
Abstract
A series of new indolizine 9a–c, 10a–c, diazepinoindolizine 7a–c, 8a–c, and pyrimidoindolizine 11 derivatives were synthesized and structures of the newly synthesized compounds were confirmed by spectral and elemental analyses. Antitumor activity evaluation was carried out using sulphorhodamine-B assay method against lung adenocarcinoma (A549), breast (MCF7), hepatoma (Hep3B) cancer cell lines and normal fibroblast cells. Compounds 7a, 9c, 10a,c and 11 showed to be the most active against the lung cancer cell line with IC50 in nanomole range (16–85 nmol/ml) and compound 11 was the best selective one (S. I. = 19). The most potent compounds against MCF7 are 8c, 9b,c, 10a,b, and 11. Their IC50 range is 4–46 nmol/ml and the best selectivity was assigned for compound 11 (S. I. = 133). As for the hepatoma cancer cell line, compounds 7a, 8a–c, 9a–c, and 10a,b were found to be the most potent with IC50 range 3–90 nmol/ml and compound 8c was the most selective one (S. I. = 42), the rest of the new compounds showed IC50 value >100 nmol/ml. Compound 10a showed a broad spectrum activity and selectivity against the tested cell lines with a much lesser effect on normal fibroblast cells (IC50 > 200 nmol/ml).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Indolizine is an important N-containing heterocyclic nucleus. It has several previous names in the chemical literature such as pyrindole, pyrrodine, pyrrocoline, and pyrrole[1,2-a]pyridine, and it is also considered as an indole isomer due to the ten delocalized Π-electrons. The indolizine system consists of two fused 5- and 6-membered rings with one nitrogen bridge at the ring junction. Its numbering system is shown in Fig. 1 [1]. The aromatic indolizine system does not appear to occur naturally; however, its perhydro derivatives, named indolizidines, represent a scaffold for several alkaloids [2]. The biological potential of indolizine derivatives was observed through their reported pharmacological activities, e.g., anti-inflammatory and anticonvulsant [3], antioxidant [4], antimicrobial [5], antitubercular [5] and antidiabetic [6].

Structure and numbering of indolizine nucleus
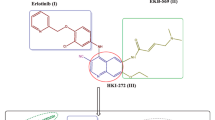
The role of indolizine scaffolds in cancer treatment was also proven through the observed anticancer activity by several indolizine derivatives, e.g., indolizine derivative-bearing cyano group I showed good antiproliferative activity against the human hepatocellular liver carcinoma (Hep-G2) cell line at IC50 value equal 0.20 µg/ml [7]. 6,7-diphenyl-2,3,8,8a-tetrahydro-1H-indolizin-5-one II, an analogue of septicine, showed cytotoxic activity against eight cancer cell lines with GI50 values in the range of 0.4–9 µM in addition to its good pharmacokinetic properties [8]. In addition, piperidin-1-yl-containing compounds IIIa,b showed greater in vitro cytotoxic activity than perillyl alcohol. They showed inhibitory properties against α-1 Na/K-ATPase and Ras oncogene activity in cancerous cells [9]. Schiff bases showed different biological activities and proved to be a versatile pharmacophore useful in designing new bioactive agents [10]. Moreover, the p-nitro benzylidine derivative IV revealed significant anticancer activity against human ovarian cancer cell line A2780, Co rectal cancer cell line HCT116 and human liver cancer cell line HepG2 [11]. All these facts guided us to synthesize our target compounds that represent hybrid molecules of indolizine scaffold with piperidin-1-yl moiety 9a–c or with the active pharmacophore p-nitro substituted Schiff base 10a–c (Fig. 2).

Indolizine, piperidine, and p-nitro Schiff base derivatives (I–IV) as antitumor active agents and our target compounds (9a–c and 10a–c)
The activity of thienodiazepinediones Va,b to antagonize the p53-Mdm2 interaction was proven using FP screening assay [12]. This encouraged us to apply isosteric replacement strategy and synthesize diazepinindolizine system 7a–c, 8a–c, which have not been investigated yet as a potential pharmacophore (Fig. 3). In 2014, Sagara et al. [13] invented novel derivatives VI with a pyrimidoindolizine scaffold useful as anticancer agents due to their ability of inhibiting EGFR tyrosine kinases. Based on this finding and for further exploration of anticancer activity of this biologically important scaffold, it was of our interest to construct the pyrimidoindolizine system 11 (Fig. 3) aiming to obtain new compounds with potential activity as anticancer.

Anticancer active agents (Va,b and VI) and our target compounds (7a–c, 8a–c and 11)
Results and discussion
Chemistry
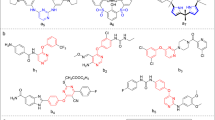
The present investigation involves the preparation of four classes of novel indolizine derivatives; (1) diazepinoindolizines 7a–c, 8a–c, (2) 6-(2-(piperidin-1-yl) acetamido) derivatives 9a–c, (3) p-nitro Schiff base compounds 10a–c, (4) pyrimidoindolizine derivative 11. The starting materials 2, 4a–c, 5a, and 6a were prepared according to the previously reported methods [14], equimolar amount of compounds 2 and 4a–c was refluxed in dry acetone for 24 h in presence of dry K2CO3 to afford compounds 5a–c, respectively. Chloroacylated indolizine derivatives 6a–c were obtained through reaction of 5a–c with chloroacetyl chloride (Scheme 1).

Synthesis of diazepinoindolizine derivatives 7a–c and 8a–c
Stirring these compounds 6a–c in DMF in the presence of dry potassium carbonate afforded diazepinoindolizines 7a–c. The IR spectra of the cyclized products 7a–c showed the presence of a stretching band at 3,280–3,217 cm−1 corresponding to the NH group, a sharp absorption band at 2,230–2,221 cm−1 attributed to the cyano group. 1H-NMR spectra of compounds 7a–c revealed the presence of a singlet signal at 4.24–4.28 ppm corresponding to the –CH2 group of the diazepine ring and a singlet exchangeable signal at 10.92–10.98 ppm assigned for the amidic proton. Additionally, 13C-NMR spectrum of compounds 7a–c was used as a tool in confirming their structures. Moreover, the mass spectrum of compound 7a revealed the molecular ion at 320 m/z.
