
Abstract
Two series of 4-phenyl-5-cyanopyrimidin-6-one derivatives bearing various S-alkyl or S-acyl moieties at position 2 were prepared as cytotoxic agents. All compounds were tested for possible anti-cancer activity on two cell lines (MCF-7 and HCT-116). The MCF-7 cell line was found to be more sensitive than the HCT-116 cell line to the action of the compounds. Compound 8g was the most potent on the MCF-7 cell line with IC50 18.3 nM/mL, whereas its IC50 on the normal cell line (MRC-5) was 64.38 nM/mL, indicating its safety and selectivity towards the MCF-7 cell line. On the other hand, compound 8d was the most potent compound on the HCT-116 cell line with IC50 23.8 nM/mL. Compound 8g was screened against five kinases. The compound showed selective inhibitory activity against pim1 kinase with IC50 11.62 µM.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is considered the second leading cause of death worldwide after cardiovascular diseases [1]. In spite of the presence of a large number of chemotherapeutic agents, they cause severe side effects and development of resistance by cancer cells even against drugs developed by targeted therapy such as imitanib [2]. Therefore, there is a continuous need for the development of novel molecules that can overcome cellular resistance.
Since the introduction of 5-flourouracil in the late 1950s as a potent anticancer agent, many uracil and thiouracil derivatives have been reported as cytotoxic agents [3].
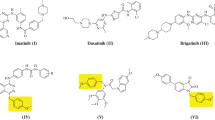
In addition, 4-aryl-5-cyanopyrimidinone derivatives were recently reported as cytotoxic agents [4–7]. In most of these derivatives, alkylation of the 2-thio group especially with alkylamino or acetanilide moieties (such as compounds I–IV, Fig. 1) was reported to enhance the cytotoxic activity of the resulting pyrimidine derivatives [4–9].

Structures of 4-phenyl-5-cyanopyrimidin-6-ones with cytotoxic activity
Prompted by these facts, we aimed in this work to prepare 4-phenyl-5-cyanopyrimidin-6-one derivatives bearing various S-(substituted amino alkyl) moieties at position 2 (compounds 6a–c and 8a–g) to act as cytotoxic agents. Comparable S-acyl derivatives (compounds 4a–c), which were poorly studied in the literature as cytotoxic agents, were also prepared. In both series, different alkyl linkers and different aliphatic and aromatic amines were used to study the effect of these variations on the cytotoxic activity. All compounds were tested for possible anti-cancer activity on two cell lines (MCF-7 and HCT-116). The most active cytotoxic compound (8g) was subjected to further studies to explore its mechanism of action.
Experimental part
General
All melting points were determined on a Stuart apparatus, and the values given were uncorrected. IR spectra were determined as KBr discs on a Shimadzu IR 435 spectrophotometer, Faculty of Pharmacy, Cairo University, and values were represented in cm−1. 1H-NMR spectra were carried out using a Mercury 300-BB 300 MHz spectrophotometer, Cairo University, Egypt, using TMS as internal standard. Chemical shift values were recorded in ppm on δ scale. 13C-NMR spectra were carried out using a Bruker 100 MHz spectrophotometer, Microanalytical Unit, Faculty of Pharmacy, Cairo University, Egypt, using TMS as internal standard. Chemical shift values were recorded in ppm on δ scale. Mass spectra were recorded on a Hewlett Packard 5988 spectrometer, or Shimadzu QP-2010 plus, Microanalytical Center, Cairo University, Egypt. Elemental analyses were carried out at the Regional Center for Mycology and Biotechnology, Azher University, Egypt. Progress of reactions was monitored by TLC using TLC sheets precoated with UV florescent silica gel Merck 60 F 254, and the sheets were visualized using a UV lamp. All reagents and solvents were purified and dried by standard techniques.
General procedure for the synthesis of S-(5-cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) 3-chloroalkanethioates (2a–c)
A mixture of compound 1 [10] (0.46 g, 2 mmol), chloroacetyl chloride, 2-chloropropionyl chloride or 3-choropropionyl chloride (2 mmol), and anhydrous K2CO3 (0.28 g, 2 mmol) in dry benzene/dry DMF (20 mL/0.5 mL) was heated under reflux for 10 h. The reaction was cooled and the separated solid was filtered, washed with water (20 mL), dried, and crystallized from DCM.
S-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) chloroethanethioate (2a)
Yield: 47 %; mp: 218–220 °C; IR (cm−1): 3444 (NH), 2997, 2943 (C–H aliphatic), 2225 (CN), 1716, 1651 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.93 (s, 2H, CH 2 Cl), 7.47–7.79 (m, 5H, Ar–H), 12.71 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C13H8ClN3O2S: C, 51.07; H, 2.64; N, 13.74. Found: C, 51.16; H, 2.71; N, 13.89.
S-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) 2-chloropropanethioate (2b)
Yield: 27 %; mp: 282–284 °C; IR (cm−1): 3186 (NH), 2978, 2935 (C–H aliphatic), 2229 (CN), 1732, 1643 (C=O); 1H NMR (DMSO-d 6) δ ppm 1.55–1.57 (d, 3H, CH 3 CH, J = 7.5 Hz), 4.47–4.55 (q, 1H, CH3 CH, J = 7.5 Hz), 7.36–7.97 (m, 5H, Ar–H), 13.10 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C14H10ClN3O2S: C, 52.59; H, 3.15; N, 13.14. Found: C, 52.64; H, 3.20; N, 13.28.
S-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) 3-chloropropanethioate (2c)
Yield: 30 %; mp: 240–242 °C; IR (cm−1): 3400 (NH), 2945, 2926 (C–H aliphatic), 2227 (CN), 1691, 1647 (C=O); 1H NMR (DMSO-d 6) δ ppm 2.72–2.76 (t, 2H, CH 2 CH2Cl, J = 6.6 Hz), 3.31–3.41 (t, 2H, CH2 CH 2 Cl, J = 6.6 Hz), 7.36–7.95 (m, 5H, Ar–H), 12.51 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 26.2 (CH 2 CH2Cl), 33.9 (CH2 CH 2 Cl), 116.4 (CN), 93.5, 128.7, 129.0, 132.2, 135.7, 161.6, 166.3 (Ar–C), 167.7 (C=O), 173.2 (SC=O); Anal. Calcd. for C14H10ClN3O2S: C, 52.59; H, 3.15; N, 13.14. Found: C, 52.65; H, 3.18; N, 13.23.
