
Abstract
The carbon-coated Ni (Ni@C) and Ni–In intermetallic compounds (IMCs) (Ni–In@C) catalysts were synthesized via combining one-pot hydrothermal method with glucose as carbon source and the carbonization under N2 atmosphere. Their reactivities were compared in in situ aqueous phase selective hydrogenation of methyl palmitate to hexadecanol using methanol as a H2 donor. The Ni@C catalyst dominatingly catalyzes decarbonylation/decarboxylation, accompanying with the serious C–C bond hydrogenolysis and methanation. In contrast, hexadecanol is mainly generated on Ni–In@C, where C–C bond hydrogenolysis and methanation are remarkably inhibited. This is ascribed to the geometric and electronic property of Ni–In IMCs. The hexadecanol yield reaches 84.0% under an optimal condition on Ni–In@C. The structure of NiIn IMC is hydrothermally stable even at 330 °C, and the Ni–In IMCs particles highly resist to sintering and leaching under harsh hydrothermal condition due to the confinement of carbon. Catalyst deactivation is mainly due to the carbonaceous deposition, and the catalyst reactivity is mostly recovered by the regeneration with CO2.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Higher fatty alcohols are important basic materials for producing surfactants, plasticizers, food additives and cosmetics. Generally, they are produced from the selective hydrogenation of fatty acid/esters derived from renewable vegetable oils and animal fats [1, 2].
Conventionally, the selective hydrogenation of fatty acid/esters is carried out with external H2 supply under a pressure. However, there is a safety problem during the storage and transportation of hydrogen, and the transportation of hydrogen is also high- cost. To circumvent this, it is very attractive to in situ generate H2 (for instance, aqueous phase reforming of methanol [3,4,5]). The in situ aqueous phase hydrogenation of lauric acid with methanol as the hydrogen donor has been investigated at 330 °C, and the yield of lauryl alcohol reaches 99.2 and 45.8% on Cu/Al2O3 and Cu/ZrO2 catalysts [2] and 62.3% on the CuCo4/C catalyst [6].
It is well known that subcritical water is highly corrosive, which can destroy the catalyst structure, leach the active components and promote the sintering of catalysts (including support and metal) [7]. To solve this problem, a feasible approach is to coating the metal particles with a high hydrothermal stable support. Al2O3 and SiO2 are not hydrothermally stable, and ZrO2 and TiO2 possess good hydrothermal stability but low surface areas [7]. Carbon has good hydrothermal stability and high surface area. But then, the interaction between metal and carbon is weak, and the carbon-supported metals are subjected to sintering and leaching [8, 9]. Recently, by virtue of the confinement effect of carbon, carbon-coated metal catalysts have been proved to effectively suppress metal sintering and leaching [10,11,12,13,14]. For instance, in the hydrogenation of acetone to isopropanol in an aqueous phase, the uncoated Pd/SiO2 catalyst is rapidly deactivated within 7 h, while the carbon-coated Pd/SiO2 catalyst is stable in a run time of nearly 60 h [12]. Nitrogen-doped carbon-encapsulated cobalt catalysts exhibits excellent activity in the aqueous hydrogenation of vanillin, ascribed to reducing the leaching of cobalt nanoparticles embedded in the framework of nitrogen-doped carbon materials [15]. The carbon-coated metal catalysts can be prepared with polydopamine, phenol–formaldehyde resins, sugars and metal–organic framework compounds as carbon sources [16,17,18,19]. Thereinto, surges are biomass-derived, and they can be converted to the carbon particles via a hydrothermal process, and have been widely used for preparing the carbon-coated metals or oxides catalysts [20]. For example, Co@C NPs has been prepared by a two-step process using glucose as a carbon source [21]. Compared with the two-step one, the one-pot process has the advantages of simplicity, efficiency and resource saving. Co@NC-600 and Fe2O3@C have been prepared by one-pot method using sucrose and glucose as carbon sources [18, 22]. It is very significant to extend the one-pot hydrothermal process method with sugars as carbon sources to prepare carbon-coated metal catalysts.