In the present work, diazepinoindolizines were treated with ethyl chloroacetate in dry acetone and in the presence of potassium carbonate as an acid-binding agent to afford the corresponding ethyl ester derivatives 8a–c. The IR spectra revealed the disappearance of the NH group absorption band and the appearance of another absorption band at 1,751–1,742 cm−1, indicating the additional carbonyl group of the ethyl esters. Also, 1H-NMR spectra of compounds 8a–c revealed the disappearance of the exchangeable NH signal found in the starting materials and the presence of a triplet signal at 1.26–1.27 ppm assigned for methyl group of the ethyl moiety, a quartet signal at 4.22–4.23 ppm assigned for methylene group of the ethyl moiety, and a singlet signal at 4.71–4.75 ppm assigned for methylene group of the acetate moiety. Additionally, 13C-NMR spectrum of compounds 8a–c demonstrated the presence of a signal at 14.08–14.09 ppm attributed to the methyl group of the ethyl moiety, 48.32 ppm attributed to the methylene group of the acetate group, 61.91–61.99 ppm attributed to the methylene group of the ethyl moiety, and 167.56–167.63 ppm attributed to the carbonyl carbon of the acetate group.
Reacting the chloroacylated indolizines 6a–c with piperidine in absolute ethanol and in the presence of sodium bicarbonate afforded the piperidin-1-yl indolizines 9a–c. The 1H-NMR spectrum of compounds 9a–c confirmed the substitution of the piperidine nucleus for the chlorine atom as it shows two signals at 1.47–1.67 ppm attributed to six protons belonging to the three methylene groups in the piperidine nucleus and a signal at 2.58–2.63 ppm attributed to four protons belonging to the two methylene groups adjacent to the nitrogen atom in the piperidine nucleus. Furthermore, 13C spectrum of compounds 9a–c revealed the presence of new signals at 23.43–23.47 ppm, 26.09–26.11 ppm, and 55.02–55.03 ppm attributed to methylene groups of piperidine moiety.
In the present work, the target compounds 10a–c were prepared via refluxing the aminonitriles 5a–c with an equimolar amount of aromatic aldehyde in absolute ethanol and a few drops of glacial acetic acid to catalyze the reaction. The structure of Schiff bases 10a–c was confirmed using spectral and elemental analysis. The IR spectra revealed the disappearance of the primary amino group absorption band. Also, 1H-NMR spectra of compounds 10a–c show two signals at 8.09–8.41 ppm attributed to four protons belonging to additional aromatic protons and a singlet signal appeared at 9.23–9.28 ppm assigned for CH group. Furthermore, 13C spectrum of the compounds revealed the presence of six new aromatic carbons and a signal at 157.06–157.30 ppm assigned for C=N.
Herein, compound 11 was prepared via refluxing the aminonitriles 5a in excess triethyl orthoacetate where cyclization occurred between the imidate carbon and nitrogen atom of the amino group generating the fused pyrimidine ring directly (Scheme 2). The structure of compound 11 was confirmed using spectral and elemental analysis. The IR spectrum revealed the disappearance of the primary amino group absorption band. 1H-NMR spectrum of compound 11 revealed the disappearance of the exchangeable NH 2 and NH signals found in the starting materials and the presence of a singlet signal at 2.24 ppm assigned for methyl group of triethyl orthoacetate moiety. Furthermore, 13C spectrum of the compound revealed the presence of new signal at 24.35 ppm attributed to methyl group and a signal at 154.39 ppm assigned for C=N.

Synthesis of the target compounds 9a–c, 10a–c and 11
Pharmacological screening
The four novel classes of indolizines were screened against lung adenocarcinoma (A549), breast (MCF7), hepatoma (Hep3B) cancer cell lines, and normal fibroblast cells (non-tumorous cell line) using sulphorhodamine-B assay method [15]. IC50 values in µmol/ml and selectivity index values that were calculated by dividing the IC50 value of the tested compound against normal fibroblasts over that against the cancer cell line [16] are represented in Table 1. Nine compounds (7a, 8a–c, 9a–c, and 10a,b) out of 13 are highly potent against Hep3B cancer cell line with IC50 range (3–90 nmol/ml) and all these nine compounds are selective to Hep3B. Six compounds (8c, 9b,c, 10a,b, and 11) are highly potent against MCF7 cancer line with IC50 range equal 4–46 nmol/ml, and all are selective to MCF7 except for compounds 9b,c. Five compounds of the tested compounds (7a, 9c, 10a,c, and 11) are potent against the A549 cell line. Their IC50 range is 16–85 nmol/ml, all are selective to A549 except for 9c. Compound 7a is the best active compound against Hep3B, IC50 = 3 nmol/ml and S. I. = 5.7 and also the best active one against A549 with IC50 value = 16 nmol/ml and S.I. = 1.1. The best active compound against MCF7 is compound 11, IC50 = 4 nmol/ml and S. I. = 132.
Conclusions
According to the results obtained during this work, we can conclude that: (1) indolizine represents a hopeful scaffold for designing new potent and selective anticancer agents; (2) diazepinoindolizines showed to be highly potent and selective anticancer agents; compounds 8a–c are active and selective to Hep3B. In addition, compound 8c showed good activity and selectivity to MCF7 (IC50 = 18 nmol/ml, S. I. = 63). Moreover, compound 7a was the most active against A549 and Hep3B; (3) pyrimidoindolizine 11 is the most potent and selective compound to MCF7. Its IC50 and S.I. are 4 nmol/ml and 132, respectively, in addition to its activity and selectivity to A549 with IC50 = 28 nmol/ml and S.I. = 19; (4) both hybrid molecules (indolizine with piperidine) 9c and (indolizine with p-nitro Schiff base) 10a exhibited broad spectrum activity against all three cancer cell lines. However, hybridization with p-nitro Schiff base leads to obtaining more selective compound 10a than 9c.
