorbed on the surface of the compound. The DTG result for the PBDS–SCMNPs reveals the mass loss of organic groups as they decompose with increasing temperature (Fig. 3). Therefore, in the temperature range between 100 and 500 °C, around 10 % weight loss is observed, confirming a loading of 0.2 mmol/g of organic groups on the surface of the SCMNPs. Both curves display considerable losses in four steps, and the material decomposed after 530 °C.

The TGA and DTA curves of the PBDS–SCMNPs
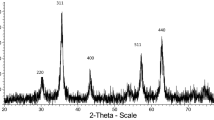
Furthermore, the size, shape, and morphology of the PBDS–SCMNPs were investigated by different methods, including XRD patterns (Fig. 4) and SEM and TEM images (Fig. 5). The XRD diagram of the MNPs displayed patterns consistent with those of spinel ferrites as explained in literature [49, 50]. The same peaks were identified in both of the PBDS–SCMNP XRD diagrams, indicating retention of the crystalline spinel ferrite core structure after the reaction process. The positions and relative intensities of all peaks confirmed the standard XRD pattern of Fe3O4 MNPs [Joint Committee on Powder Diffraction Standards (JCPDS) card no. 85-1436], indicating retention of the crystalline cubic spinel structure of the MNPs. The XRD patterns of the particles showed nine characteristic peaks linked to cubic iron oxide phase (2θ = 30.10°, 35.50°, 43.10°, 53.00°, 57.00°, 62.80°) with corresponding indices of (2 2 0), (3 1 1), (4 0 0), (3 3 1), (4 2 2), and (5 1 1). We conclude that the resultant nanoparticles were pure Fe3O4 with spinel structure and that the grafting process did not induce any phase change of Fe3O4. Also, reported MNPs coated with silica showed the same XRD pattern with increased intensities in the 2θ range between 20° and 30° [50]. Considering the XRD patterns, the main peak observed at 2θ is equal to 35.60°; therefore, the crystalline structure of the catalyst can obviously be identified, as presented in Table 1. The average crystallite size (D) is also given, as determined using the Scherrer formula
where λ is the X-ray wavelength, K is the Scherrer constant, β is the full-width at half-maximum (FWHM) of the peak, and θ is the Bragg diffraction angle. The catalyst average size obtained from this equation was found to lie in the range of 7.38–33.67 nm. SEM and TEM images of PBDS–SCMNPs were further studied to determine the particle size. Figure 5 clearly shows the dimensions and shapes of the particles. The particle sizes obtained from SEM and TEM images (7.38–33.67 nm) support the particle size calculations presented above. Both SEM and TEM revealed XRD patterns with characteristic peaks of the nanocatalyst. These results confirm that our method was efficient for synthesis of PBDS–MNPs.

The XRD spectrum of MNPs (a); SCMNPs (b); SCMNPs–Pr–Cl (c); pi-SCMNPs (d); PBDS–SCMNPs (e)

SEM (a) and TEM (b) images of PBDS–SCMNPs

The EDX data of the PBDS–SCMNPs
The EDX data for the PBDS–SCMNPs (Fig. 6) confirmed the presence of the expected elements iron, silicon, carbon, oxygen, nitrogen, and sulfur in the catalyst.
To fully describe the catalyst, the magnetic properties of the MNPs and PBDS–SCMNPs were characterized by VSM. The room-temperature magnetization curves of the MNPs and PBDS–SCMNPs are shown in Fig. 7. The MNPs showed superparamagnetic properties with saturation magnetization of about 60 emu g−1, while the saturation magnetization of the PBDS–SCMNPs was 36 emu g−1. Compared with the MNPs, the saturation magnetization of the PBDS–SCMNPs clearly decreased since the diamagnetic influence of the organic moieties resulted in lower mass fraction of MNPs. Even with this reduction in saturation magnetization, the solid retained sufficient magnetization to enable its separation from the reaction mixture using a permanent magnet.