Our previous work has shown that Ni–In IMCs possess good performance in the selective hydrogenation of fatty acid esters to fatty alcohols [23], while their hydrothermal stability and performance in in situ aqueous phase selective hydrogenation is unclear. In the present work, we investigated the reactivity of Ni–In IMCs in in situ aqueous phase selective hydrogenation of methyl palmitate using methanol as the hydrogen donor. To restrain the sintering and leaching, carbon-coated Ni–In IMCs were successfully prepared by the one-pot hydrothermal synthesis with renewable glucose as a carbon source. It has been found that NiIn IMC is hydrothermally stable and the carbon-coating prevents NiIn IMC particles from leaching and sintering. The hexadecanol yield can reach 84% on the as-prepared catalyst.
Experimental
Materials
All chemicals were analytically pure and used directly without further purification. Glucose, Tianjin Yuanli Chemical Co., Ltd.; Urea (≥ 99.0 wt%), Tianjin Guangfu Technology Co., Ltd.; Ni(NO3)2·6H2O (≥ 99 wt%), Adamas-beta Co., Ltd.; In(NO3)3·4H2O (≥ 99.999 wt%), Guangxi Shimei Ceramic Products Co., Ltd.; Methanol (≥ 99.5 wt%), Kermel Co., Ltd.; Ethanol (≥ 95 wt%), Cyclohexane (≥ 99.7 wt%), Ethyl acetate (≥ 99 wt%), Real & Lead Chemical Co., Ltd.; Ethyl palmitate (≥ 97 wt%), Octadecane (≥ 99 wt%), Tetralin (≥ 97 wt%), Aladdin Bio-Chem Technology Co., Ltd.
Catalyst preparation
Carbon-coated Ni and Ni–In IMCs were synthesized by the one-pot hydrothermal method with glucose as carbon source, followed by the carbonization in the N2 atmosphere. In a typical process, glucose, urea, Ni(NO3)2·6H2O and/or In(NO3)3·4H2O (detail amounts in Table S1 in Supplementary information) were dissolved in deionized water (50 mL), and then the mixture was ultrasonicated to form a solution. Subsequently, the solution was sealed into a 100 mL Teflon-lined stainless-steel autoclave and heated to 160 °C maintained for 12 h. After naturally cooling to room temperature, the solid was collected by centrifugation, washed with deionized water and alcohol, and dried at 80 °C for 24 h. Finally, the obtained powder was carbonized on a quartz fixed-bed reactor under a N2 flow according to the following procedure: (1) heated to 600 °C at the heating rate of 2 °C min−1 and maintained for 2 h; (2) heated to 800 at the heating rate of 3 °C min−1 and maintained for 1 h. The as-prepared carbon-coated metallic Ni and Ni–In bimetallic catalysts are labeled as wNi@C and wNixIn@C, where w and x denote nominal Ni/C mass ratio and Ni/In atomic ratio, respectively.
For comparison, the activated carbon (AC) supported Ni–In bimetallic catalyst (Ni–In/AC, nominal Ni content 27.8 wt% and Ni/In atomic ratio of 2) was prepared by the impregnation method. Firstly, the AC support (< 0.074 μm) was impregnated by the mixed aqueous solution of Ni(NO3)2·6H2O and In(NO3)3·4H2O for 48 h. Afterward, the sample was dried at 120 °C for 12 h, and then reduced on a quartz fixed-bed reactor under a H2 flow at 500 °C for 2 h. After dropping to room temperature, the obtained powders were passivated under an atmosphere of 0.5 V%O2/N2 mixed gas for 3 h.
Catalyst characterization
Fourier transform infrared (FT-IR) and Thermogravimetry (TG) were carried out on a Thermo Nicolet Nexus and a Hitachi TG/DTA7300 instrument. Raman spectra was recorded on a LabRAM HR Evolution. The X-ray diffraction (XRD) pattern was obtained on a D8 X-ray diffractometer with Cu Kα radiation (λ = 0.1541 nm). Scanning electron microscopy (SEM) was used to examine the catalyst morphology on a Hitachi regulus 8100 electron microscope. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were got on a JEM-F200 field-emission electron microscope. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific ESCALAB 250Xi with Mono Al Kα radiation (1486.6 eV). Binding energy was calibrated by C 1 s (284.8 eV). Before the measurement, the catalysts were sputtered by Ar+ ion beam to remove the oxide layer. The Ni and In contents in catalysts were measured on a ICPE-9800 by inductively coupled plasma optical emission spectroscopy (ICP-OES).