Experimental
Chemistry
Melting points were uncorrected and were carried out by open capillary tube method using IA 9100MK-Digital Melting Point Apparatus. Microanalyses were carried out at the microanalytical Center, Faculty of Science, Cairo University. Infrared spectra were made on a BRUKER Vector 22 (Japan) infrared spectrophotometer and were expressed in wavenumber (cm−1) using a potassium bromide disc at the Microanalytical Center, Faculty of Science, Cairo University. The 1H-NMR spectra were recorded in CDCl3 and DMSO-d6 on a Varian Mercury spectrometer (400 MHz) (NMR Lab. Faculty of Pharmacy, Beni-Suef University). Chemical shifts were reported on the δ scale and were related to that of the solvent and J values are given in Hz. 13C NMR spectra were obtained on a Bruker APX400 at 100 MHz at the Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt. Mass spectra were recorded on Finnigan MAT, SSQ 7000, mass spectrometer at 70 eV (EI) at the Microanalytical Center, Faculty of Science, Cairo University and Waters Micromass Q-Tof Micro mass spectrometer (ESI) and Waters Acquity Ultra Performance LC with ZQ detector in ESI mode. IUPAC chemical nomenclature were assigned using CS Chemdraw ultra version 5.0. thin-layer chromatography, using Macherey–Nagel AlugramSil G/UV254 silica gel plates and benzene-ethanol (9.5:0.5) as the eluting system.
General procedure for the preparation of 7a–7c
A mixture of compounds 6a–c (3.58 mmol) and anhydrous potassium carbonate (0.49 g, 3.58 mmol) in dry DMF (10 ml) was stirred at room temperature for 48 h. The reaction mixture was poured onto ice-cooled water; the obtained precipitate was filtered, washed with water, and recrystallized from ethanol/acetone.
2,5-Dioxo-4-phenyl-2,3,4,5,7,8,9,10-octahydro-1H-[1, 4] diazepino[5,6-b] indolizine-11-carbonitrile 7a
Compound 7a was prepared from 6a and anhydrous potassium carbonate. Yellowish crystals, 0.53 g; 47 % yield, m.p. 268–270 °C. IR (KBr, cm−1): ν max. = 3,217 (NH), 3,049 (CH aromatic), 3,008, 2,952 (CH2), 2,214 (CN), 1,688, 1,644 (COs), 1,553, 1,540 (C=C, NH), 1,490, 1,443, 1,326 (C–N, C–O). 1H-NMR (DMSO-d6, 400 MHz): δ 1.81 (m, 2H, CH2-9) 1.91 (m, 2H, CH2-8), 2.86 (t, 2H, J = 6 Hz, CH2-10), 4.19 (t, 2H, J = 6 Hz, CH2-7), 4.28 (s, 2H, CH2-3), 7.29–7.45 (m, 5H, aromatic protons) and 10.93 (s, H, NH, which disappeared on deuteration). 13C NMR (DMSO-d6, 100 MHz): δ 18.8 (CH2), 22.4 (CH2), 22.5 (CH2), 45.9 (CH2), 54.8 (CH2), 82.7 (C), 114.3 (CN), 115.7 (C), 126.1 (2CH), 126.9 (CH), 129.4 (2CH), 130.3 (C), 142.7 (C), 143.3 (C), 159.8 (CO), 168.4 (CO). Anal. Calcd. for C18H16N4O2 (320.35): C, 67.49; H, 5.03; N, 17.49. Found C, 67.62; H, 5.30; N 17.19.
2,5-Dioxo-4-(p-tolyl)-2,3,4,5,7,8,9,10-octahydro-1H-[1, 4] diazepino [5,6-b] indolizine-11-carbonitrile 7b
Compound 7b was prepared from 6b and anhydrous potassium carbonate. Yellow crystals, 0.69 g; 58 % yield, m.p. 267–269 °C. IR (KBr, cm−1): ν max. = 3,280 (NH), 3,077 (C–H aromatic), 2,961, 2,924 (CH3, CH2), 2,215 (CN), 1,689, 1,640 (COs), 1,591, 1,554, 1,534 (C=C,NH), 1,444, 1,367, 1,305 (C–N, C–O).1H-NMR (DMSO-d6, 400 MHz): δ 1.81 (m, 2H, CH2-9) 1.90 (m, 2H, CH2-8) 2.31 (s, 3H, CH 3 Ph), 2.85 (t, 2H, J = 6 Hz, CH2-10), 4.18 (t, 2H, J = 6 Hz, CH2-7), 4.24 (s, 2H, CH2-3), 7.22 and 7.24 (2 × d, 2 × 2H, J = 8.4 Hz, para-substituted phenyl ring) and 10.92 (s, H, NH, which disappeared on deuteration).13C NMR (DMSO-d6, 100 MHz): δ 23.5 (CH2), 25.7 (CH3), 27.2 (CH2), 27.3 (CH2), 50.6 (CH2), 59.7 (CH2), 87.4 (C), 119.1 (CN), 120.5 (C), 130.7 (2CH), 134.5 (2CH), 135.0 (C), 141.0 (C), 145.0 (C), 147.9 (C), 164.5 (CO), 173.1 (CO). Anal. Calcd. for C19H18N4O2 (334.37): C, 68.25; H, 5.43; N, 16.76. Found C, 68.55; H, 5.73; N 16.73.
