
Abstract
The oxidation of cyclohexylamine with molecular oxygen over a heterogeneous catalyst is an attractive one-step route to prepare a commercially important cyclohexanone oxime. The effect of the reaction conditions on the conversion of cyclohexylamine during its oxidation by molecular oxygen and on the selectivity of cyclohexanone and cyclohexanone oxime formation was investigated over heterogeneous catalysts (alumina and alumina supported silicotungstic acid). The present work is aimed at the enhancement of the cyclohexanone oxime yield, for which the optimized conditions were found as: reaction temperature of 180 °C, LHSV of cyclohexylamine 1 ml g−1 h−1, oxidant 33 % O2 in nitrogen with a total GHSV of 24 ml min−1. Cyclohexanone, formed from cyclohexylamine, reacts with cyclohexylamine to N-cyclohexylidenecyclohexylamine, their Schiff base. The most intense formation of cyclohexanone was observed in the initial period of the catalytic tests. Carbonaceous deposits, responsible for the catalyst deactivation, are formed mainly from the Schiff base; their amount increases by time on stream, and the rate of their formation correlates with the concentration of oxygen in the reaction mixture, as well as with the formation rate of cyclohexanone and the Schiff base. Injection of water to the reactant stream prolongs the catalyst lifetime and reduces the tar formation. However, it deteriorates the catalyst´s activity and selectivity. The supposed mechanism of cyclohexylamine oxidation is discussed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
ε-Caprolactam, the precursor of Nylon-6, is currently manufactured from benzene mainly through cyclohexanone as an intermediate, which is then converted into cyclohexanone oxime and finally rearranged to the lactam in a multistep process [1, 2]. Cyclohexanone can be obtained by the hydrogenation of phenol, but the preferred industrial route involves the oxidation of cyclohexane. However, the aerobic oxidation of cyclohexane to cyclohexanone limits the overall efficiency of the process owing to the low yields per pass achieved (~4 %) using homogeneous catalysts [3, 4]. Furthermore, in the traditional industrial process, hydroxylamine is used as a reagent to produce cyclohexanone oxime, which represents an additional technological step. This complication is partly solved by in situ generation of hydroxylamine [5].
The selective oxidation of cyclohexylamine by molecular oxygen is one of the alternative routes for the preparation of cyclohexanone oxime (Fig. S1). γ-Alumina, in combination with other metal oxides was shown to be a suitable catalyst for this one-step oxidation [6]. Alumina is an easily available metal oxide, and is widely used industrially as a filler, adsorbent, drying agent, catalyst support and reagent [7]. In contrast to clays and zeolites, alumina does not typically contain channels or cavities, but has a large geometrical surface area available for hosting active catalytic centers [8].
In the oxidations of organic compounds by molecular oxygen, the heterogeneous catalysts usually represent a redox system, reducible by the organic substrate and reoxidizable by oxygen; another possible way of action is the activation of the chemisorbed oxygen on the catalyst’s surface. This type of catalytic action, however, has a low probability in the case of Al2O3, not being a typical redox catalyst itself. Reduction of alumina by amines or an activation of molecular oxygen on the alumina surface was not reported in a scale suitable for practical application.
However, surprisingly, γ-alumina itself was found to be catalytically active in the oxidation of cyclohexylamine by molecular oxygen [9]; a conversion of cyclohexylamine of 15 % at 40 % selectivity to cyclohexanone oxime was reported. The catalytic activity was attributed to acidic centers. Further modification of alumina by aminic promoters leads to an increase in its catalytic activity; 60 % selectivity to cyclohexanone oxime at 35 % cyclohexanone conversion was observed. The main reaction products—beside cyclohexanone oxime—were cyclohexanone and a Schiff base formed by the reaction of cyclohexanone and cyclohexyl amine (N-cyclohexylidene–cyclohexylamine) [10].
Oxides of Mo and W can enhance the activity and selectivity of alumina and other supports toward cyclohexanone oxime. However, oxides of V, Cu, Ag and other transition metals typically occurring in redox catalysts, exhibit only a very low activity in terms of cyclohexanone oxime yields [6]. Along with the redox properties of the supported tungsten-based catalysts, the role of the acidic sites have been reported as crucial in terms of the catalytic activity [10]. On the surface of alumina-, titania- or zirconia-supported tungsten oxide, a dominant occurrence of mono-oxo wolframyl species was reported [11]. However, the W=O bond length, the Lewis acidity of the wolframyl species, the charge transfer transition energies and the reducibility of the WOx species were found to be strongly dependent on the nature of the support. In particular, the wolframyl groups on the alumina support—compared to the titania support—exhibit higher acidity, higher charge transfer transition energies, and are less easily reducible. The wolframyl species on zirconia show properties in between that of alumina and titania [11]. The importance of the acidity of the supported tungsten oxide was recently reported for a non-redox application, in the glycerol carbonate formation, catalyzed by WO3 [12]. The alumina supported tungsten oxide or silicotungstic acid catalysts were reported to exhibit as high as 70 % selectivity of cyclohexanone oxime formation at around 20 % conversion of cyclohexylamine [10].
The aim of the present work was the study of the influence of reaction conditions and water content on the formation of oxidation products of cyclohexylamine over alumina and alumina supported silicotungstic acid catalyst, and to identify different reaction pathways leading to different reaction products, or leading to the deactivation of the catalyst.
