
Abstract
This article presents new kinetic studies of the disproportionation of I(+ 3) and of its oxidation by H2O2. It also provides an update of the previously proposed model for reactions of iodine compounds with oxidation numbers from − 1 to + 5 with each other and with H2O2. This model explains the kinetics of several reactions, including the oxidation of iodine by H2O2. We show that the reduction of HOI by H2O2 results from \({\text{HOI }} + {\text{ H}}_{{2}} {\text{O}}_{{2}} \to {\text{HOOI }} + {\text{ H}}_{{2}} {\text{O}}\) followed by the reversible reaction \({\text{HOOI}} \rightleftharpoons {\text{I}}^{ - } + {\text{ H}}^{ + } + {\text{ O}}_{{2}}\). An analysis of previous measurements of the kinetic constant k(HOI + H2O2) explains the large differences between the values proposed in the literature and gives k(HOI + H2O2) = 6 M−1 s−1. The reversibility of the reaction \({\text{HOOI}} \rightleftharpoons {\text{I}}^{ - } + {\text{ H}}^{ + } + {\text{ O}}_{{2}}\) suggests a new explanation for the effect of oxygen on the Bray–Liebhafsky reaction. H2O2 would oxidize HOOI by a radical mechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Studies of systems involving iodine reactions, such as periodic and chaotic reactions [1,2,3,4], the consequences of a nuclear accident [5,6,7] and the marine boundary layer chemistry [8,9,10] require the understanding of simpler sub-systems. The models of these complicated systems involve a large number of reactions with unknown or poorly known rate constants and the aim of our work is to reduce their number. We present new measurements of the kinetics of I(+ 3) reactions in acidic solutions as well as a new analysis of previous results for inorganic reactions of iodine compounds between them and with H2O2. Adopting the terminology Up and Down reactions proposed by Liebhafsky, we continue to explore the iodine house [11] with 6 floors from I(− 1) to I(+ 5) and an elevator, H2O2.
Table 1 is an update of the model proposed in 2010 [12] taking into account our new study of the kinetics of I(+ 3) reactions. The observed effect of [H+] has consequences for other reactions. A model of iodine reactions must also explain the very unusual kinetics of the oxidation of I2 by H2O2 (reaction O) discussed below. Some kinetic constants were known. Others were adjusted in 2010 to explain this kinetics and we have updated them to take into account the results presented in this work.
The former explanation of the effects of oxygen on the Bray–Liebhafsky (BL) oscillations is also modified. Sharma and Noyes [13] had studied these effects and had even proposed, without success, that they could explain the oscillations. This explanation has been discarded but the importance of oxygen reactions has been confirmed [14,15,16]. To take these effects into account, the 2010 model included the global reaction \({\text{I}}^{ - } + {\text{ H}}^{ + } + {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2}{\text{O}}_{{2}} \to {\text{HOI}}\) with an empirical rate law. This reaction seems simple but is actually a complicated light-catalyzed radical reaction that deserves further experimental study. In the meantime, other studies suggest that it involves the HOOI intermediate proposed by Ball and Hnatiw [17] to explain the kinetics of the reduction of I(+ 1) by H2O2 in buffered solutions. The existence of this intermediate has been confirmed later [18, 19]. HOOI is also an important intermediate for the explanation of the reduction of iodate by H2O2 [20]. The classical rate law of this reduction is no longer valid when the concentration of H2O2 is larger than about 0.2–0.3 M and a radical reaction path appears. The reaction \({\text{HOI }} + {\text{ H}}_{{2}} {\text{O}}_{{2}} \to {\text{I}}^{ - } + {\text{ H}}^{ + } + {\text{ O}}_{{2}} + {\text{ H}}_{{2}} {\text{O}}\) must be split into \({\text{HOI }} + {\text{ H}}_{{2}} {\text{O}}_{{2}} \rightleftharpoons {\text{HOOI }} + {\text{ H}}_{{2}} {\text{O }}\left( {{\text{R5}}} \right)\) and \({\text{HOOI}} \rightleftharpoons {\text{I}}^{ - } + {\text{ H}}^{ + } + {\text{ O}}_{{2}} \left( {{\text{R6}}} \right)\) and a radical reaction path would be initiated by a reaction between HOOI and H2O2 [21, 22]. Reaction R6 could also explain the observation of E. Szabo and P. Ševčik [23]. They measured accurately the rate of O2(g) production during the BL reaction and identified two precursors. One is O2(aq). The concentration of the other precursor increases with [H2O2] and HOOI explains this observation. All these works suggest our new explanation of the effect of oxygen on the BL reaction: R6 is highly reversible and followed by reaction R12. We conclude that HOOI is a member of the iodine house with consequences in many systems.
Some rate constants in Table 1 are calculated using the relation Keq = k+/k- between an equilibrium constant and the kinetic constants in the forward and backward directions. It is sometimes justified by the principle of microscopic reversibility. However, many reactions of Table 1 are not elementary and the use of this principle may be open to criticism. We have offered another proof introducing the concept of quasi-elementary reaction [24]. It is defined as a reaction with a well-defined stoichiometry and rate orders corresponding to this stoichiometry. Take reaction R4 as an example. It is the sum of reactions (4a) and (4b) [25].
The quasi-stationarity of [I2OH−] gives r4a = rab and
The rate law in Table 1 is observed when \({\text{k}}_{{{\text{4b}}}} \left[ {{\text{H}}^{ + } } \right] \gg {\text{k}}_{{ - {\text{4a}}}}\). At equilibrium k4ak4b [HOI]eq[I−]eq[H+]eq = k-4ak-4b [I2]eq. This kinetic expression must be equivalent to the thermodynamic expression of the equilibrium. It follows that k4ak4b/k−4ak−4b = Keq and, with numbering of Table 1, k4/k−4 = K4. We have applied this principle of equivalence between the kinetic and thermodynamic expressions of the equilibrium to other quasi-elementary reactions in Table 1.