4-(4-chlorophenyl)-2,5-dioxo-2,3,4,5,7,8,9,10-octahydro-1H-[1, 4] diazepino [5,6-b] indolizine-11-carbonitrile 7c
Compound 7c was prepared from 6c and anhydrous potassium carbonate. Yellow crystals, 0.57 g; 45 % yield, m.p. 293–294 °C. IR (KBr, cm−1): ν max. = 3,274 (NH), 3,114 (C–H aromatic), 2,963, 2,901 (CH2), 2,220 (CN), 1,693, 1,639 (COs), 1,561, 1,536 (C=C,NH), 1,490, 1,441, 1,370 (C–N, C–O). 1H-NMR (DMSO-d6, 400 MHz): δ 1.80 (m, 2H, CH2-9) 1.89 (m, 2H, CH2-8), 2.85 (t, 2H, J = 6 Hz, CH2-10), 4.18 (t, 2H, J = 6 Hz, CH2-7), 4.28 (s, 2H, CH2-3), 7.47–7.49 (m, 4H, aromatic protons) and 10.98 (s, H, NH, which disappeared on deuteration).13C NMR (DMSO-d6, 100 MHz): δ 23.5 (CH2), 27.2 (CH2), 27.3 (CH2), 50.6 (CH2), 59.3 (CH2), 87.5 (C), 119.0 (CN), 120.3 (C), 132.1 (2CH), 134.0 (2CH), 136.0 (C), 144.0 (C), 146.2 (C), 148.2 (C), 169.2 (CO), 173.0 (CO). Anal. Calcd. for C18H15ClN4O2 (354.79): C, 60.94; H, 4.26; N, 15.79. Found C, 60.64; H, 4.44; N, 15.49.
General procedure for the preparation of 8a–8c
A mixture of compounds 7a–c (3.12 mmol), ethyl chloroacetate (0.4 g, 3.12 mmol), and anhydrous potassium carbonate (0.45 g, 3.26 mmol) in dry acetone (50 ml) was stirred under reflux for 6 h. The mixture was filtered, concentrated, and left to cool, whereby white crystals were formed, collected, dried, and recrystallized from ethanol.
Ethyl 2-(11-cyano-2,5-dioxo-4-phenyl-2,3,4,5,7,8,9,10-octahydro-1H- [1, 4] diazepino[5,6-b]indolizin-1-yl)acetate 8a
Compound 8a was prepared from 7a and ethyl chloroacetate. White crystals, 0.69 g; 55 % yield, m.p. 263–265 °C. IR (KBr, cm−1): ν max. = 3,060 (C–H aromatic), 2,977, 2,937 (CH2), 2,222 (CN), 1,742, 1,693, 1,644 (COs), 1,595, 1,549 (C=C), 1,468, 1,373, 1,307 (C–N, C–O).1H NMR (CDCl3, 400 MHz): δ 1.26 (t, 3H, J = 7.2 Hz, CH 3 CH2), 1.93 (m, 2H, CH2-9), 2.07 (m, 2H, CH2-8), 2.94 (t, 2H, J = 6.4 Hz, CH2-10), 4.22 (q, 2H, J = 7.2 Hz, CH 2 CH3), 4.54 (t, 2H, J = 6.4 Hz, CH2-7), 4.62 (s, 2H, CH2-3), 4.75 (s, 2H, NCH 2 CO) and 7.28-7.43 (m, 5H, aromatic protons).13C NMR (CDCl3, 100 MHz): δ 14.0 (CH3), 18.8 (CH2), 22.5 (CH2), 22.7 (CH2), 45.9 (CH2), 48.3 (CH2), 54.3 (CH2), 61.9 (CH2), 83.6 (C), 114.0 (CN), 117.0 (C), 125.9 (2CH), 127.3 (CH), 129.3 (2CH), 131.7 (C), 141.6 (C), 143.0 (C), 159.4 (CO), 166.8 (CO), 167.6 (COO). Anal. Calcd. for C22H22N4O4 (406.43): C, 65.01; H, 5.46; N, 13.78. Found C, 65.30; H, 5.37; N, 13.48.
Ethyl 2-(11-cyano-2,5-dioxo-4-(p-tolyl)-2,3,4,5,7,8,9,10-octahydro-1H-[1, 4] diazepino[5,6-b]indolizin-1-yl)acetate 8b
Compound 8b was prepared from 7b and ethyl chloroacetate. White crystals, 0.81 g; 62 % yield, m.p. 190–192 °C. IR (KBr, cm−1): ν max. = 3,074 (C–H aromatic), 2,966, 2,938 (CH3, CH2), 2,218 (CN), 1,742, 1,691, 1,644 (COs), 1,591, 1,549, 1,511 (C=C), 1,470, 1,373, 1,306 (C–N, C–O).1H-NMR (CDCl3, 400 MHz): δ 1.27 (t, 3H, J = 7.2 Hz, CH 3 CH2), 1.92 (m, 2H, CH2-9), 2.07 (m, 2H, CH2-8), 2.36 (s, 3H, CH 3 Ph), 2.93 (t, 2H, J = 6.4 Hz, CH2-10), 4.23 (q, 2H, J = 7.2 Hz, CH 2 CH3), 4.54 (t, 2H, J = 6.4 Hz CH2-7), 4.62 (s, 2H, CH2-3), 4.71 (s, 2H, NCH 2 CO) and 7.14-7.29 (m, 4H, aromatic protons).13C NMR (CDCl3, 100 MHz): δ14.0 (CH3), 18.8 (CH2), 21.0 (CH3), 22.5 (CH2), 22.6 (CH2), 45.8 (CH2), 48.3 (CH2), 54.4 (CH2), 61.9 (CH2), 83.5 (C), 114.1 (CN), 117.1 (C), 125.7 (2CH), 129.9 (2CH), 131.7 (C), 137.2 (C), 139.0 (C), 142.9 (C), 159.5 (CO), 166.9 (CO), 167.6 (COO). Anal. Calcd. for C23H24N4O4 (420.46): C, 65.70; H, 5.75; N, 13.33. Found C, 66.00; H, 6.02; N, 13.03.