Catalytic test
The catalyst performance was tested using a 100 mL autoclave (Beijing Century Senlong Experimental Apparatus Co., Ltd.). In a typical run, 4 g of methyl palmitate, 8 g of deionized water, a certain amount of methanol and catalyst were loaded into the autoclave, followed by sealing the autoclave and purging the inside air with N2 three times. Afterward, the autoclave was pressurized with N2 to 1.0 MPa (as internal standard for analyzing gaseous products), and then heated to the reaction temperature under stirring of 500 rpm. After the reaction, the autoclave was cooled down to 50 °C by charging water through internal cooling coils. The gaseous products were collected by an air bag for gas chromatography analysis. Cyclohexane was added to autoclave to extract the organic compounds. The carbon balance was above 92%.
H2 and C1 (CO, CH4 and CO2) gaseous products were quantitatively analyzed on a 102 gas chromatograph (GC) equipped with a TDX-101 packed column and a thermal conductivity detector (TCD) using Ar as the carrier gas, and N2 was used as the internal standard. The C1–C5 alkanes in the gaseous product were quantitatively analyzed on a SP-3420 GC equipped with HP-AL/S capillary column and hydrogen flame ionization detector (FID), and methane, whose amount was determined on a 102 GC, was used as the internal standard.
The liquid phase products were quantitatively analyzed on a SP-3420 GC equipped with HP-5 capillary column and a FID. For the organic phase, ethyl acetate and tetralin was used as the internal standard to quantitatively analyze methanol and C6–C14 alkanes. Octadecane was used as the internal standard to quantitatively analyze pentadecane, hexadecane, hexadecanal, hexadecanol, palmitic acid, and methyl palmitate. Ethanol was used as internal standard to quantitatively analyze the compounds in the aqueous phase.
The conversions of methyl palmitate and methanol and the yield of product i (C2–C16 hydrocarbons, hexadecanol, palmitic acid) were calculated by the following formulas:
The yield of CH4 was defined as follows:
Here MP denotes methyl palmitate; nMP and nmethanol denote the initial amount of substance of methyl palmitate and methanol; n′methyl palmitate and n′methanol denote the amounts of sbstance of unreacted methyl palmitate and methanol, respectively; \({\text{n}}_{i}\) and nc denote the mole of the product i and the number of carbon atom in product i.
Results and discussion
Catalyst characterization
FT-IR and TG
Here, 0.30Ni2In@C is selected to investigate the structure and the carbonization process of the hydrothermal products. In the FT-IR spectra (Fig. S1a), several characteristic bands are observed at ~ 3400 cm−1 (stretching vibrations of O–H), 1612 and 1446 cm−1 (the stretching vibrations of aromatic C=C and C=N bonds), 823 cm−1 (triazine ring modes), 1384 cm−1 (sp3 C–C bond or disordered sp2 graphitic domains) and 1184 cm−1 (C–OH/C–O–C bonds) [24,25,26]. The results indicate that glucose undergoes aromatization during the hydrothermal process, and abundant oxygen-containing functional groups still remain [27]. Especially, nitrogen is introduced in the precursor due to the Schiff base reaction between aldehyde groups of 5-hydroxymethylfurfural (hydrolysis compound of glucose) and the amine groups of urea [28]. TG profile (Fig. S1b) shows three main weight loss stages: (1) below 220 °C, the escape of physically adsorbed water and bound water; (2) 220–458 °C, the intramolecular dehydration and decarbonylation/decarboxylation of oxygen-containing functional groups [18]; (3) 458–800 °C, the pyrolysis of the sample, during which the Ni2+ and In3+ species are reduced by carbon forming NiIn IMCs, and the carbon-coated Ni or Ni–In IMCs particles are synthesized. Meanwhile, the reduced Ni and In in turn promote the carbonization process [29].