The VSM analysis of the PBDS–SCMNPs in comparison with the MNPs
N2 adsorption–desorption measurements were carried out for Brunauer–Emmett–Teller (BET) specific surface area analysis of the materials (Fig. 8). The BET surface area of the PBDS–SCMNP catalyst was 79.4 m2/g (total pore volume = 0.246 cm3 g−1; mean pore diameter = 12.37 nm), and its pore size distribution is plotted in Fig. 8. The surface area obtained for the Fe3O4 MNPs was 10.9 m2/g (total pore volume = 0.047 cm3 g−1; mean pore diameter = 17.25 nm), and their pore size distribution is plotted in Fig. 8. The BET surface area of the PBDS–SCMNP catalyst was higher in comparison with the Fe3O4 MNPs. The specific surface area of the PBDS–SCMNP catalyst increased because of the coating of the Fe3O4 MNPs by grafting organic tags. The higher surface area of the synthesized catalyst in comparison with the Fe3O4 MNP cores can be considered as one of the advantages of this catalyst. The pore size of both samples mainly lay between 1 and 10 nm, revealing that the samples included both micropores and mesopores.

N2 adsorption–desorption (BET) isotherms of Fe3O4 MNPs (a, c) and PBDS–SCMNPs (b, d)
Application of PBDS–SCMNPs as catalyst in synthesis of pyranodipyrimidine 230 and xanthene derivatives
The catalytic activity of PBDS–SCMNPs was tested in the model reaction of N,N-dimethylbarbituric acid with pyridine-2-carbaldehyde; different amounts of nanomagnetic catalyst were investigated at room temperature to 70 °C in EtOH (Table 2). As shown in Table 2, the best results were achieved when the reaction was run using 20 mg nanomagnetic catalyst at 70 °C (Table 2, entry 9). On increasing the amount of nanomagnetic catalyst or changing the temperature, the reaction yield did not change significantly. Table 2 obviously shows that, in the absence of nanomagnetic catalyst, the product was produced with low yield (Table 2, entries 1 and 2).
To optimize the reaction conditions, the reaction of pyridine-2-carbaldehyde and N,N-dimethylbarbituric acid was performed using 20 mg catalyst in numerous solvents including H2O, EtOH, CH3CN, EtOAc, acetone, CH2Cl2, and toluene at 70 °C. The results are summarized in Table 3. As illustrated in Table 3, ethanol was the best choice for this reaction, especially as a benign and green solvent compared with the other organic solvents.
To investigate the generality of the optimized reaction, a wide range of substituted aldehydes were applied to this pseudo-three-component condensation with catalytic amount (20 mg) of PBDS–SCMNPs in EtOH at 70 °C. The results are summarized in Table 4. The reaction time of aromatic aldehydes having electron-withdrawing groups was shorter with higher product yields than for electron-donating groups. Furthermore, meta- and para-substituted aromatic aldehydes produced the products with excellent yields in shorter reaction times, while owing to the steric effects of ortho-substituted aromatic aldehydes, rather lower yields of their products were obtained.
In the next test, condensation of N,N-dimethylbarbituric acid with 2-chlorobenzaldyde was tested in the presence of recovered PBDS–SCMNP catalyst to investigate its reusability. As shown in Fig. 1, no significant loss of catalytic activity was observed after five cycles of the reaction (Scheme 2).

Synthesis of pyranodipyrimidines 3a–f and xanthene derivatives 5a–f in presence of PBDS–SCMNPs
A mechanism for the synthesis of pyranodipyrimidines is proposed in Scheme 3. Firstly, PBDS–SCMNPs activate the carbonyl group of the aldehyde (1), and correspondingly, deprotonated PBDS–SCMNPs convert N,N-dimethylbarbituric acid (2) to its enolic form (7). Then, through a Knoevenagel condensation, intermediate 7 attacks the activated carbonyl group 6, and after removal of a water molecule, compound 8 is produced. In the next step, the second molecule of intermediate 7 attacks compound 8 via Michael addition to afford the final product 3 when a water molecule is removed.