General procedure for the synthesis of 6-oxo-4-phenyl-2-(substituted amino)-1,6-dihydropyrimidine-5-carbonitriles (3a–e)
A mixture of compound 2a (0.61 g, 2 mmol), the appropriate secondary aliphatic amine (2 mmol) and triethyl amine (0.28 mL, 2 mmol) in dry benzene/dry DMF (20 mL/0.5 mL) was heated under reflux for 16 h. The reaction was cooled, and the separated solid was filtered, dried, and crystallized from DCM.
6-Oxo-4-phenyl-2-(piperidin-1-yl)-1,6-dihydropyrimidine-5-carbonitrile (3a)
Yield: 15 %; mp: 238–240 °C; IR (cm−1): 3400 (NH), 2947, 2862 (C–H aliphatic), 2212 (CN), 1626 (C=O); 1H NMR (DMSO-d 6) δ ppm 1.54–1.57 (m, 2H, CH2 CH 2 CH2, J = 4.8 Hz), 1.61–1.62 (m, 4H, CH 2 CH2 N, J = 4.8 Hz), 2.97–2.99 (t, 4H, CH2 CH 2 N, J = 4.8 Hz), 7.44–7.74 (m, 5H, Ar–H), 11.60 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 24.1 (CH2 CH 2 CH2), 25.6 (CH 2 CH2 N), 46.0 (CH2 CH 2 N), 117.8(CN), 128.7, 128.8, 131.4, 137.1 (Ar–C), 170.1 (C=O); MS m/z: 280 [M+, 97.0 %], 251 [100 %], 77 [C6H5 +, 36.4 %]; Anal. Calcd. for C16H16N4O: C, 68.55; H, 5.75; N, 19.99. Found: C, 68.69; H, 5.73; N, 20.13.
2-(Morpholin-4-yl)-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (3b)
Yield: 28 %; mp: 220–222 °C; IR (cm−1): 3392 (NH), 2922, 2858 (C–H aliphatic), 2208 (CN), 1647 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.66–3.69 (m, 4H, CH2N, J = 4.8 Hz), 3.77–3.78 (m, 4H, CH2O, J = 4.8 Hz), 7.49–7.87 (m, 5H, Ar–H), 11.95 (s, 1H, NH, D2O exchangeable); MS m/z: 282 [M+, 45.2 %], 77 [C6H5 +, 11.3 %], 71 [100 %]; Anal. Calcd. for C15H14N4O2: C, 63.82; H, 5.00; N, 19.85. Found: C, 63.90; H, 5.04; N, 20.02.
6-Oxo-4-phenyl-2-(4-phenylpiperazin-1-yl)-1,6-dihydropyrimidine-5-carbonitrile (3c)
Yield: 15 %; mp: 240–242 °C; IR (cm−1): 3363 (NH), 2916, 2989 (C–H aliphatic), 2191 (CN), 1635 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.17 (m, 4H, CH2N), 3.87 (m, 4H, CH2N), 6.77–7.81 (m, 10H, Ar–H), 12.10 (s, 1H, NH, D2O exchangeable); MS m/z: 357 [M+, 19.3 %], 132 [100 %], 77 [C6H5 +, 42.0 %]; Anal. Calcd. for C21H19N5O: C, 70.57; H, 5.36; N, 19.59. Found: C, 70.74; H, 5.41; N, 19.72.
2-[4-(4-Chlorophenyl)-piperazin-1-yl]-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (3d)
Yield: 11 %; mp: 230–232 °C; IR (cm−1): 3406 (NH), 2943, 2881(C–H aliphatic), 2214 (CN), 1654 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.12–3.14 (m, 4H, CH2N), 3.91–3.92 (m, 4H, CH2N), 6.97–7.01 (d, 2H, Ar–H, J = 9 Hz), 7.23–7.26 (d, 2H, Ar–H, J = 9 Hz), 7.52–7.88 (m, 5H, Ar–H), 10.14 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C21H18ClN5O: C, 64.37; H, 4.63; N, 17.87. Found: C, 64.42; H, 4.68; N, 17.98.
2-[4-(4-Methoxyphenyl)-piperazin-1-yl]-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (3e)
Yield: 13 %; mp: 270–272 °C; IR (cm−1): 3394 (NH), 2962, 2897 (C–H aliphatic), 2198 (CN), 1631 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.10–3.11 (m, 4H, CH2N, J = 5 Hz), 3.69 (s, 3H, CH3O), 3.94 (m, 4H, CH2N, J = 5 Hz), 6.82–6.86 (d, 2H, Ar–H, J = 9 Hz), 6.92–6.96 (d, 2H, Ar–H, J = 9 Hz), 7.51–7.89 (m, 5H, Ar–H), 12.10 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 44.8 (CH2N), 49.8 (CH2N), 55.6 (CH3O), 114.7 (CN), 93.5, 117.7, 118.4, 128.7, 128.8, 131.5, 137.0, 145.3, 153.8, 161.0, 166.0 (Ar–C), 170.1 (C=O); MS m/z: 387 [M+, 55.1 %], 231 [100 %]; Anal. Calcd. for C22H21N5O2: C, 68.20; H, 5.46; N, 18.08. Found: C, 68.28; H, 5.49; N, 18.21.
General procedure for the synthesis of S-(5-cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) (substituted phenylamino) alkanethioates (4a–c)
A mixture of compounds 2a–c (2 mmol), the appropriate primary aromatic amine (2 mmol) and triethyl amine (0.28 mL, 2 mmol) in dry DMF (20 mL) was heated under reflux until completion of the reaction (24 h for 4c and 35 h for 4a and 4b). The mixture was concentrated under reduced pressure, cooled, poured onto ice-cold water (20 mL), and the separated solid was filtered, dried, and crystallized from ethanol.