XRD and Raman
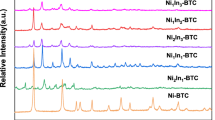
As shown in the XRD patterns (Fig. 1), the diffraction peaks assigned to metallic Ni (PDF#65-2865) and graphitic carbon (PDF#25-0284) are visible for 0.25Ni@C. Both Ni3In IMC (PDF#65-9308) and NiIn IMC (PDF#07-0178) form in 0.25Ni3In@C. NiIn IMC is dominating in 0.25Ni2In@C, accompanying with minor Ni2In3 IMC (PDF#65-7718). There is main Ni2In3 IMC in 0.25NiIn@C. Also, In2O3 exists in all 0.25NixIn@C. In conclusion, Ni2+ and partial In3+ species are reduced by carbon during the carbonization, forming metallic Ni in 0.25Ni@C and different Ni–In IMCs in 0.25NixIn@C. In 0.25NixIn@C, the phase structure of Ni–In IMCs depends on the nominal Ni/In atomic ratio. With the decrease in the nominal Ni/In atomic ratio, the proportion of In in Ni–In IMCs increases. In addition, no graphitic carbon is detected in 0.25NixIn@C, i.e., the formation of Ni–In IMCs prevents the graphitization, also confirmed by Raman results (Fig. S2). As shown in the XRD patterns of wNi2In@C catalysts (Fig. S3), 0.15Ni2In@C, 0.25Ni2In@C and 0.35Ni2In@C contain Ni2In3 IMC, NiIn IMC and In2O3. Interestingly, NiIn IMC and In2O3 exist in 0.30Ni2In@C.

XRD patterns of 0.25Ni@C and 0.25NixIn@
SEM and TEM
In the as-prepared precursor after the hydrothermal process, the agglomerated nanospheres and nanorods are observed (Fig. S4), and they become slightly smaller after the carbonization due to the loss of C/H/O. In addition, their morphology does not obviously change after the in situ aqueous phase selective hydrogenation.
Figs. 2 and S5 show the TEM/HRTEM images of 0.25Ni@C, 0.25Ni2In@C and 0.30Ni2In@C. Clearly, the metal particles are embedded in the carbon particles. The metallic Ni particle size in 0.25Ni@C is about 18.5 nm, in contrast, there are smaller NiIn IMC particles (13.5 nm) in 0.25Ni2In@C. Thus, the presence of metal in causes the metal particles small; moreover, the dispersibility of metal particles in the carbon particles is also improved. 0.30Ni2In@C has larger NiIn IMC particles (19.4 nm) than 0.25Ni2In IMC, ascribed to its higher nominal metal loading. The lattice spacing value of 0.310 nm corresponds to the (100) crystal plane of NiIn IMC) (Fig. 2) [23]. As shown by the TEM-EDS mapping and line scanning of 0.30Ni2In@C, In and Ni appear in the same area. This is due to the formation of NiIn IMC, where the Ni and In atoms are uniformly separated each other.

TEM-EDS mapping and line scanning images of 0.30Ni2In@C
XPS
Fig. 3 shows the XPS spectra of Ni 2p and In 3d in 0.25Ni@C and 0.30Ni2In@C. A pair of doublet peaks at around 870.4 and 853.2 eV for Ni 2p1/2 and Ni 2p3/2 are ascribed to Ni0 on 0.25Ni@C [30, 31]. In comparison, the Ni 2p1/2 and Ni 2p3/2 peaks of Ni0 shift to lower binding energies (870.1 and 852.8 eV, respectively) on 0.30Ni2In@C. The In 3d5/2 and In 3d3/2 peaks at the binding energies of 444.2 and 451.7 eV are attributed to In0, which are higher than that of elemental In (444.0 and 451.4 eV) [32]. Therefore, a charge transfer occurs from In to Ni, consistent with the higher electronegativity of Ni (1.98) than that of In (1.78). In addition, the peaks ascribed to Ni2+ and In3+ are observed. This is related to the exposure of the catalysts to air before XPS measurement, and the Ar+ ions sputtering did not completely remove the oxidized layer. In addition, In2O3, detected by XRD, also contributes to the presence of In3+.

XPS spectra of Ni 2p and In 3d in 0.25Ni@C and 0.30Ni2In@C
Catalytic performance
Herein, using methyl palmitate as the model reactant and methanol as the hydrogen donor, the catalysts were tested for in situ aqueous phase selective hydrogenation of methyl palmitate. Palmitic acid, n-pentadecane (n-C15H32), n-hexadecane (n-C16H34), hexadecanol and C6–C14 alkanes were detected in the liquid products.
Effect of Ni/In atomic ratios on catalyst performance
First, the performances of 0.25Ni@C and 0.25NixIn@C are compared (Fig. 4). The conversions of methyl palmitate and methanol on 0.25Ni@C are 99.7 and 94.0%, respectively. For 0.25NixIn@C, as the Ni/In atomic ratio decreases from 3 to 1, the conversion of methyl palmitate decreases from 98.9 to 95.7%, while the conversion of methanol first increases and then decreases, and 0.25Ni2In@C gives the highest conversion (81.5%) of methanol.

