
Abstract
The controlled synthesis of transition metal phosphides has been pursued to obtain excellent performances in application. Herein, we report a simple and effective method to synthesize nickel phosphide nanoparticles with target phases. Pure-phase nickel phosphide nanoparticles were obtained in different crystalline states (Ni2P and Ni12P5), and the crystalline phase of nickel phosphide could be controlled by varying the reaction conditions such as the temperature and duration of thermal treatment or the ratio between Ni and P. In addition, the nickel phosphide particles after thermal treatment maintained their sizes without serious agglomeration. In the hydrogenation of nitrobenzene, the phosphides with pure-phase (Ni2P or Ni12P5) and high crystallinity showed high catalytic activities. This proves that the crystalline phase of nickel phosphide plays an important role in the catalytic activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transition metal phosphides are a large class of compounds formed by phosphorous atoms entering metal lattices. Their unique physical and chemical properties make them promising candidates for wide applications in many fields, such as hydrotreating catalysis [1, 2], water splitting [3], battery [4, 5], and so on. Therefore, the synthesis of transition-metal phosphides has become a hot topic in recent years.
Among various phosphides (iron, cobalt, molybdenum, nickel and tungsten phosphides), nickel phosphide exhibits the most promising application prospect. Several crystalline phases of nickel phosphides (Ni3P, Ni5P2, Ni7P3, Ni12P5, Ni2P Ni5P4, NiP etc.) with different exposed crystal surfaces were synthesized successfully, and gave different applications [6,7,8,9]. For example, Ni5P4 and NiP2 showed higher performances as the lithium ion battery electrode materials [10, 11], while Ni2P and Ni12P5 were considered to be the perfect hydrotreating catalysts [12,13,14]. So, researchers have been trying to control the synthesis of nickel phosphide in the target phase, especially in the pure phase [15,16,17,18,19,20]. However, the controlled synthesis mostly depended on expensive reagents (trioctylphosphine, TOP and Ni(II)acetylacetonate, Ni(acac)2) [15,16,17] or harsh reaction conditions [18]. Moreover, many synthesis methods had to be carried out in small doses in an autoclave [19, 20]. Therefore, a simple, facile and low-cost route is important for the phase-controlled synthesis of nickel phosphide in large scale.
In our previous work [21], we developed a facile and simple method to synthesize small amorphous and crystalline Ni–P particles using inexpensive materials of nickel chloride hexahydrate and sodium hypophosphite. The production scale was theoretically free from the limitations of reactor and pressure because the process was carried out in an ordinary round bottom flask at ambient pressure. Based on this method, we further explored how to control synthesis of nickel phosphide in different crystalline phases. In the present work, the target pure-phases (Ni2P and Ni12P5) were made via thermal treating the crystalline nickel phosphides in N2 flow. Additionally, the phosphides particles maintained nanosizes even after the thermal treatment. The catalytic performances of as-synthesized nickel phosphides were investigated in the hydrogenation of nitrobenzene.
Experimental
Synthesis
The crystalline nickel phosphide (Ni–P) was synthesized according to our previous work [21]. The synthesis conditions were as follows: temperature 170 °C, time 5 h, initial Ni/P ratios 1/2, 1/3, 1/4 and 1/5. The as-synthesized nickel phosphide was then thermal treated in N2 to make controlled phases Ni2P and Ni12P5. In detail, crystalline Ni–P was placed in a tube furnace and purged in 20 mL/min N2 at room temperature for 30 min. Then, the sample was heated to a set temperature (100–500 °C) at the rate of 2.5 °C/min and kept in the terminal temperature for a certain time (1–10 h). Finally, the thermal treated phosphide was cooled to room temperature in the N2 flow.
Characterization
X-ray diffraction (XRD) was performed by a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation, employing a scanning rate of 2° min−1 and scanning range of 5–80°. SEM images of various samples were collected by a QUANTA 200 (FEI) equipment. X-ray photoelectron spectroscopy (XPS) was recorded on a RBD upgraded PHI-5000C ESCA system (Perkin Elmer) with Mg or Al Kα radiation.
Reactivity studies
The catalytic activities of the samples were tested in a 50 mL stainless steel autoclave for the hydrogenation of nitrobenzene. After 20 mg catalyst, 0.5 mL nitrobenzene and 10 mL solvent ethanol were added into the reactor, argon was passed to remove the air. Finally, 1.0 MPa hydrogen was injected and the activity tests were carried out at 100 °C for 1 h. After reaction, the catalyst was separated from the solution by centrifugation and the products were analyzed by GC with a SE-54 capillary column using toluene as the internal standard.