Ethyl 2-(4-(4- chlorophenyl)-11-cyano-2,5-dioxo-2,3,4,5,7,8,9,10-octahydro-1H- [1, 4] diazepino[5,6-b]indolizin-1-yl)acetate 8c
Compound 8c was prepared from 7c and ethyl chloroacetate. White crystals, 0.72 g; 53 % yield, m.p. 215–217 °C. IR (KBr, cm−1): ν max. = 3,093 (C–H aromatic), 2,976 (CH2), 2,222 (CN), 1,751, 1,685, 1,648 (COs), 1,595, 1,594 (C=C), 1,472, 1,375, 1,306 (C–N, C–O). 1H-NMR (CDCl3, 400 MHz): δ 1.27 (t, 3H, J = 7 Hz, CH 3 CH2), 1.94 (m, 2H, CH2-9), 2.10 (m, 2H, CH2-8), 2.95 (t, 2H, J = 6.4 Hz, CH2-10), 4.23 (q, 2H, J = 7 Hz, CH 2 CH3), 4.54 (t, 2H, J = 6.4, Hz CH2-7), 4.62 (s, 2H, CH2-3), 4.73 (s, 2H, NCH 2 CO), 7.38 and 7.43 (2xd, 2x2H, J = 8.8 Hz, para-substituted phenyl ring).13C NMR (CDCl3, 100 MHz): δ14.0 (CH3), 18.7 (CH2), 22.5 (CH2), 22.7 (CH2), 45.9 (CH2), 48.3 (CH2), 54.2 (CH2), 61.9 (CH2), 83.8 (C), 113.9 (CN), 116.9 (C), 127.2 (2CH), 129.4 (2CH), 131.9 (C), 132.9 (C), 140.0 (C), 143.2 (C), 159.3 (CO), 166.7 (CO), 167.5 (COO). Anal. Calcd. for C22H21ClN4O4 (440.88): C, 59.93; H, 4.80; N, 12.71. Found C, 60.14; H, 4.76; N, 12.41.
General procedure for the preparation of 9a–9c
A mixture of compounds 6a–c (2.8 mmol), sodium bicarbonate (0.5 g, 5.9 mmol) and piperidine (0.5 g, 5.6 mmol) in absolute ethanol (10 ml) was refluxed for 8 h. The reaction mixture was filtered while hot, concentrated, cooled, and the separated crystals were collected, dried, and recrystallized from ethanol.
1-Cyano-N-phenyl-2-(2-(piperidin-1-yl)acetamido)-5,6,7,8-tetrahydroindolizine-3-carboxamide 9a
Compound 9a was prepared from 6a and piperidine. White crystals, 0.74 g; 66 % yield, m.p. 230-232 °C. IR (KBr, cm−1): ν max. = 3,306, 3,240 (NHs), 3,054 (C–H aromatic), 2,929, 2,856 (CH2), 2,220 (CN), 1,654 (COs), 1,570, 1,505 (C=C,NH), 1,444, 1,323 (C–N, C–O).1H-NMR (CDCl3, 400 MHz): δ 1.48 (m, 2H, CH2-4″), 1.67 (m, 4H, CH2-3″–5″), 1.89 (m, 2H, CH2-7), 1.98 (m, 2H, CH2-6), 2.63 (t, 4H, J = 5.2 Hz, CH2-2″–6″), 2.91 (t, 2H, J = 6.2 Hz, CH2-8), 3.22 (s, 2H, COCH 2 ), 4.29 (t, 2H, J = 5.8 Hz, CH2-5), 7.12 (t, 1H, J = 7.4 Hz, CH-4′), 7.34 (t, 2H, J = 7.6 Hz, CH-3′–5′), 7.63 (d, 2H, J = 8 Hz, CH-2′, CH-6′), 9.42 (s, H, NHCOCH2, which disappeared on deuteration) and 9.83 (s, H, CONH phenyl, which disappeared on deuteration).13C NMR (CDCl3, 100 MHz): δ 19.0 (CH2), 22.7 (CH2), 22.8 (CH2), 23.4 (CH2), 26.1 (2CH2), 45.8 (CH2), 55.0 (2CH2), 62.0 (CH2), 88.3 (C), 113.8 (CN), 119.3 (2CH), 121.6 (C), 123.5 (C), 124.1 (CH), 129.0 (2CH), 138.3 (C), 140.4 (C), 157.6 (CO), 173.5 (CO). Anal. Calcd. for C23H27N5O2 (405.49): C, 68.13; H, 6.71; N, 17.27. Found C, 68.40; H, 6.89; N, 17.24.
1-Cyano-2-(2-(piperidin-1-yl)acetamido)-N-(p-tolyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide 9b
Compound 9b was prepared from 6b and piperidine. Yellowish crystals, 0.8 g; 69 % yield, m.p. 226–228 °C. IR (KBr, cm−1): ν max. = 3,262, 3,188 (NHs), 3,065 (C–H aromatic), 2,929, 2,852 (CH3, CH2), 2,219 (CN), 1,659 (COs), 1,601, 1,545, 1,516 (C=C,NH), 1,450, 1,392, 1,324 (C–N, C–O). 1H-NMR (CDCl3, 400 MHz): δ 1.47 (m, 2H, CH2-4″), 1.65 (m, 4H, CH2-3″–5″), 1.89 (m, 2H, CH2-7), 1.98 (m, 2H, CH2-6), 2.33 (s, 3H, CH 3 Ph), 2.58 (t, 4H, J = 5.2 Hz, CH2-2″–6″), 2.91 (t, 2H, J = 6 Hz, CH2-8), 3.15 (s, 2H, COCH 2 ), 4.27 (t, 2H, J = 5.6 Hz, CH2-5), 7.13 and 7.5 (2 × d, 2 × 2H, J = 8 Hz, para-substituted phenyl ring), 9.34 (s, H, NHCOCH2, which disappeared on deuteration) and 9.81 (s, H, CONH phenyl, which disappeared on deuteration).13C NMR (CDCl3, 100 MHz): δ 19.0 (CH2), 20.9 (CH3), 22.7 (CH2), 22.7 (CH2), 23.4 (CH2), 26.1 (2CH2), 45.7 (CH2), 55.0 (2CH2), 62.0 (CH2), 88.2 (C), 113.9 (CN), 119.2 (2CH), 121.5 (C), 123.6 (C), 129.5 (2CH), 133.8 (C), 135.8 (C), 140.3 (C), 157.4 (CO), 173.4 (CO). Anal. Calcd. for C24H29N5O2 (419.52): C, 68.71; H, 6.97; N, 16.69. Found C, 68.45; H, 7.18; N, 16.39.
