
A composite material based on the B4C–SiC system with different initial carbon contents was obtained by reaction sintering (impregnation of a porous billet with liquid silicon). Carbon reacted with silicon to form a secondary silicon carbide that filled the space in the porous workpiece until a monolithic material was obtained. Boron carbide acted as the source of carbon if it was initially absent or its content was low. The obtained materials had the following characteristics: bending strength up to 320 MPa, density up to 2.85 g/cm3, microhardness up to 30 GPa.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Structural materials based on SiC with high strength and wear and chemical resistance during operation in oxidizing media are currently widely used in industry [1,2,3].
High-density silicon carbide (SiC) ceramics are produced by solid-phase and liquid-phase sintering and reaction sintering (silicylation), which is recommended particularly for manufacturing frictional and lining materials. The last is based on impregnation of porous billets of SiC and C with a Si melt, resulting in the space between SiC particles in the billet being filled with secondary SiC until a monolithic material is obtained. The process temperature is above the Si melting point, which is much lower than the sintering temperature of SiC materials prepared using hot pressing and the standard method. These materials are sintered with practically no shrinkage.
The main advantage of SiC over metals is the combination of properties such as long-term strength and low density that enable the ceramic to be used at elevated temperatures of gas-turbine engine parts and other centrifugal structures requiring a reduction of the structure mass.
Boron carbide (density 2.52 g/cm3) is known to possess high-temperature strength because of the formation of a protective viscous film of boron glass on the particle surface upon reaction with oxygen. Boron carbide forms a continuous series of solid solutions with SiC that are wetted well by liquid Si. Therefore, they are promising for use as composite materials [4,5,6,7].
The present work reports results from studies of the microstructure and physicomechanical properties of B4C–SiC composite materials produced by reaction sintering.
Experimental
A mechanical mixture of boron carbide (50 wt.%, F230 brand, particle size 50 – 60 μm) and SiC (50 wt.%, M5 brand, particle size 3 – 5 μm) was prepared to form ceramic billets with dense packing of the particles.
Next, different amounts of C powder were also added to the composite ceramic powder. Mixtures of four compositions were prepared: without additive (composition 1) and with 5, 10, and 15 wt.% C (compositions 2, 3, and 4, respectively). The powders were mixed in a drum mixer for 5 h. The C source was technical C of K-154 brand.
Ceramic samples (5 × 5 × 35 mm) were prepared on a uniaxial hydraulic press at 100 MPa pressure. An aqueous solution of poly(ethylene glycol) (2 wt.%) was used as a plasticizer. The samples were dried at 110°C for 5 h. Then, Si (70 wt.% of the billet) was added. The mixture was sintered in a high-temperature furnace at 1500°C for 10 min. Excesses of Si were removed from the sintered sample surface by sand blasting.
The density and porosity of the sintered samples were measured by hydrostatic weighing according to GOST 473.4–81. The elasticity modulus was measured by resonance vibrations on a ZVUK-130 system. The flexural strength of the samples was determined by testing for three-point bending according to GOST 24409–80. The hardness was determined by the Vickers method on a PMT-3M microhardness tester.
The microstructure and elemental composition of the samples were studied by scanning electron microscopy and x-ray spectral microanalysis on a TESCAN Vega research system.
The phase composition of sintered ceramic samples was studied on a Rigaku Ultima IV multifunctional x-ray diffractometer using filtered Cu Kα1-radiation and a Bragg – Brentano goniometer. The diffractometer was equipped with a suite of controlling programs and PDXL processing software (x-ray Powder Diffraction Software). Diffraction patterns were solved using the COD and PDF-2 databases.
The experiments used equipment at the Center for Common Use Composition, Structure, and Properties of Structural and Functional Materials at NRC Kurchatov Institute, CRISM Prometey.
Results and Discussion
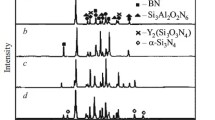
Let us examine characteristic diffraction patterns of the surface of sintered composite ceramic samples for the boundary conditions with changing content of C in the bulk of previously formed billets (Fig. 1).

Diffraction patterns of sintered ceramic samples with various starting carbon contents.