Section “I(+ 3) disproportionation” proposes a new interpretation of our kinetic study of the reaction \({\text{2HOIO}} \to {\text{IO}}_{{3}}^{ - } + {\text{ HOI }} + {\text{ H}}^{ + }\) (R13) published previously [26]. Section “I(+ 3) autocatalytic disproportionation” presents a new kinetic study of the reaction \({\text{HOI }} + {\text{ HOIO}} \to {\text{IO}}_{{3}}^{ - } + {\text{ I}}^{ - } + {\text{ 2H}}^{ + }\) (-R1) and the next section presents a new kinetic study of the oxidation of I(+ 3) by H2O2 (R9). Section “Oxidation of I2 by H2O2 with iodate added initially (Reaction O)” shows that the model in Table 1 with updated values of some kinetic constant explains also our former results. If there is no iodate initially, the I2 + H2O2 reaction begins with a non-reproducible induction period. It is discussed in section “Oxidation of I2 by H2O2 without iodate added initially” on the basis of our recent calculations [27] of the nullclines corresponding to the model in Table 1. Section “I(+ 1) reduction by H2O2” shows that this induction period allows measurements of the kinetic constant of the reaction \({\text{HOI }} + {\text{ H}}_{{2}} {\text{O}}_{{2}} \to {\text{HOOI }} + {\text{ H}}_{{2}} {\text{O}}\) (R5). Appendix (in the Supplementary Information) gives details about the calculations of the kinetic constants given in Table 1.
Experiments and calculations
The I(+ 3) solutions are prepared as explained before [28] by the reaction of weighed amounts of I2 and KIO3 in concentrated H2SO4. They contain about 1 to 2% I(+ 1). The calculation of the initial composition of the experimental solutions takes this into account. The other reagents are of the best purity commercially available and are used without further purification. The 18 MΩ water is supplied by a Barnstead Micropore ST model. The initial concentrations [H+] are calculated using the Pitzer model of the H2SO4 solutions. The absorbance measurements are made with an Agilent Cary 60 scanning spectrophotometer. The addition of the samples of I(+ 3) in H2SO4 to the aqueous phase (of composition depending on the kind of experiment) being exothermic, the aqueous phase is cooled before mixing so that the temperature after mixing is close to 25 °C. The spectrophotometer cell is thermostatically controlled at 25 °C and contains a small stirrer.
The sample of I(+ 3) (20 to 30 mg) must be added quickly to the aqueous phase (3 to 5 cm3). Mixing in the reverse order causes a local overheating giving some fast initial reactions and wrong results. Two mixing methods were used. The “syringe” method consists of placing the aqueous phase in a cell with a small circular opening, injecting the sample of I(+ 3) with a fast mixing syringe, closing the cell with a Teflon stopper, inverting the cell to mix well and place it in the spectrophotometer. This cell is tight and allows absorbances measurements up to large conversions, but the first measurements can only be made about 15–20 s after the injection of the I(+ 3) sample. The “paddle” method consists of placing the cooled aqueous phase in a cell with a wide opening (1 cm2) and weighing the I(+ 3) sample on a small paddle. It is introduced quickly into the cell, shaken briefly to mix well, and a Teflon cover is placed over the cell. The cell also contains a small stirrer. This method allows absorbances measurements after less than 5 s and gives accurate values at small conversions, but the cell is not tight and the absorbances are sometimes too small at large conversion. Both method were used for the measurements of the rate of disproportionation of I(+ 3) in the presence of crotonic acid, which is relatively slow, and for the measurements of the rate of autocatalytic disproportionation which begins with an induction period. They gave similar results. Oxidation of I(+ 3) by H2O2 is faster and we used the “paddle” method.
The spectrophotometer results are transferred to a computer and the analytical calculations are done with Excel. The differential equations associated with the model are integrated using the ode15s function of Matlab specially adapted to stiff differential equations. The main program calls the fminsearch function which determines the kinetic constants minimizing the sum of squares of the difference between the absorbances measured and calculated by ode15s. Calculations usually take less than 2 min and the relative error on the iodine mass balance is less than 10–13.
I(+ 3) disproportionation
The I(+ 3) disproportionation is autocatalytic as explained in the next section. To study the kinetics of reaction 2HOIO → IO3− + HOI + H+ (R13), we carried out experiments with crotonic acid (CA) [26]. It reacts very quickly with HOI according to reaction (1) to form an iodohydrin HOICA.
Reaction (R13) becomes the rate determining step giving the kinetic law (2) when the concentration of CA is large enough. We used [CA]0 = 5 × 10–3 to 1.2 × 10–2 M and [I(+ 3)]0 = 5 × 10–4 to 2 × 10–3 M. The kinetic constant of reaction (1), kCA = 4730 M−1 s−1, has been measured previously [29].
[I(+ 3)] represents the total concentration [I(+ 3)] = [HOIO] + [H2OIO+] = (1 + K19 [H+])[HOIO] so that k13 = (1 + K19 [H+])2 kexp, disp. The integration of the kinetic law (2) gives the equation below which makes it possible to calculate kexp, disp by making as only assumption that the absorbance measured at 275 nm varies linearly with the extent of the reaction [26]. However, it is necessary to estimate the value of A∞ by extrapolation of the A values at long term.