N-(4-chlorophenyl)-1-cyano-2-(2-(piperidin-1-yl)acetamido)-5,6,7,8-tetrahydro indolizine-3-carboxamide 9c
Compound 9c was prepared from 6c and piperidine. Yellowish crystals, 0.78 g; 64 % yield, m.p. 253–255 °C. IR (KBr, cm−1): ν max. = 3,235, 3,181 (NHs), 3,055 (C–H aromatic), 2,933, 2,854 (CH2), 2,222 (CN), 1,662 (COs), 1,601, 1,547 (C=C,NH), 1,486, 1,454, 1,391 (C–N, C–O). 1H-NMR (CDCl3, 400 MHz): δ 1.47 (m, 2H, CH2-4″), 1.65 (m, 4H, CH2-3″–5″), 1.90 (m, 2H, CH2-7), 1.99 (m, 2H, CH2-6), 2.59 (t, 4H, J = 5.2 Hz, CH2-2″–6″), 2.92 (t, 2H, J = 6 Hz, CH2-8), 3.15 (s, 2H, COCH 2 ), 4.26 (t, 2H, J = 5.4 Hz, CH2-5), 7.3 and 7.6 (2 × d, 2 × 2H, J = 8.4 Hz, para-substituted phenyl ring), 9.39 (s, H, NHCOCH2, which disappeared on deuteration) and 10.03 (s, H, CONH phenyl, which disappeared on deuteration).13C NMR (CDCl3, 100 MHz): δ 18.9 (CH2), 22.7 (CH2), 22.7 (CH2), 23.4 (CH2), 26.0 (2CH2), 45.8 (CH2), 55.0 (2CH2), 62.0 (CH2), 88.3 (C), 113.7 (CN), 120.5 (2CH), 121.8 (C), 123.2 (C), 129.0 (2CH), 129.0 (C), 136.9 (C), 140.6 (C), 157.5 (CO), 173.6 (CO). Anal. Calcd. for C23H26ClN5O2 (439.94): C, 62.79; H, 5.96; N, 15.92. Found C, 62.85; H, 6.09; N, 15.62.
General procedure for the preparation of 10a–10c
A mixture of compounds 5a–c (3.56 mmol) and p-nitrobenzaldehyde (3.56 mmol) in absolute ethanol (20 ml) in the presence of glacial acetic acid (0.5 ml) was refluxed for 4 h. The reaction mixture was then concentrated, set aside to cool, where orange crystals were formed, collected, and recrystallized from ethanol.
1-Cyano-2-((4-nitrobenzylidene)amino)-N-phenyl-5,6,7,8-tetrahydroindolizine-3-carboxamide10a
Compound 10a was prepared from 5a and p-nitrobenzaldehyde. Orange crystals, 1.03 g; 70 % yield, m.p. 272–274 °C. IR (KBr, cm−1): ν max. = 3,278 (NH), 3,073 (C–H aromatic), 2,937 (CH2), 2,208 (CN), 1,659 (CO), 1,545, 1,478 (C=C,NH), 1,434, 1,339 (C–N, C–O).1H-NMR (CDCl3, 400 MHz): δ 1.94 (m, 2H, CH2-7), 2.06 (m, 2H, CH2-6), 2.99 (t, 2H, J = 6.2 Hz, CH2-8), 4.59 (t, 2H, J = 6 Hz, CH2-5), 7.16 (t, 1H, J = 7.8 Hz, CH-4′), 7.39 (t, 2H, J = 7.6 Hz, CH-3′, -5′), 7.60 (d, 2H, J = 8 Hz, CH-2′, -6′), 8.1 and 8.41 (2 × d, 2 × 2H, J = 8.4 Hz, CH-2″, -3″, -4″, -5″, -6″), 9.25 (s, H, N=CH) and 10.62 (s, 1H, NH, which disappeared on deuteration).13C NMR (CDCl3, 100 MHz): δ 18.6 (CH2), 22.9 (CH2), 23.0 (CH2), 47.2 (CH2), 81.6 (C), 115.7 (CN), 119.7 (2CH), 120.6 (C), 124.2 (CH), 124.4 (2CH), 129.2 (2CH), 129.2 (2CH), 135.7 (C), 138.1 (C), 140.7 (C), 143.6 (C), 149.7 (C), 157.2 (CH), 158.6 (CO). Anal. Calcd. for C23H19N5O3 (413.43): C, 66.82; H, 4.63; N, 16.94. Found C, 67.12; H, 4.70; N, 16.91.