S-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) (phenylamino)ethanethioate (4a)
Yield: 22 %; mp: 120–122 °C; IR (cm−1): 3304, 3269 (NH), 2991, 2953 (C–H aliphatic), 2225 (CN), 1716, 1670 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.15 (s, 2H, CH2CO), 7.24–7.82 (m, 10H, Ar–H), 9.40 (s, 1H, NH, D2O exchangeable), 12.71 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C19H14N4O2S: C, 62.97; H, 3.89; N, 15.46. Found: C, 63.04; H, 3.94; N, 15.62.
S-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) 2-[(4-chlorophenyl)amino]propanethioate (4b)
Yield: 29 %; mp: 252–254 °C; IR (cm−1): 3435, 3246 (NH), 2926, 2954 (C–H aliphatic), 2210 (CN), 1651, 1624 (C=O); 1H NMR (DMSO-d 6) δ ppm 1.12-1.23 (d, 3H, CH 3 CH), 3.30–3.35 (m, 1H, CH3 CH), 7.23–7.52 (m, 5H, Ar–H), 7.61–7.69 (d, 2H, Ar–H),7.81–7.88 (d, 2H, Ar–H), 10.40 (s, 1H, NH, D2O exchangeable), 12.50 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 37.6 (CH 3 CH), 83.1 (CH3 CH), 119.7 (CN), 121.1, 128.3, 128.4, 128.5, 128.5, 128.6, 129.0, 129.2, 130.3, 130.7, 138.1 (Ar–C), 160.2 (C=O), 165.5 (SC=O); Anal. Calcd. for C20H15ClN4O2S: C, 58.46; H, 3.68; N, 13.64. Found: C, 58.61; H, 3.72; N, 13.81.
S-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl) 3-[(4-methylphenyl)amino]propanethioate (4c)
Yield: 17 %; mp: 172–174 °C; IR (cm−1): 3344, 3298 (NH), 2974, 2935 (C–H aliphatic), 2225 (CN), 1689, 1647 (C=O); 1H NMR (DMSO-d 6) δ ppm 2.27 (s, 3H, CH3), 2.72-2.76 (t, 2H, CH 2 CH2N, J = 7.2 Hz), 3.07–3.11 (t, 2H, CH2 CH 2 N, J = 7.2 Hz), 7.16–7.67 (m, 5H, Ar–H), 7.84–7.87 (d, 2H, Ar–H, J = 7 Hz), 7.92–7.95 (d, 2H, Ar–H, J = 7 Hz), 9.97 (s, 1H, NH, D2O exchangeable), 12.20 (s, 1H, NH, D2O exchangeable); MS m/z: 390 [M+, 0.5 %], 302 [100 %], 77 [C6H5 +, 24.3 %]; Anal. Calcd. for C21H18N4O2S: C, 64.60; H, 4.65; N, 14.35. Found: C, 64.68; H, 4.73; N, 14.51.
General procedure for the synthesis of 2-(5-cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-ylsulfanyl)-N-(substituted phenyl) alkanamides 6a–c and 6-oxo-2-{[2(3)-oxo-2-(substituted amino)alkyl]sulfanyl}-4-phenyl-1,6-dihydropyrimidine-5-carbonitriles (8a–g)
Method A
A mixture of compound 1 (0.46 g, 2 mmol), chloro derivatives 5a, 7c or 7f [ 11, 15, 17] (2 mmol) in 10 % KOH solution (1 g KOH in 10 mL water) was stirred at room temperature for 48 h. The separated solid was filtered, dried, and crystallized from ethanol.
Method B
A mixture of compound 1 (0.46 g, 2 mmol), chloro derivatives 5a–c [ 11–13] or 7a–g [11, 14–17] (2 mmol), and anhydrous K2CO3 (0.55 g, 4 mmol) in dry DMF (20 mL) was heated under reflux for 16 h. The mixture was concentrated under reduced pressure, cooled, and poured slowly onto ice-cold water (15 mL). The separated solid was filtered, dried, and crystallized from ethanol.
2-(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-ylsulfanyl)-N-p-tolyl-acetamide (6a)
Yield: 47 % (method A), 45 % (method B); mp: 232–234 °C; IR (cm−1): 3263, 3180 (NH), 2922, 2858 (C–H aliphatic), 2220 (CN), 1695, 1660 (C=O); 1H NMR (DMSO-d 6) δ ppm 2.27 (s, 3H, CH3), 4.16 (s, 2H, CH2CO), 7.10–7.13 (d, 2H, Ar–H), 7.34–7.55 (m, 5H, Ar–H), 7.85–7.88 (d, 2H, Ar–H), 9.41 (s, 1H, NH, D2O exchangeable), 10.22(s, 1H, NH, D2O exchangeable); MS m/z: 376 [M+, 0.4 %], 107 [100 %], 106 [CH3C6H4NH+, 99.4], 77 [C6H5 +, 54.5 %]; Anal. Calcd. for C20H16N4O2S: C, 63.81; H, 4.28; N, 14.88. Found: C, 63.90; H, 4.31; N, 15.02.
2-[(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl)sulfanyl]-N-phenylacetamide (6b)
Yield: 25 % (method B); mp: 138–140 °C; IR (cm−1): 3304, 3197 (NH), 2953, 2924 (C–H aliphatic), 2210 (CN), 1668, 1637 (C=O); 1H NMR (DMSO-d 6) δ ppm 1.44–1.46 (d, 3H, CH 3 CH, J = 7.2 Hz), 4.43–4.46 (q, 1H, CH3 CH, J = 7.2 Hz), 6.98–7.95 (m, 10H, Ar–H), 10.31 (s, 1H, NH, D2O exchangeable), 11.07 (s, 1H, NH, D2O exchangeable); MS m/z: 376 [M+, 20.6 %], 77 [C6H5 +, 41.5 %], 57 [100 %]; Anal. Calcd. for C20H16N4O2S: C, 63.81; H, 4.28; N, 14.88. Found: C, 63.94; H, 4.26; N, 15.03.