Performance of 0.25Ni@C and 0.25NixIn@C in in situ aqueous phase selective hydrogenation of methyl palmitate. Reaction condition: 330 °C, 4 g methyl palmitate, 3 g methanol, 8 g H2O, 0.4 g catalyst and 6 h
Under hydrothermal condition, methyl palmitate is easily hydrolyzed to palmitic acid and methanol [11, 33]. And the conversion of palmitic acid is the key. The yield of palmitic acid is 19.2% on 0.25Ni@C. n-Pentadecane (the yield of 36.2%) is the main product, while the yields of n-hexadecane and hexadecanol are only 1.7 and 0.4%, respectively. In addition, there is a high yield (22.2%) of C6–C14 alkanes. That is, the metallic Ni is active for decarbonylation/decarboxylation and C–C bond hydrogenolysis [23, 34]. In contrast, hexadecanol, derived from the hydrogenation pathway, is dominating on 0.25NixIn@C. This is attributed to Ni–In IMCs where the presence of oxyphilic metal In favors the adsorption mode of η2(C, O)-aldehyde. As indicated by XPS, there is an electron transfer from In to Ni on Ni–In IMCs. Thus, the negative charged Ni and the positively In atoms preferentially adsorb the positively charged C atoms and the negatively charged O [23], and so the hydrogenation of C–O/C=O is promoted. Moreover, the In atoms isolate the continuous Ni atoms in Ni–In IMCs, restraining the C–C bond hydrogenolysis. As a result, there are a very low yield (< 1%) of C6–C14 hydrocarbons on 0.25NixIn@C. As indicated by XRD, 0.25Ni3In@C, 0.25Ni2In@C and 0.25NiIn@C have different Ni–In IMCs, while they give similar yield of hexadecanol (~ 65.0%). In addition, the yields of n-hexadecane and palmitic acid are all around 5%. Thus, the structure of Ni–In IMCs shows a little effect on the yields of the main liquid products.
Influence of Ni/C mass ratios on performance of Ni2In@C
As shown in Fig. 5, with increasing the Ni/C mass ratio, the conversions of methyl palmitate and methanol and the yield of hexadecanol first increase and then decrease, while the yield of palmitic acid tends to decrease. 0.30Ni2In@C has the highest conversions of methyl palmitate (98.3%) and methanol (83.6%), the highest yield of hexadecanol (73.6%) and the lowest yield of palmitic acid (3.1%). In all, 0.30Ni2In@C has the best performance.

Performance of wNi2In@C in in situ aqueous phase selective hydrogenation of methyl palmitate. Reaction condition: 330 °C, 4 g methyl palmitate, 3 g methanol, 8 g H2O, 0.4 g catalyst and 6 h
Effect of reaction condition on performance of 0.30Ni2In@C
As indicated above, 0.30Ni2In@C shows good performance in in situ aqueous phase selective hydrogenation of methyl palmitate to hexadecanol. The effect of reaction condition on its performance is investigated to get high yield of hexadecanol.
The effect of reaction temperature is illustrated in Fig. 6a. The yields of hexadecanol are 72.1, 81.3 and 80.7% at the temperature of 320, 330 and 340 °C, i.e., first increase and then remain unchanged, while the yields of n-pentadecane and n-hexadecane increase. That is, raising the reaction temperature promotes decarbonylation/decarboxylation and the dehydration of hexadecanol [35]. Additionally, the yields of CH4 and C6–C14 are all lower than 0.2% at each reaction temperature, further indicative of low activity of NiIn IMC for the C–C bond hydrogenolysis and methanation.