We also analyzed the experimental results using Matlab to integrate the differential equations associated with the model in Table 1 without the H2O2 reactions but with reaction (1) added. The fminsearch function adjusts the kinetic constant k13 to minimize the sum of squares \(\sum {\left( {{\text{A}}_{{{\text{calc}}}} - {\text{ A}}_{{{\text{exp}}}} } \right)^{{2}} }\) where Acalc = εI(+3) [I(+ 3)] + εI(+5) [I(+ 5)] + εCA [CA] + εHOICA [HOICA]. New measurements gave \(\varepsilon_{{{\text{I}}\left( { + {3}} \right)}} = {121},\quad \varepsilon_{{{\text{I}}\left( { + {5}} \right)}} \, = \,{11}.{4},\quad \varepsilon_{{{\text{CA}}}} \, = \,{6}.{4}0{\text{ and}}\,\varepsilon_{{{\text{HOICA}}}} \, = \,{399}\,{\text{at}}\,{275}\,{\text{nm}}\). The agreement between the rate constants calculated with the order two rate law or with Matlab is excellent.
The rate constant kexp,disp increases very quickly with decreasing acidity [26]. We had explained this effect by reaction (R20) followed by reaction (R21).
The acidity constant of HOIO is unknown but its order of magnitude is K20 = 10–5 to 10–6 M [30] so that [OIO−] = K20 [HOIO]/[H+] is much smaller than [HOIO] under our experimental conditions.
and [HOIO] = [I(+ 3)]/(1 + K19 [H+]) give
The large effect of [H+] on kexp,disp could be explained by the factor (1 + K19 [H+])2 if K19 ~ 3 M−1. However, the study of the autocatalytic disproportionation rate of I(+ 3) in the next section shows that K19 is much smaller so that this explanation must be discarded and we propose a new explanation of our 2013's results. We assume [H2OIO+] ≪ [HOIO] under our experimental conditions and explain the effect of [H+] on kexp,disp by the formation of the intermediate I2O4H−, similar to the oxide I2O3 known in the gas phase. Its formation will be supported by our new study of the I(+ 3) oxidation by H2O2 discussed below.
Reaction R13 is obtained by combining these reactions with the acid–base quasi-equilibria,. Assuming \([{\text{I}}_{{2}} {\text{O}}_{{4}} {\text{H}}^{ - } ] \, = {\text{K}}_{{{22}}} \left[ {{\text{HOIO}}} \right]\left[ {{\text{OIO}}^{ - } } \right]\), we obtain \({\text{r}}_{{{13}}} = \left( {{\text{k}}_{{{23}}} + {\text{k}}_{{{24}}} \left[ {{\text{OH}}^{ - } } \right]} \right){\text{ K}}_{{{22}}} \left[ {{\text{HOIO}}} \right]\left[ {{\text{OIO}}^{ - } } \right]\) or \({\text{r}}_{{{13}}} = \left( {{{{{k^{\prime}}}_{{{13}}} } \mathord{\left/ {\vphantom {{{{k^{\prime}}}_{{{13}}} } {\left[ {{\text{H}}^{ + } } \right]}}} \right. \kern-\nulldelimiterspace} {\left[ {{\text{H}}^{ + } } \right]}} + {{{{k^{\prime\prime}}}_{{{13}}} } \mathord{\left/ {\vphantom {{{{k^{\prime\prime}}}_{{{13}}} } {\left[ {{\text{H}}^{ + } } \right]^{{2}} }}} \right. \kern-\nulldelimiterspace} {\left[ {{\text{H}}^{ + } } \right]^{{2}} }}} \right) \, \left[ {{\text{HOIO}}} \right]^{{2}}\) where \({{k^{\prime}}}_{{{13}}} = {\text{k}}_{{{23}}} {\text{K}}_{{{2}0}} {\text{K}}_{{{22}}}\) and \({{k^{\prime\prime}}}_{{{13}}} = {\text{k}}_{{{24}}} {\text{K}}_{{\text{w}}} {\text{K}}_{{{2}0}} {\text{K}}_{{{22}}}\). The experiments give kexp,disp = r13/[HOIO]2 and the plot of kexp,disp [H+] as a function of 1/[H+] in Fig. 1 shows that this equation explains our former results. The intercept and slope give the values of \({{k^{\prime}}}_{{{13}}} \,{\text{and}}\,{{k^{\prime\prime}}}_{{{13}}}\).

I(+3) autocatalytic disproportionation
Effect of the acidity on the rate constant of reaction (R13). Experimental values (×) and linear regression line (—) giving \({{k^{\prime}}}_{{{13}}} \, = \,0.0{45}\,{\text{s}}^{{ - {1}}} \,{\text{and}}\,{{k^{\prime\prime}}}_{{{13}}} \, = \,0.0{65}\,{\text{M}}\,{\text{s}}^{{ - {1}}}\) .
I(+ 3) autocatalytic disproportionation
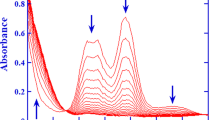
The kinetic of the I(+ 3) disproportionation without added CA is not simple. At the beginning, the main reaction is (R-1) followed by the fast reactions (R2) and (R3) giving the autocatalytic reaction \({\text{2HOIO}} + {\text{HOI}} \to {\text{IO}}_{{3}}^{ - } + {\text{H}}^{ + } + {\text{2HOI}}\). The induction period depends on the concentration of I(+ 1) in the preparation of I(+ 3) and overlaps the period required for the solution to be well mixed so that there are no mixing problems. The concentration [HOI] increases and can become as large as 20% of [I(+ 3)]0 at high acidities. Then, it goes through a maximum shown in Fig. 2 and the main reaction becomes \({\text{HOIO}} + {\text{2HOI}} \to {\text{IO}}_{{3}}^{ - } + {\text{H}}^{ + } + {\text{ I}}_{{2}} + {\text{ H}}_{{2}} {\text{O}}\). When [HOIO] becomes small, some HOI remains and the direction of reactions (R2) and (R3) is reversed. [HOI] decreases very slowly according to \({\text{5HOI}} \to {\text{IO}}_{{3}}^{ - } + {\text{ H}}^{ + } + {\text{ 2I}}_{{2}} + {\text{ 2H}}_{{2}} {\text{O}}\). The overall stoichiometry is \({\text{5I}}\left( { + {3}} \right) \to {\text{3IO}}_{{3}}^{ - } + {\text{ 3H}}^{ + } + {\text{ I}}_{{2}} + {\text{ H}}_{{2}} {\text{O}}\) but the experimental values of [I2] at the end of the experiments is always lower than [I(+ 3)]0/5 because the disproportionation of I(+ 1) is very slow under our experimental conditions. Fig. 2 gives examples of evolutions over time.