1-Cyano-2-((4-nitrobenzylidene)amino)-N-(p-tolyl)-5,6,7,8-tetrahydroindolizine-3-carboxamide10b
Compound 10b was prepared from 5b and p-nitrobenzaldehyde. Orange crystals, 1.11 g; 73 % yield, m.p. 280–282 °C. IR (KBr, cm−1): ν max. = 3,279 (NH), 3,069 (C–H aromatic), 2,925, 2,857 (CH3, CH2), 2,213 (CN), 1,659 (CO), 1,546, 1,518 (C=C,NH), 1,410, 1,338 (C–N, C–O).1H-NMR (CDCl3, 400 MHz): δ 1.93 (m, 2H, CH2-7), 2.05 (m, 2H, CH2-6), 2.36 (s, 3H, CH 3 Ph), 2.98 (t, 2H, J = 5.8 Hz, CH2-8), 4.59 (t, 2H, J = 5.4 Hz, CH2-5), 7.20, 7.48 (2 × d, 2 × 2H, J = 8 Hz, CH-2′, -3′, -4′, -5′, -6′), 8.09 (d, 2H, J = 8 Hz, CH-2″, -6″), 8.37 (d, 2H, J = 8.4 Hz, CH-3″, -5″), 9.23 (s, H, N=CH) and 10.55 (s, 1H, NH, which disappeared on deuteration).13C NMR (CDCl3, 100 MHz): δ 18.6 (CH2), 20.9 (CH3), 22.9 (CH2), 23.0 (CH2), 47.2 (CH2), 81.5 (C), 115.8 (CN), 119.7 (2CH), 120.8 (C), 124.3 (2CH), 129.2 (2CH), 129.7 (2CH), 133.9 (C), 135.4 (C), 135.5 (C), 140.7 (C), 143.5 (C), 149.6 (C), 157.0 (CH), 158.5 (CO). Anal. Calcd. for C24H21N5O3 (427.46): C, 67.44; H, 4.95; N, 16.38. Found C, 67.40; H, 4.85; N, 16.35.
N-(4-chlorophenyl)-1-cyano-2-((4-nitrobenzylidene)amino)-5,6,7,8-tetrahydro indolizine-3-carboxamide 10c
Compound 10c was prepared from 5c and p-nitrobenzaldehyde. Yellow crystals, 1.11 g; 70 % yield, m.p. 255–257 °C. IR (KBr, cm−1): ν max. = 3,274 (NH), 3,064 (C–H aromatic), 2,952, 2,873 (CH2), 2,209 (CN), 1,666 (CO), 1,539, 1,482 (C=C,NH), 1,404, 1,340 (C–N, C–O). 1H-NMR (CDCl3, 400 MHz): δ 1.96 (m, 2H, CH2-7), 2.07 (m, 2H, CH2-6), 3.01 (t, 2H, J = 5.8 Hz, CH2-8), 4.58 (t, 2H, J = 5.4 Hz, CH2-5), 7.35 (d, 2H, J = 8 Hz, CH-3′, -5′), 7.57 (d, 2H, J = 8 Hz, CH-2′, -6′), 8.10 (d, 2H, J = 8.4 Hz, CH-2″, -6″), 8.41 (d, 2H, J = 8.4 Hz, CH-3″, -5″), 9.28 (s, H, N=CH) and 10.67 (s, 1H, NH, which disappeared on deuteration).13C NMR (CDCl3, 100 MHz): δ 18.7 (CH2), 22.9 (CH2), 23.0 (CH2), 47.3 (CH2), 81.5 (C), 115.9 (CN), 120.5 (2CH), 120.8 (C), 125.1 (2CH), 130.4 (2CH), 131.2 (2CH), 134.3 (C), 135.8 (C), 135.5 (C), 140.7 (C), 143.7 (C), 149.7 (C), 157.3 (CH), 158.8 (CO). Anal. Calcd. for C23H18ClN5O3 (447.87): C, 61.68; H, 4.05; N, 15.64. Found C, 61.98; H, 4.10; N, 15.34.
2-Methyl-4-oxo-3-phenyl-3,4,6,7,8,9-hexahydropyrimido[4,5-b]indolizine-10-carbonitrile 11
A mixture of compound 5a (3.56 mmol) and excess triethylorthoacetate was refluxed for 12 h. The mixture was then evaporated under reduced pressure and the residue was crystallized from ethanol. 0.62 g; 58 % yield, m.p. 244–246 °C. IR (KBr, cm−1): ν max. = 3,062 (C–H aromatic), 2,954 (CH3, CH2), 2,215 (CN), 1,688 (CO), 1,555 (C=C), 1,422, 1,314 (C–N, C–O).1H-NMR (CDCl3, 400 MHz): δ 1.96 (m, 2H, CH2-8), 2.07 (m, 2H, CH2-7), 2.24 (s, 3H, CH 3 C=N), 3.08 (t, 2H, J = 6.2 Hz, CH2-9), 4.45 (t, 2H, J = 6 Hz, CH2-6), 7.24 (d, 2H, J = 7.6 Hz, CH-2′, -6′) and 7.56 (t, 3H, J = 7 Hz, CH-3′, -4′, -5′).13C NMR (CDCl3, 100 MHz): δ 18.9 (CH2), 22.4 (CH2), 23.0 (CH2), 24.3 (CH3), 45.6 (CH2), 85.3 (C), 113.9 (CN), 115.1 (C), 128.0 (2CH), 129.4 (CH), 130.0 (2CH), 137.4 (C), 144.8 (C), 146.5 (C), 154.3 (C), 154.68 (CO). Anal. Calcd. for C18H16N4O (304.35): C, 71.04; H, 5.30; N, 18.41. Found C, 71.17; H, 5.39; N, 18.41.
Biological activity
Cytotoxic activity of the newly synthesized compounds was evaluated using sulphorhodamine-B (SRB) assay method and all the target compounds were screened against three cancer cell lines in addition to the non-tumorous cell line. Hep3B, MCF7, and A549 cancer cell lines were obtained from the American Type Culture Collection (ATCC, Minnesota, USA) through the Tissue Culture Unit, The Egyptian Organization for Biological Products and Vaccines (Vacsera, Egypt). Reagents and chemicals were purchased from Sigma Aldrich Chemical Company (St. Louis, MO, USA). Anticancer activity evaluation was performed at the Center for Genetic Engineering, Al-Azhar University, Cairo, Egypt.