Effect of a reaction temperature; b reaction time; c methanol dosage; and d catalyst dosage on performance (conversions of methyl palmitate and methanol; carbon balance; yields of methane and main liquid phase products) of 0.30Ni2In@C in in situ selective hydrogenation of methyl palmitate. Reaction condition: a 4 g methyl palmitate, 3 g methanol, 8 g H2O, 0.4 g catalyst and 4 h; b 4 g methyl palmitate, 3 g methanol, 8 g H2O, 0.4 g catalyst and 330 °C; c 4 g methyl palmitate, 8 g H2O, 0.4 g catalyst, 330 °C and 4 h; and d 4 g methyl palmitate, 3 g methanol, 8 g H2O, 330 °C and 4 h
With prolonging the reaction time from 0 to 10 h (Fig. 6b), the yield of hexadecanol first increases and then decreases, and the highest hexadecanol yield of 81.3% reaches at the 4th h. The yield of n-hexadecane increases from 1.5 to 7.6%, while the yields of n-pentadecane are all lower than 3.6%, and the yields of CH4 and C6–C14 are both lower than 0.2%. The change in the product distribution along with the time indicates that the main reaction pathway on 0.30Ni2In@C is hydrogenation pathway, that is, methyl palmitate → palmitic acid → hexadecanal → hexadecanol → n-hexadecane.
In addition, the increased methanol and catalyst dosages favor the selective hydrogenation (Fig. 6c, d). Reasonably, the increased methanol dosage generates more hydrogen, while the increased catalyst dosage provides more active sites.
In all, the hexadecanol yield of 84.0% is obtained on 0.30Ni2In@C at 330 °C, 4 g methyl palmitate, 3 g methanol, 0.6 g catalyst and reaction time 4 h. It is higher than that (73.1%) on Ni–In/AC with the same nickel content.
Catalyst stability and regeneration
To investigate the stability of 0.30Ni2In@C, the used catalyst is recycled after washing with cyclohexane three times. As shown in Fig. S6 on the fresh catalyst (i.e., 1st test), the yield of hexadecanol is 81.3%. In the first and second recycling, the yields of hexadecanol are 73.9 and 46.3%, respectively.
In order to explore the reason for the catalyst deactivation, the used 0.30Ni2In@C catalyst is characterized. NiIn IMC and In2O3 still exist after reaction (Fig. S7a). That is, NiIn IMC is hydrothermally stable even at 330 °C, and the average NiIn IMC particle sizes are 19.4 and 22.7 nm before and after reaction (Fig. S5). The Ni and In contents are 27.8 and 35.1% in the fresh catalyst, respectively, and they are 26.5 and 33.8% in the used one. Thus, the sintering and leaching of the active phase are not obvious under the harsh hydrothermal condition, which is ascribed to the confinement of carbon. The TG profile shows about 3 wt% carbonaceous deposit on the used catalyst, and carbonaceous deposit is also confirmed by a higher ID/IG value for the used catalyst (Fig. S7b, c). Here, the catalyst (0.4 g) after the 3rd test was treated by CO2 at 500 °C and then reduced with H2 at 550 °C to regenerate it. After the regeneration, the mass of the residual catalyst deceases to about 0.343 g due to the reaction between CO2 and carbon. The residual catalyst shows similar reactivity to 2nd test, giving the yields of hexadecanol of 74.2% (Fig. S6). This further implies that the catalyst deactivation is mainly due to the carbon deposition.
Conclusion
Carbon-coated Ni and Ni–In IMCs catalysts are successfully synthesized by the one-pot hydrothermal method. The confinement of carbon remarkably prevents the Ni–In IMCs particles from sintering and leaching under harsh hydrothermal condition, and NiIn IMC is hydrothermally stable even at 330 °C. The formation of Ni–In IMCs restrains the graphitization while enhances the dispersion of Ni in the carbon nanoparticles. In Ni–In IMCs, a charge transfer occurs from In to Ni. Different from metallic Ni that dominatingly catalyze the decarbonylation/decarboxylation, Ni–In IMCs mainly give hexadecanol via selective hydrogenation with very low activity for C–C bond hydrogenolysis and methanation. This is due to the geometric and electronic property of NiIn IMC. 0.30Ni2In@C has good performance, which is remarkably affected by reaction temperature, reaction time, methanol dosage and catalyst dosage. The hexadecanol yield reaches 84.0% on 0.30Ni2In@C under a suitable reaction condition at 330 °C. The carbonaceous deposition mainly contributes to the catalyst deactivation, while the treatment with CO2 is feasible to regenerate the deactivated catalyst.