Left hand scale: Experimental (o) and calculated (—) absorbances at 462 nm at low (a) and high (b) acidities. [H2SO4] = 0.068 M and [I(+ 3)]0 = 4.34 × 10–4 M (a). [H2SO4] = 0.76 M and [I(+ 3)]0 = 5.41 × 10–4 M (—). Right hand scale: Calculated I(+ 1) concentrations (- - -)
Matlab simulates the experimental curves and the function fminsearch adjusts k−1 and the initial value of [I(+ 1)]. The electronic supplement gives the results. The effect on k−1 of likely modifications of the kinetic constants, including k13, is lower than the experimental inaccuracies. On the other hand, k-1 depends on the values of K18 and K19. The concentrations known experimentally or calculated by mass balances are the total concentrations \(\left[ {{\text{I}}\left( { + {1}} \right)} \right] = \left( {{1} + {\text{K}}_{{{18}}} \left[ {{\text{H}}^{ + } } \right]} \right) \, \left[ {{\text{HOI}}} \right]{\text{ and }}\left[ {{\text{I}}\left( { + {3}} \right)} \right] = \left( {{1} + {\text{ K}}_{{{19}}} \left[ {{\text{H}}^{ + } } \right]} \right) \, \left[ {{\text{HOIO}}} \right]\). Thus, the individual concentrations [HOI] and [HOIO] calculated by Matlab depend on K18 and K19 and, therefore, also the value of k-1. To estimate the values of K18 and K19 we apply the principle of equivalence between the kinetic and thermodynamic expressions of an equilibrium. The kinetic of the reaction (R1) in the forward direction is well known [41]: its rate is proportional to [H+]2. Consequently, k-1 must be independent of [H+]. If K19 > 0.1 M−1, the calculated values of k-1 increase with [H+] as shown in Fig. 3 and, moreover, the fit of the experimental curves becomes bad if K19 > 0.5 M−1. We therefore neglect K19. Fig. 3 also shows that k-1 is nearly independent of [H+] if K18 ~ 0.3 M−1. We had estimated previously K18 ~ 0.5 M−1 [25] and these results suggest to decrease slightly this value. These experiments give k-1 = 210 ± 6 M−1 s−1 (t-Student 95% confidence interval).

Values of k-1 adjusted by Matlab if K18 = 0.5 M−1 and K19 = 0 (O), K18 = 0.3 M−1 and K19 = 0.2 M−1 (+), K18 = 0.3 M−1 and K19 = 0 (×)
Oxidation of I(+ 3) by H2O2
When a sample of I(+ 3) is added to an acidic solution of H2O2, the main reaction is the oxidation \({\text{HOIO }} + {\text{ H}}_{{2}} {\text{O}}_{{2}} \to {\text{IO}}_{{3}}^{ - } + {\text{ H}}^{ + } + {\text{ H}}_{{2}} {\text{O}}\) (R9). The amount of iodine produced is negligible showing that the reduction of HOIO by reaction R7 is much slower. The results show that the rate of R9 is proportional to the square of the concentration [HOIO] and inversely proportional to [H+]. The rate constants k9 in Table 2 were calculated for the rate law \({\text{r}}_{{9}} = {\text{ k}}_{{9}} {{\left[ {{\text{HOIO}}} \right]^{{2}} \left[ {{\text{H}}_{{2}} {\text{O}}_{{2}} } \right]} \mathord{\left/ {\vphantom {{\left[ {{\text{HOIO}}} \right]^{{2}} \left[ {{\text{H}}_{{2}} {\text{O}}_{{2}} } \right]} {\left[ {{\text{H}}^{ + } } \right]}}} \right. \kern-\nulldelimiterspace} {\left[ {{\text{H}}^{ + } } \right]}}\) using the Matlab function fminsearch minimizing the sum of the squares of the deviations between the calculated and measured absorbances of HOIO at 275 nm over time. The electronic supplement gives examples. The average value is k9 = (1.7 ± 0.2) × 105 M−1 s−1 (t-Student 95% confidence interval). New measurements of the molar absorption coefficient of HOIO at 275 nm gave ε(HOIO) = 121 ± 2. Other values used in this work are ε(H2O2) = 5.7, ε(IO3−) = 11.4, ε(HOI) = 107 and ε(I2) = 121 at 275 nm.
We had shown that H2O2 reduces the HOI monomer and oxidizes its dimer I2O. The rate law of the HOIO disproportionation with an excess of CA discussed above and the kinetic law of HOIO oxidation by H2O2 suggest that the HOIO reactions are similar to those of HOI. HOIO also forms a dimer and H2O2 reduces its monomer and oxidizes its dimer. Also, the effect of [H+] on these reactions suggests that this dimer is I2O4H− introduced above that reacts according to (R24).
