
Abstract
The scale-up procedure of the acetone-furfural aldol condensation was studied on Mg-Al mixed oxide (molar ratio of 3:1). The catalyst’s stability was evaluated in three consecutive condensation steps at 60–70 °C, WHSV = 1.6–2/h-1 and the acetone-furfural molar ratio of 5:1 (mol:mol). After each condensation step, the catalyst’s reactivation was performed at 450 °C, ambient pressure under 50 NL/h nitrogen flow for 4 h. The experiments showed that catalyst deactivation was an effect related to the amount of treated feedstock. Moreover, our results showed the possibility of deactivation compensation by increasing reaction temperature or decreasing feeding rate. From a reaction mechanism point of view, furfural reacted first with acetone to FAc (4-(2-furyl)-3-buten-2-one), a crucial compound for heavier compounds formation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
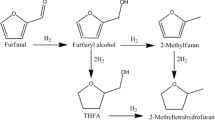
Aldehydes and ketones are components of bio-derived materials typically produced by hydrolysis of waste lignocellulose. These compounds are characterized by a high oxygen content and a high unsaturation degree [1, 2]. With a combination of a relatively low molecular weight and carbon number, it makes them not suitable for direct hydroprocessing. Their hydrodeoxygenation would result in the formation of products with a low boiling point [3]. Additionally, light hydrocarbons do not have a wide utilization in refineries and petrochemistry; thereby, their production from bio-derived compounds is not cost-efficient. One possible way to increase the value of small aldehydes and ketones is to subject them to aldol condensation. It is an initial step that helps convert these compounds to products with higher molecular weight, thereby preparing them for further processing, such as hydrodeoxygenation, that produce the components of transportation fuels [3,4,5,6,7,8,9]. The aldol condensation of acetone and furfural is a well-known complex reaction catalyzed by basic materials (Fig. 1) [10,11,12,13,14]. Mg-Al hydrotalcites (HTCs), also known as layered double hydroxides (LDHs), can be easily synthesized by precipitation at a meagre cost. These materials have high activity in aldol condensation at temperatures below 100 °C in a batch mode in a calcined form as mixed oxides and in a rehydrated form as reconstructed HTCs [15]. The rehydration can be performed by either liquid water or steam or just by a wet gas flow [16]. Thus, low reaction temperatures required for aldol condensation in the presence of low-cost, basic catalysts make this reaction step economically feasible from a heat consumption point of view. The aldol condensation of furfural and acetone provides the synthesis of larger molecules (Fig. 1) with boiling points ranging from petrol to vacuum oil. This hydrocarbon pool might be interesting for refineries as an alternative feedstock for hydrocarbons production for fuel and petrochemical production.

Most of the previously published studies on aldol condensation of furfural and acetone have been usually performed in batch reaction conditions [10,11,12,13,14]. However, the successful development of production performed on a laboratory-scale level later should be up-scaled. Transferring alternative technology through a pilot plant level is crucial for its introduction into existing industries. This stage helps test in advance critical characteristics such as the stability of catalytic systems capable of conducting long operating cycles. Nevertheless, the stage faces some difficulties related to the reaction process. A typical activity decrease in a flow aldol-condensation setup limits reactor efficiency and requires improvement. The long-term aldol condensation needs a robust catalytic system and an optimized reactor design based on the high reaction heat formation. For fuel application of this reaction, it is necessary to work with a stable catalytic system with long operating cycles.
This work continues our previous studies [10,11,12,13], where acetone-furfural aldol condensation tests have been performed in batch conditions. The study is focused on scaling-up the reaction to a pilot plant level with the emphasis of Mg-Al reconstructed HTC performance in terms of its stability, reactivation and composition of the reaction product. Mg-Al HTC with the Mg/Al molar ratio of 3:1 was selected as an optimum ratio for the acetone-furfural aldol condensation reported previously [12].