3-[(5-Cyano-6-oxo-4-phenyl-1,6-dihydropyrimidin-2-yl)sulfanyl]-N-phenyl-propionamide (6c)
Yield: 47 % (method B); mp: 212–214 °C; IR (cm−1): 3298, 3197 (NH), 2954, 2924 (C–H aliphatic), 2210 (CN), 1668, 1654 (C=O); 1H NMR (DMSO-d 6) δ ppm 2.85–2.87 (t, 2H, CH2 CH 2 CO), 3.48–3.50 (t, 2H, CH 2 CH2CO), 7.03–8.03 (m, 10H, Ar–H), 9.98 (s, 1H, NH, D2O exchangeable), 11.72 (s, 1H, NH, D2O exchangeable); MS m/z: 376 [M+, 0.7 %], 229 [100 %], 77 [C6H5 +, 17.3 %]; Anal. Calcd. for C20H16N4O2S: C, 63.81; H, 4.28; N, 14.88. Found: C, 63.89; H, 4.31; N, 14.98.
6-Oxo-2-{[2-oxo-2-(piperidin-1-yl)ethyl]sulfanyl}-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8a)
Yield: 52 % (method B); mp: 290–292 °C; IR (cm−1): 3296, (NH), 2947, 2922 (C–H aliphatic), 2198 (CN), 1675, 1637 (C=O); 1H NMR (DMSO-d 6) δ ppm 1.57–1.59 (m, 6H, CH 2 CH 2 CH 2 ), 3.31 (m, 4H, CH 2 N), 3.76 (s, 2H, CH2CO), 7.48–7.95 (m, 5H, Ar–H), 11.91 (s, 1H, NH, D2O exchangeable); MS m/z: 354 [M+, 0.5 %], 84 [C5H10N+, 100 %], 77 [C6H5 +, 49.2 %]; Anal. Calcd. for C18H18N4O2S: C, 61.00; H, 5.12; N, 15.81. Found: C, 61.12; H, 5.17; N, 16.02.
2-{[2-(Morpholin-4-yl)-2-oxoethyl]sulfanyl}-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8b)
Yield: 65 % (method B); mp: 208–210 °C; IR (cm−1): 3292, (NH), 2958, 2927 (C–H aliphatic), 2212 (CN), 1675, 1640 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.30 (s, 2H, CH2CO), 3.66–3.67 (t, 4H, CH2N, J = 4.5 Hz), 3.77–3.78 (t, 4H, CH2O, J = 4.5 Hz), 7.49–7.97 (m, 5H, Ar–H), 11.80 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C17H16N4O3S: C, 57.29; H, 4.52; N, 15.72. Found: C, 57.38; H, 4.59; N, 15.93.
6-Oxo-2-[(2-oxo-2-(4-phenylpiperazin-1-yl)-ethyl)sulfanyl]-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8c)
Yield: 53 % (method A), 46 % (method B); mp: 120–122 °C; IR (cm−1): 3387, (NH), 2953, 2920 (C–H aliphatic), 2222 (CN), 1660, 1647 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.13 (m, 4H, CH2N), 3.61 (m, 4H, CH2N), 4.03 (s, 2H, CH2CO), 6.81–7.97 (m, 10H, Ar–H), 10.26 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 37.5 (CH 2 CO), 41.8 (CH2N), 45.8 (CH2N), 116.3 (CN), 89.5, 119.7, 120.4, 128.5, 128.6, 129.4, 130.2, 138.0, 151.2, 167.4, 167.4 (Ar–C), 170.7 (C=O), 171.3 (SC=O); MS m/z: 431 [M+, 0.3 %], 229 [100 %], 77 [C6H5 +, 23.3 %]; Anal. Calcd. for C23H21N5O2S: C, 64.02; H, 4.91; N, 16.23. Found: C, 64.17; H, 4.97; N, 16.39.
2-{2-[4-(4-Methoxyphenyl)-piperazin-1-yl]-1-methyl-2-oxo-ethylsulfanyl}-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8d)
Yield: 20 % (method B); mp: 192–194 °C; IR (cm−1): 3292, (NH), 2993, 2929 (C–H aliphatic), 2214 (CN), 1683, 1640 (C=O); 1H NMR (DMSO-d 6−-) δ ppm 1.47–1.49 (d, 3H, CH 3 CH, J = 7.2 Hz), 3.11–3.13 (m, 4H, CH2N, J = 4.08 Hz), 3.69 (s, 3H, CH3O), 4.13–4.15 (m, 4H, CH2N, J = 4.08 Hz), 4.37–4.39 (q, 1H, CH3 CH, J = 7.2 Hz), 6.83–7.89 (m, 9H, Ar–H), 12.0 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 37.5 (CH 3 CH), 44.8 (CH2N), 49.8 (CH2N), 55.6 (CH3O), 83.8 (CH3 CH), 117.8 (CN), 114.7, 118.4, 128.7, 129.0, 131.5, 132.5, 137.1, 145.3, 150.5, 153.8, 161.2 (Ar–C), 163.0 (C=O), 170.0 (SC=O); Anal. Calcd. for C25H25N5O3S: C, 63.14; H, 5.30; N, 14.73. Found: C, 63.24; H, 5.28; N, 14.90.
2-{[3-(Morpholin-4-yl)-3-oxopropyl]sulfanyl}-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8e)
Yield: 48 % (method B); mp: 282–284 °C; IR (cm−1): 3265, (NH), 2950, 2924 (C–H aliphatic), 2204 (CN), 1665, 1635 (C=O); 1H NMR (DMSO-d 6−-) δ ppm 3.12 (t, 2H, CH2 CH 2 CO), 3.23 (t, 2H, CH 2 CH2CO), 3.66–3.67 (m, 4H, CH2N, J = 4.5 Hz), 3.77–3.78 (m, 4H, CH2O, J = 4.5 Hz), 7.48–7.88 (m, 5H, Ar–H), 11.80 (s, 1H, NH, D2O exchangeable); 13C NMR (CDCl3) δ ppm 38.0 (CH2 CH 2 CO), 45.2 (CH 2 CH2CO), 66.2 (CH2N), 67.1 (CH2O), 117.6 (CN), 128.8, 129.1, 131.4, 131.5, 132.3, 137.1, 155.0 (Ar–C), 163.6 (C=O), 170.0 (SC=O); MS m/z: 370 [M+, 9.1 %], 240 [100 %], 77 [C6H5 +, 50.4 %]; Anal. Calcd. for C18H18N4O3S: C, 58.36; H, 4.90; N, 15.12. Found: C, 58.44; H, 4.96; N, 15.30.