The oxidation R25 is much faster than the disproportionation of HOIO from the smallest values of [H2O2] and the quasi-stationarity of [I2O4H−] gives (k-22 + k25 [H2O2]) [I2O4H−] = k22 [HOIO][OIO−] and the rate law
This expression explains the rate law of R9 in Table 1 with k9 = k24k22K20/k-22 and α9 = k24/k−22. The term in α9 [H2O2] has no effect on the results in Table 2 because the concentration of H2O2 was small but will be important to simulate the H2O2 effect on the rate of reaction O (Fig. 4).**

Experimental rate constants of the I2 oxidation by H2O2 at 25 °C with about 0.01 mol/l iodate added initially. [HClO4] = 0.04 (open diamond), 0.10 (×), 0.20 (O), 0.40 M (+) and values calculated with the model in Table 1 (lines)
Oxidation of I2 by H2O2 with iodate added initially (Reaction O)
The oxidation of iodine by H2O2 without iodate added initially begins with a non-reproducible period of induction discussed below. It is suppressed and a fast oxidation of I2 is observed if iodate is added initially. We had carried out more than a thousand experiments in a wide range of concentrations and temperatures and published the main results previously [12, 31]. When the initial iodate concentration is about 0.005 to 0.1 M, the rates are complicated functions of acidity, [H2O2] and [I2] but are independent of the iodate concentration. To analyze these rates, it is convenient to define the function kexp = − d(ln[I2])/dt although the reaction is not exactly of order 1 with respect to [I2]. Fig. 5 shows values of kexp at 25 °C if [I2] = 4 × 10–4 M. The values of kexp do not directly give values of kinetic constants, only relations between them. A modification of one constant requires readjusting the kinetic constants of the reactions R3, R7 and R10. The previously published model [12, 31] explained our results but we had to modify the values of some kinetic constants to take into account our new results. The proposed model with updated rate constants explains not only our new experimental results but also the previous ones. Our model centered on the reactions of HOI and I2O is robust in the sense that slight modifications of it can be compensated by adjustments of some kinetic constants. Fig. 4 shows that the rate constants in Table 1 give excellent simulations of the kexp values. Note the decrease in kexp when the concentration of the reagent H2O2 increases.
Oxidation of I2 by H2O2 without iodate added initially
H2O2 does not react directly with I2. An acidic solution containing only I2 and H2O2 gives a quasi-steady state corresponding to the slow decomposition \({\text{H}}_{{2}} {\text{O}}_{{2}} \to {\text{H}}_{{2}} {\text{O }} + {\text{ O}}_{{2}}\). To obtain the oxidation of I2, the concentration in iodide must be decreased. This can be obtained by addition of iodate as in the previous section, by precipitation of AgI(s) or by formation of HgI+ but can also appear spontaneously after a more or less long time. Fig. 6 of reference [31] gave an example and the electronic supplement gives another one. Since the only concentration that changes significantly during the quasi-steady period is that of produced oxygen, we explained the transition to reaction O by the oxidation of iodide by oxygen. We had shown that the reaction \({\text{I}}^{ - } + {\text{ H}}^{ + } + {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2}{\text{O}}_{{2}} \to {\text{HOI}}\) with an empirical rate law allows to explain the experimental observations [12, 31]. The direct oxidation of iodide is much too slow but a radical pathway can be fast. Studying the nullclines calculated with the model in Table 1 [27] we concluded that the transition is explained by a saddle-node bifurcation occurring when the concentration of oxygen in solution reaches a critical value. Anything that promotes the transfer of oxygen to the gas phase delays the transition. This explains that it occurs later if the contact surface between the solution and the gas phase is increased and if the solution is stirred [32,33,34].
An essential feature of the proposed model is the competition between the Down reactions R5 followed by R6 and the Up reaction R10. The concentration of the intermediate I2O is very small in aqueous solutions, is quasi-stationary and approximately proportional to [HOI]2. The ratio between the rates r10 = k10 [I2O] [H2O2] and r5 = k5 [HOI] [H2O2] is therefore approximately proportional to [HOI]. If [HOI] is small, H2O2 mainly reacts as a reducing agent. If [HOI] is large, H2O2 mainly reacts as an oxidant. The reaction R4 being a quasi-equilibrium, [I−] must be small enough for H2O2 to act mainly as an oxidant giving reaction O.
It is difficult to quantify the effect of oxygen. The transfer reaction R14 to the gas phase is an over-simplification and the solution can become highly supersaturated in oxygen. Complicated phenomena of transfer at the interface and germination-growth of bubbles must be taken into account. Moreover, the rate of oxidation of iodide is photocatalyzed and depends on complicated radical reactions [36]. Finally, it can be noted that the reaction \({\text{2H}}_{{2}} {\text{O}}_{{2}} \to {\text{2H}}_{{2}} {\text{O }} + {}^{1}{\text{O}}_{{2}}\) is thermodynamically possible [37]. We could produce singlet oxygen which would react much faster with iodide. These complications explain that the induction period of the oxidation of I2 by H2O2 without iodate added initially seems stochastic [34, 35]. A phenomenon seems stochastic when it depends on parameters that we ignore or do not control. A qualitative explanation of the transition between the quasi-steady state and the reaction O when the oxygen concentration reaches a critical value is already a success of the proposed model.