Experimental
Catalyst synthesis and characterization
The scale-up laboratory preparation of a catalyst was carried out in a CSTR regime using a jacketed reactor Atlas (Syrris) according to a procedure described in [12]. Mg-Al HTC with the Mg/Al molar ratio of 3:1 was prepared by a co-precipitation method at T = 60 °C and pH = 9.5. To keep constant pH, an aqueous solution of Mg(No3)2·6H2O and Al(NO3)3·9H2O (CMg+Al = 1 mol/L) at 30 mL/min flow rate was mixed with an aqueous solution of KOH (2 mol/L) and K2CO3 (0.2 mol/L) at 50 mL/min. All the chemicals were supplied by Lach-Ner s.r.o. in analytical grade purity. After the co-precipitation, the resulting suspension was filtered using a laboratory filter press and washed with demineralized water until pH = 7. The filter cake (3.2 kg) was extruded using a VDE extruder (Caleva) with a shamrock matrix (2.5 mm). The extrudates were dried at 65 °C for 24 h under mild airflow. The amount of the product extrudates after the synthesis and shaping was 730 g. These extrudates were further calcined at 500 °C for 6 h with a heating rate of 1 °C/min under an oxygen atmosphere. The final particles were crushed and sieved to obtain a fraction of 3–5 mm.
After synthesis, a homogeneous sample of the catalytic material was characterized by analytical methods. The synthesized sample’s crystallographic structure was determined using X-ray powder diffraction (XRD) using a D8 Advance ECO (Bruker AXS) with CuKα radiation and λ = 1.5406 Å. Measuring step size was 0.02° using time step 0.5 s. The patterns were collected over 2θ in the range of 5–70°. DIFFRAC.EVA V 4.1.1 was used to process the measured data. The measurement was also conducted to confirm that the catalyst’s synthesis was appropriately provided compared to a reference sample [12]. The textural properties were characterized by the mercury Porosimetry using an AutoPore IV 9510 (Micromeritics Instrument Corporation, Norcross, GA, USA) and nitrogen physisorption using an Autosorb iQ (Quantachrome Instruments, Boynton Beach, FL, USA). The specific surface area was calculated from the adsorption isotherm’s linear plot using the Brunauer, Emmett and Teller (BET) method in the pressure range of 0.05–0.30 P/P0. Finally, composition analysis was determined using inductively coupled plasma spectroscopy (ICP; Agilent 725) and elemental analysis (FLASH 2000 Combustion CHNS/O analyzer). The used catalyst was characterized using the same analytical methods as employed for the fresh catalyst, except in the case of textural properties, in which residues from reaction could damage the instrument.
Experimental setup and test
The scale-up procedure of the catalytic acetone-furfural aldol condensation was carried out in a plug flow fixed bed reactor with an inner diameter of 40 mm and an overall length of 1150 mm. In the reactor axis, 10 mm thermowell with an integrated thermosond was located. The thermosond consisted of 20 thermocouples placed on the length of 570 mm (30 mm step). Reactor heating was assured by three-zone heating with an independent controlling system for each zone. A water cooler was installed at the reactor outlet to avoid volatile products evaporation.
Before a test, the reactor was loaded with the mixture of the synthesized sample (50 g) with SiC (1:1, vol:vol). The SiC particle size fraction was 1–2 mm. A space under and above the catalyst layer was filled with SiC of the same grade. Glass wool plugged the reactor’s inlet and outlet to protect the experimental unit's pumps and valves from catalyst bed dust and SiC particles. The reactor was then closed, flushed with nitrogen, followed by the reactor’s leak tests and separators. Finally, the thermosond was placed to control reaction temperature in a catalytic bed and monitor temperature in the zones above and under the catalyst bed.
The loaded catalyst was activated in situ at ambient pressure under 50 NL/h nitrogen flow at a ramp of 50 °C/h until reaching 450 °C. This temperature was kept for 4 h, followed by the reactor cooling down by increasing nitrogen flow to 1100 NL/h. After stabilizing temperature at 60 °C, the pressure was increased up to 1.0 MPa, and a nitrogen flow was adjusted to 50 NL/h. At these conditions, the catalyst's rehydration with demineralized water was performed for 8 h at the flow rate of 100 g/h.
After the activation, the rehydrated catalyst was tested in three consecutive runs (Table 1). The first run was started by feeding the reaction substrate with a rate of 100 g/h equal to weight hourly space velocity (WHSV) = 2/h. Reactant mixture was prepared by mixing acetone and furfural (Ac:F) in the molar ratio of 5:1. Such a high Ac:F molar ratio was given by assuring the liquid nature of reaction products. They can also consist of highly viscous materials soluble in acetone and flushed from the catalyst bed. Product samples were collected every 4 h after substrate feeding began. If two liquid phases were found in the product, the aqueous phase was separated and quantified. An aqueous phase was detected in the first four samples due to the fluent switching mode of the reactor from rehydration to the reaction. The oil fraction was analyzed by gas chromatography (GC) on Agilent 7890A with a flame ionization detector and HP-5 capillary column (30 m/0.32 ID/0.25 µm). The identification of the products was provided using standard reference compounds as well as additional GC-MS analyses. After 200 h on stream (TOS), substrate feeding and heating was stopped. Then, the reactor was rinsed with acetone under nitrogen flow to remove furfural and products residues. Further, the reactor was dried by increasing temperature to 80 °C for 1 h under nitrogen flow around 1000 NL/h.