6-Oxo-2-[3-oxo-3-(4-phenylpiperazin-1-yl)-propylsulfanyl]-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8f)
Yield: 59 % (method A), 55 % (method B); mp: 260–262 °C; IR (cm−1): 3365 (NH), 2956, 2926 (C–H aliphatic), 2210 (CN), 1668, 1645 (C=O); 1H NMR (DMSO-d 6) δ ppm 2.85–2.88 (t, 2H, CH 2 CH2CO), 3.30 (m, 4H, CH2N), 3.51 (m, 4H, CH2N), 3.59 (t, 2H, CH2 CH 2 CO), 6.80–7.91 (m, 10H, Ar–H), 11.80 (s, 1H, NH, D2O exchangeable); MS m/z: 445 [M+, 6.3 %], 301 [100 %], 77 [C6H5 +, 49.4 %]; Anal. Calcd. for C24H23N5O2S: C, 64.70; H, 5.20; N, 15.72. Found: C, 64.84; H, 5.24; N, 15.88.
2-{3-[4-(4-Chlorophenyl)-piperazin-1-yl]-3-oxo-propylsulfanyl}-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (8g)
Yield: 41 % (method B); mp: 148–150 °C; IR (cm−1): 3344, (NH), 2935, 2924 (C–H aliphatic), 2216 (CN), 1689, 1647 (C=O); 1H NMR (DMSO-d 6) δ ppm 3.07–3.09 (t, 2H, CH 2 CH2CO, J = 3.6 Hz), 3.27–3.28 (m, 4H, CH2N, J = 4.2 Hz), 3.59–3.60 (m, 4H, CH2N, J = 4.2 Hz), 3.40 (t, 2H, CH2 CH 2 CO, J = 3.6 Hz), 6.91–7.89 (m, 9H, Ar–H), 12.20 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C24H22ClN5O2S: C, 60.06; H, 4.62; N, 14.59. Found: C, 60.22; H, 4.59; N, 14.76.
In vitro anticancer testing
Materials and methods
The breast tumor cell line (MCF-7), the colon tumor cell line (HCT-116), and the human lung fibroblast cell line (MRC-5) were obtained frozen in liquid nitrogen (−180 °C) from the American Type Culture Collection (ATCC) and were maintained in the National Cancer Institute, Cairo, Egypt, by serial sub-culturing.
All chemicals used in this study are of high analytical grade. They were obtained from either Sigma-Aldrich or Bio-Rad.
Measurement of potential cytotoxicity
The cytotoxic activity of the newly synthesized compounds was measured in vitro on breast tumor cell line (MCF-7) and colon tumor cell line (HCT-116) using Sulforhodamine-B stain (SRB) assay applying the method of Skehan et al. [18] In addition, compound 8g was examined for its cytotoxic effect on the human lung fibroblast cell line (MRC-5).
The cells were plated in a 96-multiwell plate (104 cells/well) for 24 h before treatment with the test compounds to allow attachment of the cells to the wall of the plate. The test compounds were dissolved in DMSO and diluted with saline to the appropriate volume. Different concentrations of the test compounds (0, 1, 2.5, 5, and 10 μg/mL) were added to the cell monolayer. Triplicate wells were prepared for each individual dose. Monolayer cells were incubated with the test compounds for 48 h at 37 °C in atmosphere of 5 % CO2. After 48 h, the cells were fixed with trichloroacetic acid, washed with water, and stained for 30 min with 0.4 % (wt/vol) Sulforhodamine-B stain dissolved with 1 % acetic acid. Excess stain was removed by four washes with 1 % acetic acid and the attached stain was recovered with Tris EDTA buffer. The colour intensity was measured in ELISA reader. For each compound, the relation between the surviving fraction and the drug concentration was plotted to get the survival curve of each tumor cell line. The IC50 values (the concentration required for 50 % inhibition of cell viability) were calculated using sigmodial dose response curve-fitting models (GraphPad, Prizm software incorporated), each concentration was repeated three times. The results are given in Table 1 and represented graphically in Fig. 2.
Kinase inhibitory activity
Materials and methods
The kinase inhibitory activity of compound 8g was determined using the Kinexus compound profiling service, Canada. The protein kinases employed in the compound profiling process were GSK-3β, pim1, pim2, EGFR and CDK5/p25 enzymes. All the kinases were cloned, expressed, and purified using proprietary methods. Quality control testing was routinely performed to ensure compliance to acceptable standards. 33P-ATP was purchased from PerkinElmer. All other materials were of standard laboratory grade.
Kinase protein assays
A radioisotope assay format was used for profiling evaluation of protein kinase target, and all assays were performed in a designated radioactive working area. Protein kinase assays were performed at ambient temperature for 20–30 min in a final volume of 25 μL according to the following assay reaction components: Component 1, 5 μL of diluted active protein kinase (~10–50 nM final concentration in the assay); Component 2, 5 μL of stock solution of substrate; Component 3, 5 μL of kinase assay buffer; Component 4, 5 μL of compound 8g or 10 % DMSO; Component 5, 5 μL of 33P-ATP (250 μM stock solution, 0.8 μCi).
The assay was initiated by the addition of 33P-ATP and the reaction mixture was incubated at ambient temperature for 30 min. After the incubation period, the assay was terminated by spotting 10 μL of the reaction mixture onto a Multiscreen phosphocellulose P81 plate. The Multiscreen phosphocellulose P81 plate was washed three times for approximately 15 min each in a 1 % phosphoric acid solution. Radioactivity of the P81 plate was counted in the presence of scintillation fluid in a Trilux scintillation counter. A blank control was set up that included all the assay components except the addition of the appropriate substrate (replaced with equal volume of assay dilution buffer). The corrected activity for protein kinase target was determined by removing the blank control value. The results were displayed in terms of percent inhibition.