Results and discussion
Characterization of catalysts
XRD
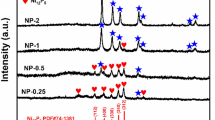
The XRD patterns of untreated and thermal treated nickel phosphides under different temperatures and times are shown in Fig. 1. As shown in Fig. 1A, the untreated nickel phosphide exhibited three diffraction peaks at around 32.8, 42.4, and 47.4°. The peaks were indexed as Ni12P5 phase (JCPDS No. 651623). However, the peaks at 42.4, and 47.4° were wider than the standard peaks, which were considered to be the Ni12P5 phases without complete crystallization. After treatment at 100 °C for 7 h and 200 °C for 2 h, the diffraction peaks of nickel phosphide hardly changed, which indicated that the phase of nickel phosphide was stable under mild conditions. At 200 °C, the crystalline phase began to change when the thermal treatment time was extended to 7 h, and new peaks at 38.4, 41.8, 44.4, 49.0 and 56.2° for Ni12P5 emerged. As further raising the temperature from 300 to 500 °C, the peaks of Ni2P phase began to appear and became dominant at high temperatures. Meanwhile, the peaks of Ni12P5 phase decreased gradually and disappeared eventually at 500 °C. Obvious phase transitions could be observed at medium temperature of 300 °C. It was noteworthy that NiO peaks appeared at high temperatures (400 and 500 °C), which may be caused by the decomposition of nickel phosphate generated during the synthesis of nickel phosphide.

XRD patterns of various samples treated under different temperatures and times. Initial Ni/P: 1/3
The temperature of thermal treatment was fixed at 300 °C to further investigate the effect of time on the phase transition. The results showed that the effect of time was significant (Fig. 1B). At short times (1 and 2 h), the mixture of Ni12P5 and Ni2P were obtained, but the major phase was Ni12P5. As prolonging the time, Ni2P became to the major phase gradually, and finally became to the pure phase at 7 h. No new phase appeared when the time was extended to 10 h, suggesting that the generated Ni2P phase was relatively stable. These results clearly indicated that lower temperature and shorter time favored the formation of Ni12P5, while higher temperature and longer time favored the formation of Ni2P. However, it was difficult to get pure-phase of Ni12P5 with high crystallinity through simply adjusting the temperature and time.
To obtain the pure Ni12P5 phase with high crystallinity, we tried to change the initial Ni/P ratio. Based on above experiences, 200 °C and 7 h were selected as the thermal treatment conditions. Fig. 2A shows the XRD patterns of untreated nickel phosphides with different initial Ni/P ratios. Amorphous structure was obtained at the high Ni/P ratio of 1/2, while crystalline structures with the same diffraction peaks were obtained during the Ni/P ratios of 1/3–1/5. After thermal treatment at 200 °C for 7 h, all samples gave the similar peaks corresponding to the pure-phase of Ni12P5. The only difference was that the intensity of peaks was weaker at higher Ni/P ratios (1/2 and 1/3) and higher at lower Ni/P ratios (1/4 and 1/5). It indicated that increasing the amount of phosphorus source can promote the crystallization of Ni12P5.

XRD patterns of nickel phosphides with different initial Ni/P ratios. A without treatment; B thermal treatment under 200 °C and 7 h
SEM
Fig. 3 shows the SEM results of untreated and thermal treated nickel phosphides under different conditions. The untreated nickel phosphide gave small and uniform particles around 30-80 nm. There was no change for the particles when nickel phosphide was treated at 100 °C for 7 h. It was consistent with the result of XRD, proving that nickel phosphide particles were stable at 100 °C. With raising the treatment temperature and prolonging the time, the particle sizes of nickel phosphides increased slightly. However, all nickel phosphide particles treated under different conditions (even at a high temperature of 400 °C) still maintained nanosizes (< 200 nm) without serious agglomeration. It suggested that the method in this work can effectively control the particle sizes of nickel phosphide. In addition, under the same treatment conditions (200 °C, 7 h), nickel phosphide with initial Ni/P of 1/4 showed smaller particles (30–70 nm) than that (40–100 nm) with initial Ni/P of 1/3. This might be because more phosphorus sources produced more PH3 gas during the preparation process, which was conducive to the generation of smaller particles of nickel phosphide.