Cells were seeded for 24 h in 96-well microtiter plates at a concentration of 1,000–2,000 cells/well, 100 µl/well, then cells were incubated for 48 h with various concentrations (0, 6.25, 12.5, 25, 50, 100 µg/ml) of the tested compounds, three wells were used for each concentration, after incubation for 48 h the cells were fixed with 10 % trichloroacetic acid 150 µl/well for 1 h at 4 °C, washed by distilled water for three times. Wells were stained for 10-30 min at room temperature with 0.4 % SRB, dissolved in 1 % acetic acid 70 µl/well, washed with acetic acid 1 % to remove unbound dye until a colorless drainage was obtained. The plates were subjected to air drying for 24 h not exposed to UV. The dye was solubilized with 150 µl/well of 10 mM Trise-EDTA (pH 7.4) for 5 min on a shaker at 1,600 rpm. The optical density of each well was measured spectrophotometrically at 545 nm with an ELISA microplate reader. The percent of surviving cells was calculated and plotted against different concentrations of the tested compounds to obtain the survival curve. The IC50 values were calculated using sigmoidal concentration– response curve fitting models (SigmaPlot software). Selectivity index was calculated by dividing the IC50 value of the tested compound against the normal fibroblast cells over that against the cancer cell line (Table 1).
Abbreviations
- SRB:
-
Sulphorhodamine-B
- S. I.:
-
Selectivity index
- µM:
-
Micromole
References
F.J. Swinbourne, J.H. Hunt, G. Klinkert, Adv. Heterocycl. Chem. 23, 103–170 (1979). doi:10.1016/S0065-2725(08)60842-9
J.P. Michael, Nat. Prod. Rep. 25, 139–165 (2008). doi:10.1039/b612166g
K.M. Dawood, H. Abdel-Gawad, M. Ellithey, H.A. Mohamed, B. Hegazi, Arch. Der Pharm. 339, 133–140 (2006). doi:10.1002/ardp.200500176
O.B. Østby, B. Dalhus, L.L. Gundersen, F. Rise, A. Bast, G.R.M.M. Haenen, Eur. J. Org. Chem. 22, 3763–3769 (2000). doi:10.1002/1099-0690(200011)2000:22<3763:AID-EJOC3763>3.0.CO;2-S
L.L. Gundersen, A.H. Negussie, F. Rise, O.B. Østby, Arch. Pharm. Pharm. Med. Chem. 336(3), 191–195 (2003). doi:10.1002/ardp.200390019
W. Mederski, N. Beier, L.T. Burgdorf, R. Gericke, M. Klein, C. Tsaklakidis, US Patent no. 8,106,067 B2, 31 Jan (2012). file:///C:/Users/Toshiba/Downloads/US8106067.pdf (last accessed 5 Feb 2015)
Y.M. Shen, P.C. Lv, W. Chen, P.G. Liu, M.Z. Zhang, H.L. Zhu, Eur. J. Med. Chem. 45(7), 3184–3190 (2010). doi:10.1016/j.ejmech.2010.02.056
V.M. Sharm, K.V.A. Seshu, C.V. Krishna, P. Prasanna, V.C. Sekhar, A. Venkateswarlu, S. Rajagopal, R. Ajaykumar, D.S. Deevi, N.V.S.R. Mamidi, R. Rajagopalan, Bioorg. Med. Chem. Lett. 13(10), 1679–1682 (2003). doi:10.1016/S0960-894X(03)00263-4
F. Lefranc, Z. Xu, P. Burth, V. Mathieu, G. Revelant, M.V.E. de Faria, C. Noyon, D.G. Garcia, D. Durfour, C. Bruyère, C.F.G. de Albuquerque, P.V. Antwerpan, B. Rogister, S. Hesse, G. Kirsch, R. Kiss, Eur. J. Med. Chem. 63, 213–223 (2013). doi:10.1016/j.ejmech.2013.01.046
A. Kajal, S. Bala, S. Kamboj, N. Sharma, V. Saini, J. Catal., 2013, article ID 893512, http://dx.doi.org/10.1155/2013/893512
A.S. Abd El-All, F.A.F. Ragab, A.A. Magd El-Din, M.M. Abdalla, M.M. El-Hefnawi, A.A. El-Rashedy, Glob. J. Pharmacol. 7(2), 143–152 (2013). doi:10.5829/idosi.gjp.2013.7.2.7399
Y. Huang, S. Wolf, M. Bista, L. Meireles, C. Camacho, T.A. Holak, A.D. Miling, Chem. Biol. Drug Des. 76, 116–129 (2010). doi:10.1111/j.1747-0285.2010.00989.x
T. Sagara, S. Ito, S. Otsuki, K. Nonoshita, U.S. Patent no. 2014/0057899Al, Feb 2014
M.Y. Ebeid, S.M.E.L. Moghazy, M.M. Hanna, F.A. Romeih, F.F. Barsoum, Bull. Fac. Pharm. 35, 171 (1997). Cairo Univ
P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J.T. Warren, H. Bokesch, S. Kenney, M.R. Boyd, J. Nat. Cancer Inst. 82, 1107–1112 (1990). doi:10.1093/jnci/82.13.1107
M.-H. Jung, M.I. El-Gamal, M.S. Abdel-Maksoud, T. Sim, K.H. Yoo, C.-H. Oh, Bioorg. Med. Chem. Lett. 22, 4362–4367 (2012). doi:10.1016/j.bmcl.2012.05.004
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Belal, A., Gouda, A.M., Ahmed, A.S. et al. Synthesis of novel indolizine, diazepinoindolizine and Pyrimidoindolizine derivatives as potent and selective anticancer agents. Res Chem Intermed 41, 9687–9701 (2015). https://doi.org/10.1007/s11164-015-1958-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-015-1958-9