Results and discussion
Chemistry
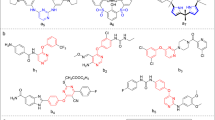
Schemes 1 and 2 outline the synthesis of the target compounds.

Preparation of the S-acylpyrimidine derivatives 2–4

Preparation of the S-alkylpyrimidine derivatives 6 and 8
4-Oxo-6-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (1) [10] was allowed to react with chloroacetyl chloride, 2-chloropropionyl chloride, and 3-chloropropionyl chloride in dry benzene using K2CO3 as a base. A small amount of DMF was added to enhance the solubility of the starting compound 1. The products were identified as S-chloroalkanethioates 2a–c based on their spectral data. Thus, the IR spectra of compounds 2a–c indicated the appearance of two C=O bands at 1691–1732 cm−1 and 1643–1651 cm−1 corresponding to the introduced acyl group and the ring carbonyl group, respectively. Also, only one NH band appeared at 3186–3444 cm−1 together with CH aliphatic band at 2926–2997 cm−1.
Moreover, 1H NMR spectra of compounds 2a–c showed an exchangeable singlet signal at δ 12.51–13.10 ppm corresponding to NH proton (which indicated selectivity for S-acylation over N-acylation). Meanwhile, the introduced aliphatic protons appeared as a singlet signal at δ 3.93 ppm (CH2Cl protons) in compound 2a, as doublet and quartet signals at δ 1.55–1.57 ppm and δ 4.47-4.55 ppm (CH3CHCl protons) in compound 2b, and as two triplet signals at δ 2.72–2.76 ppm and δ 3.31-3.41 ppm corresponding to CH2CH2Cl protons in compound 2c. On the other hand, 13C NMR spectrum of compound 2c revealed the appearance of CH2CH2Cl carbons at δ 26.2 and δ 33.9 ppm, while two C=O signals appeared at δ 167.7 and δ 173.2 ppm corresponding to the ring C=O and S–C=O carbons, respectively.
The aim of the present work was to prepare compounds 3* via nucleophilic substitution of the chloroalkylpyrimidines 2a–c with secondary amines. The reaction was carried out by refluxing equimolar amounts of compounds 2a–c and the appropriate secondary aliphatic amine in dry benzene/DMF mixture, to facilitate the work up of the reaction. However, examining the spectral data of the products indicated that the secondary amine replaced the S-acyl group to give compounds 3a–e. The IR spectra of compounds 3a–e indicated the disappearance of the thioate C=O band and the appearance of only one C=O absorption band at 1626–1654 cm−1. Further evidence was obtained from 1H NMR spectra of compounds 3a–e that indicated the disappearance of the characteristic aliphatic S-acyl protons and the appearance of the secondary amine aliphatic protons. Moreover, 13C NMR spectrum of compound 3a revealed the disappearance of S-acyl carbons [both aliphatic and C=O carbons] and the appearance of the signals of piperidine carbons at δ 24.1 ppm, δ 25.6 ppm, and δ 46.0 ppm and only one C=O signal appeared at δ 170.1 ppm [ring C=O]. Likewise, 13C NMR spectrum of compound 3e revealed the appearance of piperazinyl carbons at δ 44.8 ppm and δ 49.8 ppm. The signal of the methoxy carbon appeared at δ 55.6 ppm and the C=O signal appeared at δ 170.1 ppm. Moreover, the mass spectra of compounds 3a-c and 3e showed their corresponding molecular ion peaks at m/z 280, m/z 282, m/z 357, and m/z 387, respectively.
In order to prove this unexpected replacement, trials were done to prepare compound 3b* via reacting compound 1 with morpholine under the same reaction conditions (dry benzene/DMF, TEA and reflux for 16 h.). However, the reaction did not give any product and the starting material was obtained unreacted (No replacement took place). Thus, it seemed that the introduced S-acyl group afforded good leaving group and facilitated the nucleophilic substitution by amines.
On the other hand, reacting compounds 2a–c with appropriate primary aromatic amines in dry DMF resulted in the formation of the target compounds 4a–c. The IR spectra of compounds 4a-c indicated the appearance of two carbonyl absorption bands at 1651–1716 and 1624–1670 cm−1 and two NH absorption bands at 3246–3435 cm−1. In addition, 1H NMR spectra of compounds 4a–c showed new exchangeable singlet signal at δ 9.40–10.40 ppm corresponding to the introduced phenylamino NH proton. In addition, the alkyl protons were shifted upfield due to replacement of the highly electronegative chlorine atom by the less electronegative phenylamino group. Thus, in compound 4a, a singlet signal appeared at δ 3.15 ppm assigned to CH2CO protons, while in compound 4b, doublet and quartet signals appeared at δ 1.12–1.23 ppm and δ 3.30–3.35 ppm assigned to CH3CH protons, respectively. Finally, in compound 4c, two triplet signals appeared at δ 2.72–2.76 ppm and δ 3.07–3.11 ppm corresponding to COCH2CH2 protons. On the other hand, 13C NMR spectrum of compound 4b showed two signals at δ 37.6 ppm and δ 83.1 ppm respectively, due to CH3CH carbons, while the two C=O carbons appeared at δ 160.2 ppm and δ 165.5 ppm. The results obtained from this reaction indicated that the less nucleophilic aromatic amines did not replace the alkyl thioate group, but instead they replaced the chlorine atom.
In this work, trials were done to S-alkylate pyrimidine derivative 1 with equimolar amounts of compounds 5a–c [11–13] or 7a–g [11, 14–17] in 2N potassium hydroxide/water system at room temperature. However, only compounds 6a, 8c, and 8f were obtained after stirring for 48 h, while the rest of the compounds failed to be synthetized through the same procedure even after prolonging the stirring time up to 72 h, and the starting compounds were separated unreacted. Therefore, another base/solvent system was tried, and the reaction was carried out in anhydrous potassium carbonate/dry DMF system under reflux for 16 h. The application of these conditions afforded the target compounds 6a–c and 8a–g in poor to moderate yields.
The IR spectra of compounds 6a–c and 8a–g indicated the appearance of new amidic C=O absorption band at 1647–1695 cm−1.