SEM images of untreated and thermal treated nickel phosphides. A–G initial Ni/P: 1/3; H initial Ni/P: 1/4
XPS
The chemical states of Ni and P in the untreated nickel phosphide (Ni–P) and thermal treated nickel phosphides (Ni12P5 and Ni2P) were characterized by XPS. In the Ni 2p spectra shown in Fig. 4, the Ni 2p3/2 peaks at 852.3 and 855.4 eV for the untreated Ni–P were corresponding to the Ni 2p1/2 peaks at 873.5 and 869.7 eV, which were assigned to Niδ+ (0 < δ < 2) in phosphide and oxidized Ni species, respectively [22]. The peak at 861.3 eV was assigned to the satellite of Ni 2p3/2 [23]. The thermal treated nickel phosphides Ni12P5 and Ni2P showed the similar Ni 2p3/2 and Ni 2p1/2 peaks. However, compared to the Ni 2p spectrum of untreated nickel phosphide, Niδ+ and oxidized Ni for Ni 2p3/2 shifted to higher binding energy levels after thermal treatment. In P 2p spectra, the peaks at around 129.5 and 133.4 eV for untreated nickel phosphide were assigned to Pδ− (0 < δ < 1) in phosphide and oxidized P species on the surface of phosphide [16]. The peak at 130.2 eV was also considered to be Pδ− due to some interaction between Ni2+ and Pδ− [21]. The thermal treated nickel phosphides (Ni12P5 and Ni2P) exhibited two similar peaks at 130.2 and 133.6 eV without the peak around 129.5 eV. In addition, P 2p spectra showed that Ni12P5 and Ni2P had more oxidized P species compared with the untreated Ni–P. Apparently, the passivation reaction on the surface was easier for the thermal treated nickel phosphides when the samples were exposed to air. It also indicated that the reactivity for Ni12P5 and Ni2P would be higher than the untreated Ni–P. From the change of above Ni and P 2p spectra, it could be seen that some electron transfer ought to occur during the thermal treatment process.

XPS spectra of Ni and P 2p for Ni–P, Ni12P5 and Ni2P. Ni–P: initial Ni/P of 1/3, without treatment; Ni12P5: initial Ni/P of 1/4, thermal treatment under 200 °C and 7 h; Ni2P: initial Ni/P of 1/3, thermal treatment under 300 °C and 7 h
Catalytic hydrogenation performances
Fig. 5 shows the catalytic performances of various nickel phosphides in the hydrogenation of nitrobenzene. High selectivities to aniline (≥ 99.0%) with tiny side-product of cyclohexylamine were obtained over all nickel phosphides. The untreated Ni–P gave a much low conversion of nitrobenzene of 40.2%. A similar activity was observed over Ni–P-100 °C-7 h, because of no changes for crystalline phase (Fig. 1) and particle size (Fig. 3) by thermal treatment at 100 °C. It was noteworthy that the other thermal treated nickel phosphides all showed enhanced activities even if the particle sizes had some slight increase after thermal treatment (Fig. 3). Moreover, with raising the treated temperature and prolonging the time, the conversion of nitrobenzene increased gradually. The improvement of activity was considered to be attributed to the transition of crystalline phase of nickel phosphides according to the results of XRD. Ni2P and Ni12P5 were proved to be the highly active phases for the hydrogenation of nitrobenzene. So those nickel phosphides with better crystallinity of Ni2P or Ni12P5 gave higher catalytic activities. When the initial Ni/P ratio was 1/3, Ni–P-300 °C-7 h with almost pure Ni2P phase exhibited the highest conversion of 80.5%. Ni–P-400 °C-2 h showed a slightly lower activity than Ni–P-300 °C-7 h due to the existence of NiO species (Fig. 1) as well as the larger particle sizes (Fig. 3). When the initial Ni/P ratio was 1/4, Ni–P-200 °C-7 h with pure Ni12P5 phase (Fig. 2) gave a much high conversion of 84.2%.

Catalytic performances of thermal treated nickel phosphides in the hydrogenation of nitrobenzene. Reaction conditions: 20 mg catalyst, 0.5 mL nitrobenzene, 10 mL ethanol, 100 °C, 1.0 MPa H2 pressure, 1 h. aInitial Ni/P: 1/3. bInitial Ni/P: 1/4
It is well known that hydrogenation of nitrobenzene is an important reaction usually catalyzed by the noble metal of Pd [24, 25]. Developing cheap and efficient catalysts to replace noble metals for the hydrogenation of nitrobenzene remains a challenge. In recent years, various Co, Ni and Mo-based catalysts were fabricated. Compared to the state-of-the-art non-noble metal catalysts shown in Table S1, the Ni–P catalysts synthesized in this work gave higher reaction rates even at mild conditions.