I(+ 1) reduction by H2O2 (reactions R5 and R6)
Furrow [38] measured the absorbances at 354 and 460 nm of solutions of I2 and H2O2 without iodate during the quasi-stationary period. These absorbances allow to calculate the concentrations of I2 and I3−, then that of I− and finally that of HOI corresponding to the quasi-equilibrium R4. The sum of the reactions R5 + R6 + R11 gives the decomposition \({\text{H}}_{{2}} {\text{O}}_{{2}} \to {\text{H}}_{{2}} {\text{O }} + {\text{ O}}_{{2}}\) and Furrow assumed that their rates are equal during this period giving \({\text{k}}_{{5}} \left[ {{\text{HOI}}} \right] = \left( {{{k^{\prime}}}_{{{11}}} + {{ k^{\prime\prime}}}_{{{11}}} \left[ {{\text{H}}^{ + } } \right]} \right) \, \left[ {{\text{I}}^{ - } } \right]\). The values of \({{k^{\prime}}}_{{{11}}} \,{\text{and}}\,{{k^{\prime\prime}}}_{{{11}}}\) being known, k5 can be calculated. Furrow obtained k5 = 3 M−1 s−1 but its values were widely dispersed. We recalculated his results using updated values of the parameters (ε(I2, 354) = 17; ε(I3−, 354) = 26,250; ε(H2O2, 354); K4 = 1011 M−2 when I = 0.10 M) and plotted k11 [I−]/[HOI], where \({\text{k}}_{{{11}}} = {{k^{\prime}}}_{{{11}}} + {{ k^{\prime\prime}}}_{{{11}}} \left[ {{\text{H}}^{ + } } \right]\), as a function of [HOI]. If the above equality was verified, the values of k11 [I−]/[HOI] would be independent of [HOI]. Fig. 5 shows on the contrary a decrease when [HOI] increases. This explains why the values calculated ignoring this dependence were so dispersed.

Analysis of Furrow's results [38] explained in the text
The model in Table 1 explains this effect of [HOI]. Numerical simulations show that reactions R2 and R10 cannot be neglected. Their sum gives the same global reaction as R11, \({\text{I}}^{ - } + {\text{ H}}^{ + } + {\text{ H}}_{{2}} {\text{O}}_{{2}} \to {\text{HOI }} + {\text{ H}}_{{2}} {\text{O}}\). If R3 was an equilibrium and if we could neglect the effect of R12, we would have k5 [HOI][H2O2] = k11 [I−] [H2O2] + k10 [I2O] [H2O2] giving k11 [I−]/[HOI] = k5 – k10 k−3/k3 [HOI]. This approximate expression explains the effect of [HOI] seen in Fig. 5 and suggests k5 ~ 5 M−1 s−1 or a little larger by extrapolation at \(\left[ {{\text{HOI}}} \right] \to 0\). The numerical simulations of these experiments taking the kexp values for reaction O into account give k5 = 6 M−1 s−1. The electronic supplement shows a comparison between the experimental values and the values calculated with the model in Table 1.
Liebhafsky [39] measured the rate of oxygen production due to the reaction \({\text{HOI }} + {\text{ H}}_{{2}} {\text{O}}_{{2}} \to {\text{I}}^{ - } + {\text{ H}}^{ + } + {\text{ O}}_{{2}} + {\text{ H}}_{{2}} {\text{O}}\) (R5 + R6) in the presence of solid iodine and an excess of Tl+ ions. The concentration of iodine in solution was equal to its solubility, the concentration of iodide was fixed by equilibrium \({\text{Tl}}^{ + } + {\text{ I}}^{ - } \rightleftharpoons {\text{TlI}}\left( {\text{s}} \right)\) and that of HOI by reaction (R4). He deduced the values of k5 = (d[O2]/dt)/([HOI] [H2O2] in Fig. 6 giving on average k5 = 37 M−1 s−1. We recalculated his results [12] using more recent values of the equilibrium constants, but keeping Liebhafsky calculations assumptions, and proposed k5 = 23 M−1 s−1, a value significantly greater than k5 = 6 M−1 s−1 adopted above.
The effect of [H+] revealed by Fig. 6 suggests that the dispersion of the k5 values obtained by Liebhafsky is not due to experimental inaccuracies and that the system is more complicated than he thought. This is confirmed by numerical simulations using the model in Table 1 supplemented by reaction (3) and with [I2] fixed by the solubility of iodine. Under these conditions, oxygen is produced not only by reaction R5 followed by R6 but also by reactions R7 and R12. d[O2]/dt > r5 explains that the values of k5 obtained by Liebhafsky are too large. The model also explains a surprising observation reported by Liebhafsky. If he added only Tl+, the iodide concentration became very small, the rate of oxygen production much too high, and iodate was produced [ref. 39, bottom of p. 3504]. To avoid this, he had to add a large amount of solid TlI(s). If k12 ~ 107 M−1 s−1, order of magnitude estimated in the appendix, an important reaction path is R2 + R3 + R4 + R5 + R12 giving overall \({\text{2I}}^{ - } + {\text{ 2H}}^{ + } + {\text{ 3H}}_{{2}} {\text{O}}_{{2}} \to {\text{I}}_{{2}} + {\text{ O}}_{{2}} + {\text{ 4H}}_{{2}} {\text{O}}\) and the observed decrease in [I−]. The addition of solid TlI(s) was necessary to avoid this decrease and to obtain the overall reaction \({\text{2TlI}}\left( {\text{s}} \right) + {\text{2H}}^{ + } + {\text{3H}}_{{2}} {\text{O}}_{{2}} \to {\text{2Tl}}^{ + } + {\text{ I}}_{{2}} + {\text{ O}}_{{2}} + {\text{ 4H}}_{{2}} {\text{O}}\). This reaction greatly contributes to the production of oxygen and varies [H+]. The model explains Liebhafsky observations, shows that the system he studied gives too large values of k5 and is too complicated to allow an exact measurement.

Shin, Lee and von Gunten [40] studied the reaction between H2O2 and I2 in buffered solutions between pH 4 and pH 12. They confirmed that this reaction is catalyzed by the buffers. The large differences between the values of the kinetic constants published in the literature can, in part, be explained by the effect of the nature and the concentration of the buffers. These authors obtained k5 = 29 ± 5 M−1 s−1 but this value was obtained in acetic buffers and the value in unbuffered solutions is certainly smaller. In addition, the extrapolation to pH 1 of results obtained above pH 4 is very approximate. The value k5 = 6 M−1 s−1 that we propose is therefore compatible with the results of Shin, Lee and von Gunten.