Reactivation of the catalyst before the second run was started with the same catalyst without opening the reactor or manipulating of the catalyst bed. The temperature was increased by following the same procedure as during activation, followed by a similar rehydration scheme. The only change was made by decreasing the reactant substrate's feeding rate (Ac:F = 5:1; mol:mol) reduce from 100 to 80 g/h (WHSV = 1.6/h). The aim for this was to increase the conversion of acetone extent. The reactivation test was finished after 160 h on stream. The catalyst was washed in situ by following the procedure applied for the first run.
Reactivation of the catalyst before the third run was done with the same procedure as after the 1st run. The 3rd run was at similar conditions to the first run (WHSV = 2.0/h, i.e. feed rate of 100 g/h), but this time was increasing the temperature from 60 to 70 °C.
Results and discussion
Catalyst characterization
The catalyst sample was synthesized based on a previous study already published [6]. Here we provided a catalyst characterization to confirm whether the synthesis was successful. The catalyst composition was determined by ICP-OES to evaluate Mg and Al’s real content on the sample. Table 2 shows the catalyst composition and the results of the BET surface.
The textural properties were characterized by mercury porosimetry. Figure 2 shows the pore diameter distribution of the catalyst before loading into the reactor. The catalytic porosimetry corresponds to the type of the used material for catalyst synthesis.

Pore diameter distribution of the hydrotalcite before loading into the reactor
Finally, XRD was used to determine the composition of the phases in the catalyst sample. Figure 3 shows XRD spectra of dried Mg-Al catalyst before calcination, where it is easy to identify the peaks which correspond to phase-pure hydrotalcite (HTC) Mg6Al2CO3(OH)16⋅4H2O [12]. A well-formed crystalline layered HTC structure of the sample was confirmed by the presence of characterizing diffraction peaks at 2Θ≈11, 23 and 34 (35) [12].

The XRD diffractogram of the hydrotalcite before loading into the reactor. XRD spectrum by D8 Advance ECO (Bruker AXS) with CuKα radiation and λ = 1.5406 Å
Catalytic test and product distribution
Acetone-furfural aldol condensation products consist of a large number of compounds (Fig. 1). The products were divided into two groups according to the number of monomer units in their structure to facilitate the results’ interpretation. The most desired oligomers with 6 and more monomer units were named “heavy” and were not individually separated. The heavy products’ yield was constantly decreasing during the 1st test from 57 to 10% (Fig. 4a). After the catalyst reactivation in the 2nd run with lower WHSV (1.6/h), heavy products’ initial concentration reached 48% and was kept almost the same for the next 40–100 h on stream (Fig. 4a). As seen from Fig. 4a, there was an increase in heavy products’ concentration after 100 h on stream. That fact was caused by the mistakable increased temperature to 80 °C. Thus, the reaction conditions changed significantly and promoted the formation of heavy products. After the reaction temperature was stabilized to the desired value during the next 24 h, heavy products’ yield followed the same trend as before the temperature increase. The second run was stopped after 160 h on stream, then the catalyst was reactivated again and restarted for the third run with standard WHSV = 2/h. The initial yield of heavy products of the third run reached 31% and fell to 12% in 150 h on stream, then the reaction temperature raised to 70 °C resulting in an increase and stabilization of heavy products yield at 14% (Fig. 4a).