Conclusions
Pure-phase nickel phosphides (Ni2P and Ni12P5) could be synthesized by simple thermal treatment of nickel phosphide nanoparticles prepared in glycol at low temperature. Lower temperature and shorter time favored the formation of Ni12P5, while higher temperature and longer time favored the formation of Ni2P. Additionally, at the low temperature 200 °C, a low Ni/P ratio is conducive to increasing the crystallinity of Ni12P5. Surprisingly, no serious agglomeration was observed when the nickel phosphides were treated under different conditions, and all the particles maintained their nanosizes. The phosphides after thermal treatment, especially those with high crystallinity and pure-phase (Ni2P and Ni12P5), showed high catalytic hydrogenation activities in the hydrogenation of nitrobenzene. It suggested that the activity of nickel phosphide was closely related to its crystalline phase.
References
Yang L, Li X, Wang AJ, Prins R, Wang Y, Chen YY, Duan XP (2014) J Catal 317:144–152
Peroni M, Lee I, Huang X, Barath E, Gutierrez OY, Lercher JA (2017) ACS Catal 7:6331–6341
Chung DY, Jun SW, Yoon G, Kim H, Yoo JM, Lee K-S, Kim T, Shin H, Shin H, Kwon SG, Kang K, Hyeon T, Sung Y-E (2017) J Am Chem Soc 139:6669–6674
Guo HN, Chen CC, Chen K, Cai HC, Chang XY, Liu S, Li WQ, Wang YJ, Wang CY (2017) J Mater Chem A 5:22316–22324
Bai J, Xi BJ, Mao HZ, Lin Y, Ma XJ, Feng JK, Xiong SL (2018) Adv Mater 30:1802310
Moon J-S, Kim E-G, Lee Y-K (2014) J Catal 311:144–152
Li HM, Lu SQ, Sun JY, Pei JJ, Liu D, Xue YR, Mao JJ, Zhu W, Zhuang ZB (2018) Chem Eur J 24:11748–11754
Wang XD, Zhou HP, Zhang DK, Pi MY, Feng JJ, Chen SJ (2018) J Power Sources 387:1–8
Liu P, Wei T, Xu J, Xue B, Zhang W, Li Y (2013) Reac Kinet Mech Cat 109:105–115
Ma JJ, Ni SB, Lv XH, Yang XL, Zhang LL (2014) Mater Lett 133:94–96
Aso K, Hayashi A, Tatsumisago M (2011) Inorg Chem 50:10820–10824
Layan Savithra GH, Muthuswamy E, Bowker RH, Carrillo BA, Bussell ME, Brock SL (2013) Chem Mater 25:825–833
Senevirathne K, Burns AW, Bussell ME, Brock SL (2007) Adv Funct Mater 17:3933–3939
Badari CA, Lonyi F, Drotar E, Kaszonyi A, Valyon J (2015) Appl Catal B 164:48–60
Li D, Senevirathne K, Aquilina L, Brock SL (2015) Inorg Chem 54:7968–7975
Pan Y, Liu Y, Zhao J, Yang K, Liang J, Liu D, Hu W, Liu D, Liu Y, Liu C (2015) J Mater Chem A 3:1656–1665
Muthuswamy E, Savithra GHL, Brock SL (2011) ACS Nano 5:2402–2411
Yun G, Guan Q, Li W (2017) RSC Adv 7:8677–8687
Deng Y, Zhou Y, Yao Y, Wang J (2013) New J Chem 37:4083–4088
Wang B, Huang X, Zhu Z, Huang H, Dai J (2012) Appl Nanosci 2:423–427
Liu P, Chang W-T, Liang X-Y, Wang J, Li Y-X (2016) Catal Commun 76:42–45
Iino A, Cho A, Takagaki A, Kikuchi R, Oyama ST (2014) J Catal 311:17–27
Huang Z, Chen Z, Chen Z, Lv C, Meng H, Zhang C (2014) ACS Nano 8:8121–8129
Liu P, Chen Y-L, Zhang Z-X, Liu H-F, Li Y-X (2018) Reac Kinet Mech Cat 125:595–603
Prekob Á, Muránszky G, Hutkai ZG, Pekker P, Kristály F, Fiser B, Viskolcz B, Vanyorek L (2018) Reac Kinet Mech Cat 125:583–593
Acknowledgements
Authors thank the National Natural Science Foundation of China (No. 21406019), Postdoctoral Science Foundation of China (No. 2016M601794), Postdoctoral Science Foundation of Jiangsu province, Jiangsu Shuangchuang Program, and Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, P., Zhang, ZX., Jun, S.W. et al. Controlled synthesis of nickel phosphide nanoparticles with pure-phase Ni2P and Ni12P5 for hydrogenation of nitrobenzene. Reac Kinet Mech Cat 126, 453–461 (2019). https://doi.org/10.1007/s11144-018-1496-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-018-1496-8