Conclusion
The model in Table 1 explains the kinetics of the reactions in acidic solutions of iodine compounds at oxidation states from − 1 to + 5 with each other and with H2O2. The new experiments confirm the existence of the HOOI intermediate compound proposed previously [17,18,19,20,21,22]. The effect of acidity on the disproportionation of HOIO and on its oxidation by H2O2 shows that OIO− is much more reactive than HOIO and suggests that the mechanism of these reactions involves an intermediate compound noted I2O4H− (reaction R22). This model, with updated kinetic constants, also quantitatively explains the kinetics of many reactions previously studied under very different conditions and especially the complicated kinetics of the oxidation of I2 by H2O2 with or without iodate added initially. This model becomes more and more likely that it allows to explain a greater number of experiments of different types. None of these results suggest radical reactions in the dark, except in the mechanism of the oxidation of iodide by oxygen. Light, including that of a spectrophotometer, can initiate radical reactions briefly discussed in the appendix (in the Supplementary Information) which deserve further work.
References
Comprehensive Chemical Kinetics 2004, Volume 40, Chapter 14, Instability, periodic reactions, and chaos.
Schuster HG, Just W (2005) Deterministic chaos—an introduction, 4th edn. Wiley, Weinheim
Čupić ŽD, Taylor AF, Horváth D, Orlik M, Epstein IR (2020) Advances in oscillating reactions. Front Chem 9:292
Ivanović-Šasić AZ, Marković VM, Anić SR, Lj Z, Kolar-Anić and Ž. D. Čupić, (2011) Structures of chaos in open reaction systems. Phys Chem Chem Phys 13:20162–20171
Bosland L, Cantrel L, Girault N, Clement B (2010) Modeling of iodine Radiochemistry in the ASTEC severe accident code: description and application to FPT-2 Phebus test. Nuclear Technol 171:88–107
Fortina C, Fèvre-Nolleta V, Cousin F, Lebègue P, Louisa F (2019) Box modelling of gas-phase atmospheric iodine chemical reactivity in case of a nuclear accident. Atmos Environ 214:116838
Saha S, Roy S, Mathi P, Mondal JA (2020) Adsorption of iodine species (I3−, I−, and IO3−) at the nuclear paint monolayer−water interface and its relevance to a nuclear accident scenario. J Phys Chem A 124:6726–6734
Mahajan AS, Shaw M, Oetjen H, Hornsby KE, Carpenter LJ, Kaleschke L, Tian-Kunze X, Lee JD, Moller SJ, Edwards P, Commane R, Ingham T, Heard DE, Plane JMC (2010) Evidence of reactive iodine chemistry in the Arctic boundary layer. J Geophys Res 115:D20303
Badia A, Reeves CE, Baker AR, Saiz-Lopez A, Volkamer R, Koenig TK, Apel EC, Hornbrook RS, Carpenter LJ, Andrews SJ, Sherwen T, von Glasow R (2019) Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem. Atmos Chem Phys 19:3161–3189
Inamdar S, Tinel L, Chance R, Carpenter LJ, Sabu P, Chacko R, Tripathy SC, Kerkar AU, Sinha AK, Venkateswaran Bhaskar P, Sarkar A, Roy R, Sherwen T, Cuevas C, Saiz-Lopez A, Ram K, Mahajan AS (2020) Estimation of reactive inorganic iodine fluxes in the Indian and Southern Ocean marine boundary layer. Atmos Chem Phys 20:12093–12114
Schmitz G, Furrow SD (2018) Kinetics of reactions of iodine inorganic compounds in acidic solutions. In: Physical Chemistry 2018, Proceedings of the 14th Int. Conf. Fundam. Appl. Aspects Phys. Chem. Belgrade, Sept 24–28, Society of Physical Chemists of Serbia pp 271–278
Schmitz G (2010) Iodine oxidation by hydrogen peroxide in acidic solutions, Bray–Liebhafsky reaction and other related reactions. Phys Chem Chem Phys 12:6605–6615
Sharma KR, Noyes RM (1976) Oscillations in chemical systems. 13. A detailed molecular mechanism for the Bray–Liebhafsky reaction of iodate and hydrogen peroxide. J Am Chem Soc 98(15):4345–4361
Schmitz G (1999) Effects of oxygen on the Bray–Liebhafsky reaction. Phys Chem Chem Phys 1:4605
Ševčik P, Kissimonová K, Adamčikova L (2000) Oxygen production in the oscillatory Bray–Liebhafsky reaction. J Phys Chem A 104:3958–3963
Ševčik P, Adamčikova L (1998) Effect of a gas bubbling and stirring on the oscillating Bray–Liebhafsky reaction. J Phys Chem A 102:1288–1291
Ball JM, Hnatiw JB (2001) The reduction of l2 by H2O2 in aqueous solution. Can J Chem 79:304–311
Schmitz G (2008) Buffers catalysis of the iodine(+1) reduction by hydrogen peroxide In Physical Chemistry 2008, Proceedings of the 9th Int. Conf. Fundam. Appl. Aspects Phys. Chem. Society of Physical Chemists of Serbia, Belgrade pp. 219–224
Schmitz G (2009) Iodine(+1) reduction by hydrogen peroxide, Russian. J Phys Chem 83(9):1447
Schmitz G, Furrow SD (2012) Kinetics of the iodate reduction by hydrogen peroxide and relation with the Briggs–Rauscher and Bray–Liebhafsky oscillating reactions. Phys Chem Chem Phys 14:5711–5717
Schmitz G, Furrow SD (2014) Iodine Inorganic Reactions in Acidic Solutions and Oscillating Reactions, Physical Chemistry 2014, Proceedings of the 12th Int. Conf. Fundam. Appl. Aspects Phys. Chem. Society of Physical Chemists of Serbia, Belgrade pp 320–326
Schmitz G, Furrow SD (2016) Bray–Liebhafsky and non-catalyzed Briggs–Rauscher oscillating reactions. Russ J Phys Chem A 90(2):271–275
Szabo E, Ševčík P (2013) Reexamination of gas production in the Bray−Liebhafsky reaction: what happened to O2 pulses? J Phys Chem A 117:10604–10614
Schmitz G, Lente G (2020) Fundamental concepts in chemical kinetics. ChemTexts 6(1):1
Schmitz G (2004) Inorganic reactions of iodine(+1) in acidic solutions. Int J Chem Kinet 36:480
Schmitz G, Furrow SD (2013) Kinetics of iodous acid disproportionation. Int J Chem Kinet 48(8):525–530
Schmitz G, Nullclines (2021) A Simple Explanation of Complicated Phenomena, In Physical Chemistry 2021, Proceedings of the 15th Int. Conf. Fundam. Appl. Aspects Phys. Chem., pp. 222–229, Belgrade, Sept 20–24, Society of Physical Chemists of Serbia, 2021. ISBN 978-86-82475-40-8.