Distribution of heavy compounds and F2Ac3 in condensation tests during experiments. Run 1: WHSV 2.0/h; T 60 °C; Ac-F ratio 5:1; Run 2: WHSV 1.6/h; T 60 °C; Ac-F ratio 5:1; Run 3: WHSV 2.0/h; T 60–70 °C; Ac-F ratio 5:1. Products analysis by GC-FID and HP-5 capillary column
The heaviest identified product was pentameric F2Ac3 consisting of two furfural and three acetone units. In the initial test, F2Ac3 concentration did not exceed 5% concentration (Fig. 4b). The concentration was practically constant during the test, while during the 2nd run, the yield was growing linearly up to 8% in 160 h on stream. After another reactivation, in the 3rd run, the F2Ac3 concentration showed identical concentration as detected in the initial run, although the yield of heaviest products was significantly affected by catalyst deactivation. This fact points to F2Ac3 formation on different active sites or active sites located in smaller pores than heavy products. In the same way, it shows that the catalyst deactivation is directly related to feedstock amount treated as an analogy to some cumulative effect. One of these effects might be a gradual raising of mass transport properties limitations by the heaviest products, which could block transport pores due to their large structures. Another way of transport pores blocking is potential clogging by solid products and intermediates.
Another group of lighter adducts was named tetramers. This group was represented by minor products having exclusively two furfural and two acetone units (Fig. 1). The difference, in this case, was in the dehydration degree of tetramer (F2Ac2–OH vs. F2Ac2). Yields of both products in the first experiment were constant for 150 h on stream, reaching 3.5% of the dehydrated product and less than 1% of the hydrated one (Fig. 5). Then the yield of F2Ac2 started to decline. The catalyst reactivation and test restart with lower WHSV = 1.6/h showed a little higher yield of the hydrated product with a slightly increasing trend (Fig. 5). The concentration of the dehydrated product was constant over 160 h of the experiment. The second reactivation followed with the third run at WHSV = 2/h showed an identical initial concentration of hydrated product till reaction temperature increase after 150 h on stream (Fig. 5b). The dehydrated product’s initial concentration was identical to the initial test as well, but gradually decreased to 2.2% and little increased after reaction temperature increase to around 2.8%.

Tetramers concentration during the aldol condensation tests. Run 1: WHSV 2.0/h; T 60 °C; Ac-F ratio 5:1; Run 2: WHSV 1.6/h; T 60 °C; Ac-F ratio 5:1; Run 3: WHSV 2.0/h; T 60–70 °C; Ac-F ratio 5:1. Products analysis by GC-FID and HP-5 capillary column
Hydrated FAc2OH and dehydrated F2Ac represented the trimeric compounds. At the beginning of the first run, the FAc2OH product was yielded in higher concentrations (Fig. 6a). This fact was given by high initial catalyst activity resulting in the fast conversion of furfural into heavier products and high molar acetone to the furfural molar ratio (5:1). After gradual catalyst deactivation, trimers’ concentration became identical, reaching 3% of each product after 90 h on stream. The concentration of F2Ac then gradually increased to 6% after 200 h on stream. The catalyst reactivation followed with the 2nd run at WHSV 1.6/h showed an identical initial concentration of both trimers with a linear decrease of hydrated FAc2OH with no significant influence of reaction temperature change after 100 h on stream (Fig. 6a).

Trimers concentration during the aldol condensation tests. Run 1: WHSV 2.0/h; T 60 °C; Ac-F ratio 5:1; Run 2: WHSV 1.6/h; T 60 °C; Ac-F ratio 5:1; Run 3: WHSV 2.0/h; T 60–70 °C; Ac-F ratio 5:1. Products analysis by GC-FID and HP-5 capillary column
On the other hand, longer contact time evoked a more intensive concentration of dehydrated F2Ac in the product (Fig. 6b). This compound’s yields were probably slightly affected by reaction temperature rise after 100 h on stream, as shown in Fig. 6a. However, the effect was not as significant as in the case of heavy products at the same moment. The third test after the second reactivation showed a higher initial F2Ac concentration than detected in the first experiment (Fig. 6b). However, the concentration increment rate was also linear but not as progressive as in the 1st run. Reaction temperature increase after 150 h on stream resulted in stabilization of catalyst activity and the yield of F2Ac around 5.7% and FAc2OH 1.3%.
Based on the proposed reaction scheme (Fig. 1), the dimeric compounds are supposed to be just primary reaction intermediates, and for this reason, these compounds should be detected in trace amounts. At the beginning of the first experiment, the initial concentration of both was lower than 10%. In 200 h of reaction, the FAc concentration raised to 23%, while the DAA remained nearly the same (Fig. 7). Reactivation and test with lower WHSV showed that contact time almost did not affect selectivity to dimeric intermediates. Temperature increase during the second experiment evoked a decrease in FAc concentration (Fig. 7a), which correlated with the increase of heavy products at the same time (Fig. 4a). These changes pointed to the reaction mechanism of acetone-furfural aldol condensation, where dimeric compounds were formed first and then interact with other products into larger oligomers.