Fused Si diffusing into the bulk of a billet in the three-component system B4C–SiC–C was primarily consumed by the reaction with C to form secondary SiC, which crystallized on grains of B4C and SiC. The masses of components for the reaction forming SiC could be theoretically calculated. However, it was difficult in practice to calculate accurately the exact amount of required starting Si so that residual Si formed in the sintered material (up to 10 vol.%). Previously, the reaction of Si with C was reported to be exothermic with the temperature rise in the reaction zone having a significant influence on the growth rate of SiC crystals [8]. The actual temperature of the ceramic being sintered in local zones was extremely difficult to determine. It could reach 2000 – 2500°C [8, 9].
According to the literature [10, 11], B4C dissolves partially in a Si melt. This was confirmed in all x-ray patterns by the presence of a nonstoichiometric phase that could be interpreted as a solid solution on the B4C particle surface (core-shell type structure). The detected compound had crystal-lattice parameters much like those for dissolved B4C. This result hindered an assessment of the true phase composition of the synthesized compound.
The new phase SiB6 formed if the C content in the billet being sintered was increased to >10 wt.%. The physicochemical characteristics of this compound have been described [12]. In particular, it was found to be highly resistant to thermal shock. SiB6 was a stable high-temperature modification and, according to the Si–B phase diagram, was formed at temperatures over 1864°C (Fig. 2). This was considerably greater than the sintering temperature of the samples studied in the present work. The production of SiB6 was most probably related to decomposition of B4C with free B reacting with the excess of Si to form SiB6. In general, the phase transitions did not allow a relationship with the strength characteristics of the sintered ceramic materials to be established.

Phase diagram of Si–B system [13].
Let us examine the microstructure in the transverse section of the ceramic sample surfaces (Fig. 3). Grains of B4C dissolved more rapidly during the silicylation without the starting C. As a result, the grain boundaries could be etched (dark zones, Fig. 3a ) although the grain boundary for materials of other compositions with previously added C had clear outlines. The microhardness of the dark phase with a high B4C content ranged from 11 to 30 GPa (Table 1). The microhardness of B4C grains in the sample structures of other compositions reached a maximum of 33 GPa.

Microstructure of ceramic materials: composition 1 (a) and composition 4 (b ).
Phase-formation processes during sintering had a substantial effect on the physicomechanical characteristics of the ceramic.
Table 1 shows that the density of the ceramic samples increased and the porosity decreased as a result of the formation of secondary SiC and subsequent saturation by it of the material volume upon reaction of liquid Si with C. This sub-stantially increased the flexural strength. It is noteworthy that the density of the B4C–SiC ceramic was slightly less than that of reaction-sintered SiC (380 MPa). However, the addition of B4C significantly increased the hardness of the composite ceramic material. Table 1 presents for comparison the physicomechanical properties of standard ceramic materials based on SiC and B4C.
Addition of starting C and silicylation of the B4C–SiC–C composite led to the formation of secondary SiC, filling of the whole pore space and; therefore, production of a highly dense monolithic material with a high set of physicomechanical characteristics.
Conclusion
A composite material based on B4C–SiC with various contents of starting C was produced by reaction sintering (impregnation by liquid Si of a porous billet of B4C–SiC–C). The phase composition of the sintered material was determined. Compounds of composition B12.97Si0.03C2 (solid solution of Si in B4C) and SiB6 were shown to form during the sintering. An analysis of the microstructures of the sintered materials showed that B4C was more rapidly dissolved in the Si melt without starting C. In this instance, B4C acted as a source of C for the synthesis of SiC; the released B, most probably for the formation of SiB6.
Addition of starting C (15 wt.%) for the synthesis of the composite materials was found experimentally to increase the strength up to 320 MPa; density, 2.85 g/cm3; and microhardness of the resulting solid solution, 30 GPa.
References
T. Abraham, “Powder market update: Nanoceramic applications emerge,” Am. Ceram. Soc. Bull., 83(8), 23 – 25 (2004).
F. L. Matthews and R. D. Rawlings, Composite Materials: Engineering and Science, CRC Press, Boca Raton, Fla., 2003 [Russian translation, Tekhnosfera, Moscow, 2004, 408 pp].