Schmitz G, Noszticzius Z, Hollo G, Wittmann M, Furrow SD (2018) Reactions of iodate with iodine in concentrated sulfuric acid. Formation of I(+3) and I(+1) compounds. Chem Phys Lett 691:44
Furrow SD, Cervellati R, Amadori G (2002) New substrates for the oscillating Briggs–Rauscher reaction. J Phys Chem A 106:5841–5850
Schmitz G (2008) Inorganic reactions of iodine(III) in acidic solutions and free energy of iodous acid formation. Int J Chem Kinet 40:647–652
Schmitz G (2011) Iodine oxidation by hydrogen peroxide and Bray–Liebhafsky oscillating reaction: effect of the temperature. Phys Chem Chem Phys 13:7102–7111
Olexová A, Mrákavová M, Melicherčík M, Treindl L (2006) The autocatalytic oxidation of iodine with hydrogen peroxide in relation to the Bray–Liebhafsky oscillatory reaction. Collect Czech Chem Commun 71:91–106
Olexová A, Mrákavová M, Melicherčík M, Treindl L (2010) Oscillatory system I−, H2O2, HClO4: the modified form of the Bray–Liebhafsky reaction. J Phys Chem A 114:7026–7029
Stanisavljev DR, Stevanović KZ, Bubanja IM (2018) Outsized stochasticity of iodine oxidation with hydrogen peroxide and its implications on the reaction mechanism. Chem Phys Lett 706:120–126
Stevanović KZ, Bubanja IM, Stanisavljev DR (2019) Is iodine oxidation with hydrogen peroxide coupled with nucleation processes? J Phys Chem C 123:16671–16680
Jortner J, Ottolenghi M, Stein G (1962) The effect of oxygen on the photochemistry of the iodide ion in aqueous solutions. J Phys Chem 66:2042–2045
Nardello V, Briviba K, Sies H, Aubry J-M (1998) Identification of the precursor of singlet oxygen (1O2, 1∆g) involved in the disproportionation of hydrogen peroxide catalyzed by calcium hydroxide. Chem Commun 5:599–600
Furrow SD (1987) Reactions of iodine intermediates in iodate-hydrogen peroxide oscillators. J Phys Chem 91:2129–2135
Liebhafsky HA (1932) The catalytic decomposition of hydrogen peroxide by the iodine-iodide couple. III. The rate of oxidation, in acid solution, of hydrogen peroxide by iodine. J Am Chem Soc 54:3499–3508
Shin J, Lee Y, von Gunten U (2020) Kinetics of the reaction between hydrogen peroxide and aqueous iodine: implications for technical and natural aquatic systems. Water Res 179:115852
Schmitz G (1909) Kinetics and mechanism of the iodate-iodide reaction and other related reactions. Phys Chem Chem Phys 1999:1
Furuichi R, Liebhafsky HA (1975) Bull Chem Soc Jpn 48:745
Schmitz G (2000) Kinetics of the Dushman reaction at low I- concentrations. Phys Chem Chem Phys 2:4041
Liebhafsky HA, Mohammad A (1933) The kinetics of the reduction in acidic solution of hydrogen peroxide by iodide ion. J Am Chem Soc 55:3977–3986
Lozar J, Lafage B (1994) Kinetics of the oxidation of iodide ions by oxygenated water using spectrophotometry with a microcomputer. Bulletin de l’Union des Physiciens 88(764):895–902
Stanisavljev DR, Milenković MC, Mojović MD, Popović-Bijelić AD (2011) Oxygen centered radicals in iodine chemical oscillators. J Phys Chem A 115:7955–7958
Stanisavljev DR, Milenković MC, Popović-Bijelić AD, Mojović MD (2013) Radicals in the Bray−Liebhafsky oscillatory reaction. J Phys Chem A 117:3292–3295
Kéki S, Székely G, Beck MT (2003) The effect of light on the Bray–Liebhafsky reaction. J Phys Chem A 107:73–75
Stanbury DM (2018) Comment on the principle of detailed balancing in complex mechanisms and its application to iodate reactions. J Phys Chem A 122:3956–3957
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Schmitz, G.E., Furrow, S.D. Kinetics and mechanism of I(+ 3) reactions and consequences for other iodine reactions. Reac Kinet Mech Cat 135, 1171–1186 (2022). https://doi.org/10.1007/s11144-022-02155-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-022-02155-4