Dimeric adducts/intermediates concentration in aldol condensation samples. Run 1: WHSV 2.0/h; T 60 °C; Ac-F ratio 5:1; Run 2: WHSV 1.6/h; T 60 °C; Ac-F ratio 5:1; Run 3: WHSV 2.0/h; T 60–70 °C; Ac-F ratio 5:1. Products analysis by GC-ID and HP-5 capillary column
Catalyst deactivation
As described in the Experimental section, the used catalyst was analyzed after catalytic tests using the same methods as the fresh ones to support the discussion related to the catalyst deactivation. In this way, Table 3 shows the catalyst composition and the catalyst samples’ elemental analysis after use.
According to these results, the catalyst composition did not change significantly, with an Mg/Al ratio similar to the fresh catalyst. However, during the catalyst testing, the main effect observed was the increase of carbon content on the catalyst surface due to the carbon depositing by coking during the experiments. This result might support the catalyst deactivation effect described, which related the decrease of catalyst activity with the feedstock amount treated. To support this assumption, Fig. 8 shows the pore diameter distribution of the catalyst after testing.

Pore diameter distribution of catalyst sample after catalytic tests
As expected, the increase in carbon content on the catalyst surface affected catalyst textural properties, decreasing the pore diameter distribution. On the other hand, the XRD analysis shows similar spectra to the fresh catalyst sample, pointing that the catalyst’s crystallographic structure remained unchanged. Figure 9 shows the XRD spectra on the catalyst sample after experiments.

The XRD diffractogram of the hydrotalcite after catalytic tests
Conclusions
The long-term catalytic aldol condensation of furfural and acetone on Mg-Al mixed oxide was conducted for 560 h in total. Three runs with the following catalyst reactivations were provided in a continuous mode for 160–200 h. The fresh and used catalyst samples were analyzed by XRD, showing similarity to the reference sample used in a batch mode and no additionally formed phases or phase changes after its use. Results of reactivation experiments showed a gradually decreasing the catalytic activity of Mg-Al mixed oxide. As a decrease of the heaviest products formation, the deactivation was related to the amount of the converted feedstock. The possible reasons could be (i) formation of products that are not in a liquid state under operated reaction conditions and clogging; (ii) formation of large molecules, which cannot leave catalyst pores (transport limitations). The oligomers' formation probably did not include the interaction of furfural, but its dimeric intermediate FAc (4-(2-furyl)-3-buten-2-one). The reaction temperature increase and WHSV decrease were found as the possible compensation of catalytic activity decrease, which can also change minor (tetrameric) product formation selectivity.
References
Kanaujia PK, Sharma YK, Garg MO, Tripathi D, Singh R (2014) Review of analytical strategies in the production and upgrading of bio-oils derived from lignocellulosic biomass. J Anal Appl Pyrolysis 105:55–74. https://doi.org/10.1016/j.jaap.2013.10.004
Isikgor FH, Remzi Becer C (2015) Lignocellulosic biomass: a sustainable platform for production of bio-based chemicals and polymers. Polym Chem 6:4497–4559. https://doi.org/10.1039/C5PY00263J
Ramos R, Tišler Z, Kikhtyanin O, Kubička D (2016) Towards understanding the hydrodeoxygenation pathways of furfural-acetone aldol condensation products over supported Pt catalysts. Catal Sci Technol 6:1829–1841. https://doi.org/10.1039/c5cy01422k
Ramos R, Tišler Z, Kikhtyanin O, Kubička D (2017) Solvent effects in hydrodeoxygenation of furfural-acetone aldol condensation products over Pt/TiO2 catalyst. Appl Catal A Gen 530:174–183. https://doi.org/10.1016/j.apcata.2016.11.023
Faba L, Díaz E, Ordóñez S (2014) One-pot aldol condensation and hydrodeoxygenation of biomass-derived carbonyl compounds for biodiesel synthesis. Chemsuschem 7(10):2816–2820. https://doi.org/10.1002/cssc.201402236