Suggested mechanism for synthesis of pyran derivatives
Conclusions
Magnetically recoverable PBDS–SCMNPs were found to be a highly efficient and economically sustainable catalyst for preparing a wide range of substituted pyranodipyrimidines and xanthene derivatives through a pseudo-three-component condensation reaction. The noticeable features of this methodology are short reaction time, good to excellent product yield, use of an easily magnetically recyclable catalyst, and green and mild reaction conditions. The PBDS–SCMNP catalyst was completely characterized using various techniques, including FT-IR, TGA, XRD, SEM, TEM, VSM, and BET analysis.
Change history
06 March 2019
In the original publication of the article, the graphical abstract was incorrectly published. The correct graphical abstract should be as given below.
06 March 2019
In the original publication of the article, the graphical abstract was incorrectly published. The correct graphical abstract should be as given below.
References
A. Goel, V.J. Ram, Tetrahedron 65, 7865 (2009)
T.E. Smith, W.H. Kuo, V.D. Bock, J.L. Roizen, E.P. Balskus, A.B. Theberge, Org. Lett. 9, 1153 (2007)
C. Li, M.V. Nitka, J.B. Gloer, J. Campbell, C.A. Shearer, J. Nat. Prod. 66, 1302 (2003)
S.G. Toske, P.R. Jensen, C.A. Kauffman, W. Fenical. Nat. Prod. Lett. 6, 303 (1995)
S.B. Ferreira, F.D.C. da Silva, F.A.F.M. Bezerra, M.C.S. Lourenco, C.R. Kaiser, A.C. Pinto, V.F. Ferreira, Arch. Pharm. 343, 81 (2010)
D.R. da Rocha, A.C.G. de Souza, J.A.L.C. Resende, W.C. Santos, E.A. dos Santos, C. Pessoa, M.O. de Moraes, L.V. Costa-Lotufo, R.C. Montenegro, V.F. Ferreira, Org. Biomol. Chem. 9, 4315 (2011)
L. Bonsignore, G. Loy, D. Secci, A. Calignano, Eur. J. Med. Chem. 28, 517 (1993)
M.Z. He, N. Yang, C.L. Sun, X.J. Yao, M. Yang, Med. Chem. Res. 20, 200 (2010)
M.G. Dekamin, M. Eslami, A. Maleki, Tetrahedron 69, 1074 (2013)
A. Shahrisa, M. Zirak, A.R. Mehdipour, R. Miri, Chem. Heterocycl. Compd. 46, 1354 (2011)
R.A. Baker, J.H. Tatum, S. Nemec, Mycopathology 111, 9 (1990)
S. Banerjee, A. Horn, H. Khatri, G. Sereda, Tetrahedron Lett. 52, 1878 (2011)
M.A. ElSohly, D. Slade, Life Sci. 78, 539 (2005)
R. Patrick, K. Verhoest, R. Fonseca, X. Hou, C. Proulx-LaFrance, M. Corman, C.J. Helal, M.M. Claffey, B. Jamison, B. Tuttle, J. Karen, J. Coffman, S. Liu, F. Nelson, R.J. Kleiman, F.S. Menniti, C.J. Schmidt, M. Vanase-Frawley, S. Liras, J. Med. Chem. 55, 9045 (2012)
H. Mehrabi, H. Abusaidi, J. Iran. Chem. Soc. 7, 890 (2010)
A.H. Adbel-Fattah, A.M. Hesien, S.A. Metwally, M.H. Elnagdi, Lieb. Ann. Chem. 13, 585 (1989)
S. Srivastava, S. Batra, A.P. Bhaduri, Incl. Med. Chem. 35, 602 (1996)
J.M. Quintela, C. Peinador, M.J. Moreira, Tetrahedron 51, 5901 (1995)
Z.J. Ye, R.B. Xu, X.S. Shao, X.Y. Xu, Z. Li, Tetrahedron Lett. 51, 4991 (2010)
A. Hasaninejad, M. Shekouhy, N. Golzar, A. Zare, M.M. Doroodmand, Appl. Catal. A: Gen. 402, 11 (2011)
J. Safaei-Ghomi, R. Teymuri, H. Shahbazi-Alavi, A. Ziarati, Chin. Chem. Lett. 24, 921 (2013)