D. Sciti, A. Balbo, C. Melandri, et al., “Microstructure and properties of an electroconductive SiC-based composite,” J. Mater. Sci., 42(14), 5570 – 5575 (2007).
D. D. Nesmelov and S. N. Perevislov, “Reaction sintered materials based on boron carbide and silicon carbide,” Glass Ceram., 71(9 – 10), 313 – 319 (2015).
S. N. Perevislov, P. V. Shcherbak, and M. V. Tomkovich, “High density boron carbide ceramics,” Refract. Ind. Ceram., 59(1), 32 – 36 (2018).
S. N. Perevislov, P. V. Shcherbak, and M. V. Tomkovich, “Phase composition and microstructure of reaction-bonded boron-carbide materials,” Refract. Ind. Ceram., 59(2), 179 – 183 (2018).
M. A. Markov, A. V. Krasikov, P. A. Kuznetsov, A. D. Bykova, M. V. Khromenkov, and E. A. Samodelkin, “Method of producing ceramic structural material based on silicon carbide for articles of complex geometry,” RU Pat. 2,739,774, C04B35/573, Dec. 28, 2020; Byull., No. 1.
N. P. Bansal (ed.), Handbook of Ceramic Composites, Kluwer Academic Publishers, 2005, 554 pp.
Z. Yang, X. He, M. Wu, et al., “Infiltration mechanism of diamond/ SiC composites fabricated by Si-vapor vacuum reactive infiltration process,” J. Eur. Ceram. Soc., 33(4), 869 – 878 (2013).
S. Hayun, A. Weizmann, M. P. Dariel, et al., “Microstructural evolution during the infiltration of boron carbide with molten silicon,” J. Eur. Ceram. Soc., 30(4), 1007 – 1014 (2010).
M. P. Dariel and N. Frage, “Reaction bonded boron carbide: Recent developments,” Adv. Appl. Ceram., 111(5 – 6), 301 – 310 (2012).
T. Ya. Kosolapova, T. V. Andreeva, T. S. Barnitskaya, et al., Nonmetallic Refractory Compounds [in Russian], Metallurgiya, Moscow, 1985, 224 pp.
G. V. Samsonov, T. I. Serebryakova, and V. A. Neronov, Borides [in Russian], Atomizdat, Moscow, 1975, 375 pp.
S. N. Perevislov, A. S. Lysenkov, D. D. Titov, et al., “Liquid-sintered SiC based materials with additive low oxide oxides,” IOP Conference Series: Materials Science and Engineering, IOP Publishing, 2019, 525(1), Art. 012073.
S. N. Perevislov,M. A. Markov, A. V. Krasikov, et al., “Effect of SiC dispersed composition on physical and mechanical properties of reaction-sintered silicon carbide,” Refract. Ind. Ceram., 61(2), 211 – 215 (2020); Nov. Ogneupory, No. 4, 41 – 45 (2020).
S. N. Perevislov, M. V. Tomkovich, M. A. Markov, et al., “The influence of dispersed composition of SiC on the physico-mechanical properties of reactive-sintered silicon carbide,” J. Mach. Manuf. Reliab., 49(6), 511 – 517 (2020).
S. N. Perevislov, A. S. Lysenkov, D. D. Titov, et al., “Materials based on boron carbide obtained by reaction sintering,” IOP Conference Series: Materials Science and Engineering, IOP Publishing, 2019, 525(1), Art. 012074.
M. A. Markov, S. S. Ordan’yan, S. V. Vikhman, et al., “Preparation of MoSi2–SiC–ZrB2 structural ceramics by free sintering, Refract. Ind. Ceram., 60(4), 385 – 388 (2019).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Novye Ogneupory, No. 2, pp. 29 – 33, February, 2023
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Belyakov, A.N., Markov, M.A., Perevislov, S.N. et al. Investigation of the Structure and Physicomechanical Characteristics of Reaction-Sintered Materials B4C–SiC. Refract Ind Ceram 64, 67–70 (2023). https://doi.org/10.1007/s11148-023-00806-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11148-023-00806-0