Sutton AD, Waldie FD, Wu R, Schlaf M, ‘Pete’ Silks III LA, Gordon JC (2013) The hydrodeoxygenation of bioderived furans into alkanes. Nat Chem 5:428–432. https://doi.org/10.1038/nchem.1609
King AE, Brooks TJ, Tian YH, Batista ER, Sutton AD (2015) Understanding ketone hydrodeoxygenation for the production of fuels and feedstocks from biomass. ACS Catal 5(2):1223–1226. https://doi.org/10.1021/cs501965w
Barret CJ, Chheda JN, Huber GW, Dumesic JA (2006) Single-reactor process for sequential aldol-condensation and hydrogenation of biomass-derived compounds in water. Appl Catal B Environ 66(1–2):111–118. https://doi.org/10.1016/j.apcatb.2006.03.001
Faba L, Díaz E, Ordóñez S (2014) Hydrodeoxygenation of acetone-furfural condensation adducts over alumina-supported noble metal catalysts. Appl Catal B Environ 160–161:436–444. https://doi.org/10.1016/j.apcatb.2014.05.053
Hora L, Kelbichová V, Kikhtyanin O, Bortnovskiy O, Kubička D (2014) Aldol condensation of furfural and acetone over MgAl layered double hydroxides and mixed oxides. Catal Today 223:138–147. https://doi.org/10.1016/j.cattod.2013.09.022
Kikhtyanin O, Hora L, Kubička D (2015) Unprecedented selectivities in aldol condensation over Mg-Al hydrotalcite in a fixed bed reactor setup. Catal Commun 58:89–92. https://doi.org/10.1016/j.catcom.2014.09.002
Hora L, Kikhtyanin O, Čapek L, Bortnovskiy O, Kubička D (2015) Comparative study of physico-chemical properties of laboratory and industrially prepared layered double hydroxides and their behavior in aldol condensation of furfural and acetone. Catal Today 241:221–230. https://doi.org/10.1016/j.cattod.2014.03.010
Kikhtyanin O, Čapek L, Smoláková L, Tišler Z, Kadlec D, Lhotka M, Diblíková P, Kubička D (2017) Influence of Mg-Al mixed oxide compositions on their properties and performance in aldol condensation. Ind Eng Chem Res 56(45):13411–13422. https://doi.org/10.1021/acs.iecr.7b03367
Parejas A, Cosano D, Hidalgo-Carrillo J, Ruiz JR, Marinas A, Jiménez-Sanchidrián C, Urbano FJ (2019) Aldol condensation of furfural with acetone over Mg/Al mixed oxides. Influence of water and synthesis method. Catalysts 9(2):203. https://doi.org/10.3390/catal9020203
Kocík J, Frolich K, Perková I, Horáček J (2018) Pyroaurite-based Mg-Fe mixed oxides and their activity in aldol condensation of furfural with acetone: effect of oxide composition. J Chem Technol Biotechnol 94(2):435–445. https://doi.org/10.1002/jctb.5787
Smoláková L, Frolich K, Kocík J, Kikhtyanin O, Čapek L (2017) Surface properties of hydrotalcite-based Zn(Mg)Al oxides and their catalytic activity in aldol condensation of furfural with acetone. Ind Eng Chem Res 56(16):4638–4648. https://doi.org/10.1021/acs.iecr.6b04927
Kikhtyanin O, Lesnik E, Kubička D (2016) The occurrence of Cannizzaro reaction over Mg-Al hydrotalcites. Appl Catal A Gen 525:215–225. https://doi.org/10.1016/j.apcata.2016.08.007
Kikhtyanin O, Kelbichová V, Vitvarová D, Kubů M, Kubička D (2014) Aldol condensation of furfural and acetone on zeolites. Catal Today 227:154–162. https://doi.org/10.1016/j.cattod.2013.10.059
Acknowledgements
The work is a result of the project which was carried out within the financial support of the Ministry of Industry and Trade of the Czech Republic with institutional support for long-term conceptual development of research organisation. The result was achieved using the infrastructure included in the project Efficient Use of Energy Resources Using Catalytic Processes (LM2018119) which has been financially supported by MEYS within the targeted support of large infrastructures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Horáček, J., Tišler, Z., Akhmetzyanova, U. et al. The long-term performance of reconstructed MgAl hydrotalcite in the aldol condensation of furfural and acetone. Reac Kinet Mech Cat 133, 341–353 (2021). https://doi.org/10.1007/s11144-021-01976-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-021-01976-z