C.J. Li, L. Chen, Chem. Soc. Rev. 35, 68 (2006)
G.M. Whitesides, C.L. Hill, J.C. Brunie, Ind. Eng. Chem. Process Des. Dev. 15, 226 (1976)
P.D. Stevens, G. Li, J. Fan, M. Yen, Y. Gao, Chem. Commun. 35, 4435 (2005)
A.H. Lu, W. Schmidt, N. Matoussevitch, H. Bönnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer, F. Schüth, Angew. Chem. Int. Ed. 43, 4303 (2004)
S.C. Tsang, V. Caps, I. Paraskevas, D. Chadwick, D. Thompsett, Angew. Chem. Int. Ed. 43, 5645 (2004)
A. Farmany, S.S. Mortazavi, H. Mahdavi, J. Magn. Magn. Mater. 416, 75 (2016)
C. Han, D. Zhu, H. Wu, Y. Li, L. Cheng, K. Hu, J. Magn. Magn. Mater. 408, 213 (2016)
A. Ying, L. Wang, F. Qiu, H. Hu, J. Yang, C. R. Chim. 18, 223 (2015)
M.B. Gawande, P.S. Brancoa, R.S. Varma, Chem. Soc. Rev. 42, 3371 (2013)
A. Ying, S. Liu, Y. Ni, F. Qiu, S. Xu, W. Tang, Catal. Sci. Technol. 4, 2115 (2014)
R.B.N. Baig, R.S. Varma, Ind. Eng. Chem. Res. 53, 18625 (2014)
F. Jiang, Y. Fu, Y. Zhu, Z. Tang, P. Sheng, J. Alloys Compd. 543, 43 (2012)
P. Barbaro, F. Liguori, N. Linares, C.M. Marrodan, Eur. J. Inorg. Chem. 24, 3807 (2012)
D.P. Debecker, E.M. Gaigneaux, G. Busca, Chem. Eur. J. 15, 3920 (2009)
S.V. Nadkarni, M.B. Gawande, R.V. Jayaram, J.M. Nagarkar, Catal. Commun. 9, 1728 (2008)
Y. Ohta, H. Chiba, S. Oishi, N. Fujii, H. Ohno, J. Org. Chem. 74, 7052 (2009)
D. Arup Kumar, P. Gogoia, R. Borah, RSC Adv. 4, 41287 (2014)
A. Yinga, Z. Lib, Y. Nib, S. Xub, H. Houb, H. Hua, J. Ind. Eng. Chem. 24, 127 (2015)
A. Ying, S. Xu, S. Liu, Y. Ni, J. Yang, C. Wu, Ind. Eng. Chem. Res. 53, 547 (2014)
A. Ying, S. Liu, J. Yang, H. Hu, Ind. Eng. Chem. Res. 53, 16143 (2014)
A. Ying, Z. Li, J. Yang, S. Liu, S. Xu, H. Yan, C. Wu, J. Org. Chem. 79, 6510 (2014)
J. Yang, S. Liu, H. Hu, S. Ren, A. Ying, Chin. J. Chem. Eng. 23, 1416 (2015)
A. Ying, S. Liu, Z. Li, G. Chen, L. Yang, H. Yan, S. Xu, Adv. Synth. Catal. 358, 2116 (2016)
A. Ying, H. Hou, S. Liu, G. Chen, J. Yang, S. Xu, ACS Sustain. Chem. Eng. 4, 625 (2016)
Z. Li, H. Hu, Y. Jin, R. Li, A. Ying, S. Xu, Curr. Org. Synth. 12, 466 (2015)
A. Ying, F. Qiu, C. Wu, H. Hua, J. Yang, RSC Adv. 4, 33175 (2014)
W. Stober, A. Fink, E. Bohn, J. Colloid Interface Sci. 26, 62 (1968)
M.B. Gawande, A.K. Rathi, I.D. Nogueira, R.S. Varma, P.S. Branco, Green Chem. 15, 1895 (2013)
H. Hamadi, M. Kooti, M. Afshari, Z. Ghiasifar, N. Adibpour, J. Mol. Catal. 373, 25 (2013)
R. Ghorbani-Vaghei, Z. Salimi, S.M. Malaekehpoor, F. Eslami, S. Noori, RSC Adv. 4, 33582 (2014)
Acknowledgments
We are grateful to Bu-Ali Sina University, Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS), for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ghorbani-Vaghei, R., Izadkhah, V. & Mahmoodi, J. One-pot synthesis of pyran derivatives using silica-coated magnetic nanoparticles functionalized with piperidinium benzene-1,3-disulfonate as a new efficient reusable catalyst. Res Chem Intermed 43, 2299–2314 (2017). https://doi.org/10.1007/s11164-016-2762-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2762-x