Moreover, 1H NMR spectra of compounds 6a–c showed an exchangeable singlet signal at δ 9.41–10.31 ppm corresponding to phenylamino NH proton. Meanwhile, the introduced aliphatic protons appeared at δ 1–5 ppm. On the other hand, 13C NMR spectra of compounds 8c–e revealed the presence of two C=O signals at δ 163.6–170.7 ppm and δ 170.0–171.3 ppm corresponding to the ring C=O and SC=O carbons, respectively. In addition, the introduced aliphatic carbons appeared as one or two signals at δ 37.5–85.8 ppm. Also, the mass spectra of compounds 6a–c showed their corresponding molecular ion peaks at m/z 376, while the mass spectra of compounds 8a, 8c, 8e, and 8f showed their corresponding molecular ion peaks at m/z 354, m/z 431, m/z 370, and m/z 445, respectively.
In vitro anticancer testing
The newly synthesized compounds were evaluated for their in vitro cytotoxic activity against human breast carcinoma cell line (MCF-7) and human colon carcinoma cell line (HCT-116) using Sulforhodamine-B stain (SRB) assay applying the method of Skehan et al. [18]
The relation between the surviving fractions and the drug concentrations was plotted to obtain the survival curve of the MCF-7 and HCT-116 tumor cell lines after addition of the specified compounds. The parameter used was IC50, which corresponded to the concentration required for 50 % inhibition of the cell viability.
The IC50 of the synthesized compounds are shown in Table 1, and the results are represented graphically in Fig. 2.

IC50 in nM/mL of the synthesized compounds against breast tumor cell line (MCF-7) and colon tumor cell line (HCT-116)
From the results in Table 1, it was found that all the compounds exhibited considerable response on both cell lines; however, the MCF-7 cell line was found to be more sensitive to the action of the compounds.
Within the 2-substituted-pyrimidines, compound 8g was the most potent on the MCF-7 cell line with IC50 18.3 nM/mL, while compound 8d was the most potent compound on the HCT-116 cell line with IC50 23.8 nM/mL.
SAR analysis of the effect of substitution at position 2 of the pyrimidine ring on both cell lines indicated the following:
-
For the 2-alkylaminopyrimidines 3a–e, it was found that increasing the bulkiness of the hydrophobic group led to parallel increase in the cytotoxic activity. Thus, substituted phenyl piperazine derivatives (3c and 3d) afforded better cytotoxic activity than piperidine derivative 3a or morpholine derivative 3b on both cell lines.
-
For the 2-S-acylpyrimidines 4a–c, the increase in the length of the alkyl linker from CH2 to CH2CH2 was not accompanied by parallel increase in the cytotoxic activity. [Compare compound 4a and 4c]. However, the cytotoxic activity was significantly improved by the branching of the linker using (CH3)CH alkyl linker. [Compare compound 4a, 4b, and 4c].
-
Regarding the S-alkyl derivatives 6a–c and 8a–g, it was found that compounds substituted with cyclic aliphatic amines 8a–g (especially phenyl piperazine derivatives) gave better results on both cell lines (especially MCF-7 cell line) than those substituted with aromatic amines 6a–c. Here also, increasing both the length and the branching of the alkyl chain of the linker enhanced the cytotoxic activity on both cell lines.
-
No significant difference was noticed between the cytotoxic activity of the S-alkyl derivatives (6a–c and 8a–g) and the S-acyl derivatives 4a–c.
Finally, compound 8 g (the most potent compound in the present work) was tested for its possible cytotoxicity on the normal cell line MRC-5 (human lung fibroblast cell line). The result indicated that compound 8 g exhibited IC50 of 64.38 nM/mL, three to four times its potency on the tumor cell lines.
Kinase inhibitory activity of compound 8g
Searching the literature pointed out that analogous pyrimidine derivatives showed significant inhibitory activity against different kinases such as pim1, pim2 [19], GSK-3β [20], EGFR [21], and CDK5/p25 [22].
Compound 8g that showed the highest cytotoxic activity among the pyrimidine derivatives was subjected to further testing to explore its mechanism of action. The testing was performed using the Kinexus compound profiling service. Compound 8g was screened against five kinases, namely pim1, pim2, GSK-3β, EGFR, and CDK5/p25 at a single dose (50 µM). The results indicated that compound 8g inhibited all the kinases tested by varying extents ranging from 3 to 87 %. The highest inhibition observed was with pim1 kinase at 87 % followed by GSK-3β (24 %), pim2 (19 %), EGFR (18 %), and finally CDK5/p25 by 3 %.
Thus, the compound might act by selective inhibition of pim1 kinase. The IC50 of compound 8 g against pim1 was further estimated as 11.62 µM.
Pim1 kinase is a serine/threonine kinase that is involved in many biological processes such as cell survival, proliferation, differentiation, migration, metabolism, and apoptosis [23–27]. Overexpression of pim1 was reported in hematologic cancers and solid cancers [23–27]. Therefore, targeting pim1 kinase was expected to afford potent and selective anticancer agents. The ATP binding site of pim1 contains proline-123 amino acid in the hinge region. Thus, the enzyme lacks a key amide H-bond donor and thereby is incapable of making H-bonds with ATP. This fact enables the design and the synthesis of selective pim1 inhibitors. Although many pim1 inhibitors were reported in the literature, only a few of them contains a pyrimidine core [25–27]. Thus, the 4,6-diarylpyrimidin-2-one V (Fig. 3) was reported as patent CDC7 and pim1 inhibitor by Novartis [26]. Here, the carbonyl group bind to Lys67, and the hydroxyl group on the phenyl at position 4 made a H-bond with the hinge residue Glu121 [26].

Structures of pim1 inhibitors containing pyrimidine core
Moreover, a series of 6-phenylpyrimidinones exemplified by compounds VI and VII were published in 2008 as patented by Exelixis Company. Better pim inhibitory activity was exhibited by 4,6-diarylpyrimidinones than by 2,6-diarylpyrimidinones [26, 28]. In 2009, the S-alkylpyrimidine derivative VIII was identified through high throughput screening as pim1 inhibitor with IC50 of 5 µM [19].