References
He L, Cheng H, Liang G, Yu Y, Zhao F (2013) Effect of structure of CuO/ZnO/Al2O3 composites on catalytic performance for hydrogenation of fatty acid ester. Appl Catal A 452:88–93
Zhang Z, Zhou F, Chen K, Fu J, Lu X, Ouyang P (2017) Catalytic in situ hydrogenation of fatty acids into fatty alcohols over Cu-based catalysts with methanol in hydrothermal media. Energy Fuels 31:12624–12632
Liu X, Yang M, Deng Z, Dasgupta A, Guo Y (2021) Hydrothermal hydrodeoxygenation of palmitic acid over Pt/C catalyst: mechanism and kinetic modeling. Chem Eng J 407:126332
Wang J, Xu L, Nie R, Lyu X, Lu X (2020) Bifunctional CuNi/CoOx catalyst for mild-temperature in situ hydrodeoxygenation of fatty acids to alkanes using isopropanol as hydrogen source. Fuel 265:116913
Gilkey MJ, Xu B (2016) Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal 6:1420–1436
Gou X, Okejiri F, Zhang Z, Liu M, Liu J, Chen H, Chen K, Lu X, Ouyang P, Fu J (2020) Tannin-derived bimetallic CuCo/C catalysts for an efficient in-situ hydrogenation of lauric acid in methanol-water media. Fuel Process Technol 205:106426
Xiong H, Pham HN, Datye AK (2014) Hydrothermally stable heterogeneous catalysts for conversion of biorenewables. Green Chem 16:4627–4643
Wu C, Chen X, Tang L, Wei Q, Wei X, Liang J, Wang L (2021) Rationally constructing a nano MOF-derived Ni and CQD embedded N-doped carbon nanosphere for the hydrogenation of petroleum resin at low temperature. ACS Appl Mater Interfaces 13:10855–10869
Liu Y, Yang X, Liu H, Ye Y, Wei Z (2017) Nitrogen-doped mesoporous carbon supported Pt nanoparticles as a highly efficient catalyst for decarboxylation of saturated and unsaturated fatty acids to alkanes. Appl Catal B 218:679–689
Shi J, Zhao M, Wang Y, Fu J, Lu X, Hou Z (2016) Upgrading of aromatic compounds in bio-oil over ultrathin graphene encapsulated Ru nanoparticles. J Mater Chem A 4:5842–5848
Shi Y, Ai L, Shi H, Gu X, Han Y, Chen J (2021) Carbon-coated Ni-Co alloy catalysts: preparation and performance for in-situ aqueous phase hydrodeoxygenation of methyl palmitate to hydrocarbons using methanol as the hydrogen donor. Front Chem Sci Eng 16:443
Pham HN, Anderson AE, Johnson RL, Schwartz TJ, O’Neill BJ, Duan P, Schmidt-Rohr K, Dumesic JA, Datye AK (2015) Carbon overcoating of supported metal catalysts for improved hydrothermal stability. ACS Catal 5:4546–4555
Qian L, Lan G, Liu X, Li Z, Li Y (2021) Aqueous-phase hydrogenation of levulinic acid over carbon layer protected silica-supported cobalt-ruthenium catalysts. Chin J Chem Eng 38:114–122
Huang Q, Yu W, Lu F, Lu R, Si X, Gao J, Xu J (2019) Fabrication of highly dispersed Ru nanoparticles stabilized in coated carbon shell via one-pot co-synthesis strategy for aqueous hydrogenation of bio-based itaconic acid. Catal Today 319:197–205
Yang H, Nie R, Xia W, Yu X, Jin D, Lu X, Zhou D, Xia Q (2017) Co embedded within biomass-derived mesoporous N-doped carbon as an acid-resistant and chemoselective catalyst for transfer hydrodeoxygenation of biomass with formic acid. Green Chem 19:5714–5722
Wang Z, Zou Y, Li Y, Cheng Y (2020) Metal-containing polydopamine nanomaterials: catalysis, energy, and theranostics. Small 16:1907042
Wei J, Wang G, Chen F, Bai M, Liang Y, Wang H, Zhao D, Zhao Y (2018) Sol-Gel synthesis of metal-phenolic coordination spheres and their derived carbon composites. Angew Chem Int Ed 57:9838–9843
Yuan M, Long Y, Yang J, Hu X, Xu D, Zhu Y, Dong Z (2018) Biomass sucrose-derived Cobalt@Nitrogen-Doped carbon for catalytic transfer hydrogenation of nitroarenes with formic acid. Chemsuschem 11:4156–4165
Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM (2013) The chemistry and applications of metal-organic frameworks. Science 341:1230444