In this work, the introduction of compound 8g with comparable structural features to compounds V–VIII and with potent and selective pim1 kinase inhibition makes this compound a promising lead pim1 inhibitor. Further optimization of its structure needs to be done to improve its pim1 inhibition and cytotoxic activity.
Conclusion
Two series of S-alkyl and S-acylpyrimidinones were prepared as cytotoxic agents. All the compounds showed significant cytotoxic activity on MCF-7 and HCT-116 cell lines with higher sensitivity towards the MCF-7 cell line. Compound 8g was the most potent on the MCF-7 cell line with IC50 18.3 nM/mL, whereas its IC50 in normal cell line (MRC-5) was 64.38 nM/mL, indicating its safety and selectivity towards MCF-7 cell line, while compound 8d was the most potent compound on the HCT-116 cell line with IC50 23.8 nM/mL. Compound 8 g was screened against five kinases. The compound showed selective inhibitory activity against pim1 kinase with IC50 11.62 µM. Further studies are still needed to identify the SAR of theses derivatives as pim1 kinase inhibitors and to improve both the kinase inhibitory activity and the cytotoxic activity of these derivatives.
References
Global Burden of Disease Cancer Collaboration, JAMA Oncol. 1, 505 (2015)
D. Bixby, M. Talpaz, Leukemia 25, 7 (2010)
A. Palasz, D. Ciez, Eur. J. Med. Chem. 97, 582 (2014)
A.M. Fargualy, N.S. Habib, K.A. Ismail, A.M.M. Hassan, M.T.M. Sarg, Eur. J. Med. Chem. 66, 276 (2013)
A. Mai, A. Perrone, A. Nebbioso, D. Rotili, S. Valente, M. Tardugno, S. Massa, F. De Bellis, L. Altucci, Bioorg. Med. Chem. Lett. 18, 2530 (2008)
H.T. Abdel-Mohsen, F.A.F. Ragab, M.M. Ramla, H.I. El Diwani, Eur. J. Med. Chem. 45, 2336 (2010)
A.T. Taher, S.M. Abou-Seri, Molecules 17, 9868 (2012)
P.-J. Chen, A. Yang, Y.-F. Gu, X.-S. Zhang, K.-P. Shao, D.-Q. Xue, P. He, T.-F. Jiang, Q.-R. Zhang, H.-M. Liu, Bioorg. Med. Chem. Lett. 24, 2741 (2014)
K.-P. Shao, X.-Y. Zhang, P.-J. Chen, D.-Q. Xue, P. He, L.-Y. Ma, J.-X. Zheng, Q.-R. Zhang, H.-M. Liu, Bioorg. Med. Chem. Lett. 24, 3877 (2014)
M.M.M. Ramiz, W.A. El-Sayed, E. Hagag, A.A.H. Abdel-Rahman, J. Heterocycl. Chem. 48, 1028 (2011)
H. Rajak, P. Kumar, P. Parmar, B.S. Thakur, R. Veerasamy, P.C. Sharma, A.K. Sharma, A.K. Gupta, J.S. Dangi, Eur. J. Med. Chem. 53, 390 (2012)
E.K. Harvill, R.M. Herbst, E.G. Schreiner, J. Org. Chem. 17, 1597 (1952)
Y. Unger, T. Strassner, J. Organomet. Chem. 713, 203 (2012)
G. Sakurai, E. Matsui, Chem. Pharm. Bull. 13, 594 (1965)
H. P. Dalalian, N. J. Rutherford, S. Kushner, U.S. patent, 2,807,617 (1957)
S. Ganguly, R. Vishwanathan, R. Chandra, J. Inst. Chem. 81, 57 (2009)
V.N. Devegowda, S.H. Seo, A.N. Pae, G. Nam, K.I. Choi, Bull. Korean Chem. Soc. 33, 647 (2012)
P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J.T. Warren, H. Bokesch, S. Kenney, M.R. Boyd, J. Natl Cancer Inst. 82, 1107 (1990)
Z. Xia, C. Knaak, J. Ma, Z.M. Beharry, C. McInnes, W. Wang, A.S. Kraft, C.D. Smith, J. Med. Chem. 52, 74 (2009)
F. Uehara, A. Shoda, K. Aritomo, K. Fukunaga, K. Watanabe, R. Ando, M. Shinoda, H. Ueno, H. Kubodera, S. Sunada, K.-I. Saito, T. Kaji, S. Asano, J. Eguchi, S. Yuki, S. Tanaka, Y. Yoneyama, T. Niwa, Bioorg. Med. Chem. Lett. 23, 6928 (2013)
I.A. Al-Suwaidan, A.M. Alanazi, A.A.-M. Abdel-Aziz, M.A. Mohamed, A.S. El-Azab, Bioorg. Med. Chem. Lett. 23, 3935 (2013)
A. Chatterjee, S.J. Cutler, R.J. Doerksen, I.A. Khan, J.S. Williamson, Bioorg. Med. Chem. 22, 6409 (2014)
D. Drygin, M. Haddach, F. Pierre, D.M. Ryckman, J. Med. Chem. 55, 8199 (2012)
M.C. Nawijn, A. Alendar, A. Berns, Nat. Rev. Cancer 11, 23 (2011)
A.L. Merke, E. Meggers, M. Ocker, Expert Opin. Investig. Drugs 21, 425 (2012)
T. Morwick, Exp. Opin. Ther. Patents 20, 193 (2010)
G.M. Arunesh, E. Shanthi, M.H. Krishna, J.S. Kumar, V.N. Viswanadhan, Exp. Opin. Ther. Patents 24, 5 (2014)
Exelixis, Inc. 6-Phenylpyrimidinones as Pim modulators. WO2008133955 (2008)
Acknowledgments
The authors are grateful to all members of the department of Cancer Biology, National Cancer Institute, Cairo, Egypt, for carrying out the cytotoxicity testing. The authors are grateful to Kinexus lab, Canada for performing the kinase inhibitory assays.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Said, M.M., Taher, A.T., El-Nassan, H.B. et al. Synthesis of novel S-acyl and S-alkylpyrimidinone derivatives as potential cytotoxic agents. Res Chem Intermed 42, 6643–6662 (2016). https://doi.org/10.1007/s11164-016-2487-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2487-x