Matsagar BM, Yang R, Dutta S, Ok YS, Wu KCW (2021) Recent progress in the development of biomass-derived nitrogen-doped porous carbon. J Mater Chem A 9:3703–3728
Liu L, Gao F, Concepción P, Corma A (2017) A new strategy to transform mono and bimetallic non-noble metal nanoparticles into highly active and chemoselective hydrogenation catalysts. J Catal 350:218–225
Zhang M, Sha J, Miao X, Liu E, Shi C, Li J, He C, Li Q, Zhao N (2017) Three-dimensional graphene anchored Fe2O3@C core-shell nanoparticles as supercapacitor electrodes. J Alloys Compd 696:956–963
Wang L, Niu X, Chen J (2020) SiO2 supported Ni–In intermetallic compounds: efficient for selective hydrogenation of fatty acid methyl esters to fatty alcohols. Appl Catal B 278:119293
Ryu J, Suh YW, Suh DJ, Ahn DJ (2010) Hydrothermal preparation of carbon microspheres from mono-saccharides and phenolic compounds. Carbon 48:1990–1998
Hwang S, Lee S, Yu J (2007) Template-directed synthesis of highly ordered nanoporous graphitic carbon nitride through polymerization of cyanamide. Appl Surf Sci 253:5656–5659
Zhao Z, Dai Y, Ge G, Wang G (2015) Efficient tuning of microstructure and surface chemistry of nanocarbon catalysts for ethylbenzene direct dehydrogenation. AIChE J 61:2543–2561
Bernard TA, Kabyemela M, Malaluan RM, Arai K (1999) Glucose and fructose decomposition in subcritical and supercritical water detailed reaction pathway, mechanisms, and kinetics. Ind Eng Chem Res 38:8
Sun J, Yang Y, Wang J, Lu B, Guo J (2021) Ammonia assisted regulation of nitrogen-type in carbonaceous support applied for oxygen reduction reaction. Appl Surf Sci 558:149958
Wang J, Wei Z, Gong Y, Wang S, Su D, Han C, Li H, Wang Y (2015) Ni-promoted synthesis of graphitic carbon nanotubes from in situ produced graphitic carbon for dehydrogenation of ethylbenzene. Chem Commun 51:12859–12862
Yuan H, Yan F, Li C, Zhu C, Zhang X, Chen Y (2018) Nickel nanoparticle encapsulated in few-layer nitrogen-doped graphene supported by nitrogen-doped graphite sheets as a high-performance electromagnetic wave absorbing material. ACS Appl Mater Interfaces 10:1399–1407
He L, Wang Y, Gao H, Liu Z, Xie Z (2021) Nitrogen doped carbon for Pd-catalyzed hydropurification of crude terephthalic acid: roles of nitrogen species. RSC Adv 11:33646–33652
Hollinger G, Skheyta-Kabbani R, Gendry M (1994) Oxides on GaAs and InAs surfaces: an x-ray-photoelectron-spectroscopy study of reference compounds and thin oxide layers. Phys Rev B 49:11159–11167
Ai L, Shi Y, Han Y, Chen J (2021) In situ aqueous phase hydrodeoxygenation of methyl palmitate to hydrocarbons on Ni catalyst derived from the reduction of LaNiO3 perovskite. Reac Kinet Mech Cat 133:209–227
Yu X, Chen J, Ren T (2014) Promotional effect of Fe on performance of Ni/SiO2 for deoxygenation of methyl laurate as a model compound to hydrocarbons. RSC Adv 4:46427–46436
Mondal S, Singuru R, Chandra Shit S, Hayashi T, Irle S, Hijikata Y, Mondal J, Bhaumik A (2017) Ruthenium nanoparticle-decorated porous organic network for direct hydrodeoxygenation of long-chain fatty acids to alkanes. ACS Sustain Chem Eng 6:1610–1619
Acknowledgements
The authors gratefully acknowledge support from the National Natural Science Foundation of China (No. 21576193 and 21176177).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gu, X., Shi, H., Wang, D. et al. Glucose-derived carbon-coated Ni–In intermetallic compounds for in situ aqueous phase selective hydrogenation of methyl palmitate to hexadecanol. Reac Kinet Mech Cat 135, 1621–1634 (2022). https://doi.org/10.1007/s11144-022-02221-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-022-02221-x