Experimental
Materials
Sodium hydroxide was purchased from Merck®, ammonia, hydrogen sulfide, silicotungstic acid, cyclohexylamine form Sigma-Aldrich®. All materials were of commercial reagent grade. Before using, cyclohexylamine and silicotungstic acid were purified by redestillation and recrystallization, respectively.
N-Cyclohexylidene-cyclohexylamine was prepared by mixing equimolar amounts of cyclohexanone and cyclohexylamine, and stirring the reaction mixture at a temperature of 100 °C for 1 h. The resulting material, verified by GC, contained >95 % of N-cyclohexylidenecyclohexylamine.
A solution of the hemisodium salt of silicotungstic acid was prepared by a partial neutralization of the solution of the silicotungstic acid by NaOH.
A preparation of a typical catalyst, denoted as 15Na2H2STA/Al2O3 (the numeral 15 referring to the percentage of the tungsten precursor content in the catalyst), was carried out as follows. A mixture of a hemisodium salt of silicotungstic acid (Na2H2STA), Na2H2[Si(W3O10)4] (0.6 g) with alumina (3.4 g) have been stirred in the aqueous solution (50 ml) for 1 h at room temperature. After evaporation of water at 100 °C, the samples were dried at 150 °C, pelletized, crushed and sieved to a grain size 0.6–0.9 mm and activated by calcination in a flow of dry air (50 ml min−1) at 500 °C for 6 h. The obtained material, being sensitive towards moisture, was stored in a desiccator over P2O5. A catalyst denoted as 15H4STA/Al2O3 was prepared accordingly, using unneutralized silicotungstic acid instead of its hemisodium salt. Textural characterization of the alumina supports and the as-prepared catalysts by BET and BJH plots were reported elsewhere [13], indicating a narrow pore radius distribution, centered at 8 nm with a pore volume of 0.6 cm g−1 and a specific surface area of ~200 m2 g−1. The introduction of silicotungstic acid generally decreased the pore volume as well as the specific surface area by ~10 %. The total acidity of the alumina support was ~0.7 mmol NH3 g−1; this value raised to ~1.2 upon the introduction of silicotungstic acid.
Copper-doped catalysts were prepared by adding appropriate amounts of copper nitrate to the aqueous solution of the tungsten precursor.
Neutralized alumina was prepared by stirring a suspension of alumina in 0.5 % NaOH solution at a molar ratio Al2O3:NaOH = 1:0.05 at ambient temperature for 1 h. The alumina was then filtered, washed until neutrality and activated in air flow (50 ml min−1) at 500 °C, as described above.
Ammonia-saturated alumina was prepared by saturating the freshly activated alumina with gaseous ammonia, GHSV of 10 ml min−1, at a temperature of 180 °C for 1 h. The catalyst was then purged by dry air for 2 h before introducing the reaction mixture.
If not otherwise specified, a γ-alumina of catalyst support grade, purchased form Alfa Aesar was used. Other types of aluminas—CG-20 from Sigma-Aldrich®, and Siralox 30 from Sasol®—were tested as well. Before the catalytic tests, the aluminas were activated in air flow at 500 °C for 6 h.
The amount of the carbonaceous deposits, formed during the catalytic tests, was determined gravimetrically after purging the used catalysts in a nitrogen flow for 1 h at 260 °C.
Apparatus, procedure and analysis
The oxidation of cyclohexylamine was carried out in a fixed-bed glass reactor (i.d. = 16 mm) with a total volume of 30 ml. In a typical run, 2 g of a pelletized, freshly calcined catalyst was used. The dead volume above the catalyst bed was filled with glass spheres of a diameter of 2 mm. The catalytic tests were carried out at atmospheric pressure and a temperature of 180 °C using N2/O2 mixture as an oxidant with different oxygen contents. The chosen reaction temperature represented the optimal temperature in terms of cyclohexanone oxime yield, reported recently [13]. The catalyst, after reaching the reaction temperature, was equilibrated in air flow (20 ml min−1) for 1 h before introducing the reaction mixture.
Cyclohexanone, if formed, readily reacts with cyclohexylamine giving N-cyclohexylidene–cyclohexylamine, a Schiff base (Scheme S1). Hence, the total amount of cyclohexanone, formed by oxidation of cyclohexylamine was calculated as a sum of free cyclohexanone and the Schiff base.
The reaction products were cooled in a glass cooler, and the liquid portion was collected in an ice-cooled flask. The liquid products were analyzed in the intervals of 30 min, using a Shimadzu 2014 gas chromatograph equipped with CP Sil 5 CB column (25 m × 0.53 mm) and a FID detector using a temperature program of the column: 60 °C isothermically (3 min), from 60 to 240 °C with a gradient of 10 °C min−1. For quantitative analysis, the method of the external standard was used.
The reaction products were identified on a Shimadzu QP5000 GC/MS with EI and capillary column (HP-1, 50 m × 0.2 mm × 0.33 µm), using a temperature program: from 60 to 240 °C with gradient of 10 °C min−1.
The FTIR spectra were recorded using a Shimadzu IRAffinity-1 spectrometer, the chemisorption and TPR measurements were carried out using a Micromeritics ASAP 2020 apparatus.
Results and discussion
Catalyst characterization
The FTIR spectrum of pure alumina shows two broad absorption bands in the regions ~980–810 cm−1 and bellow ~790 cm−1, respectively. These broad bands represent the envelope for different absorption maxima belonging to Al–O and Al–OH bonds, both in tetrahedral and octahedral coordination, with absorption bands expected to occur in this region for amorphous γ-alumina [10] (Fig. S2a). This absorption background, belonging to γ-alumina, is present in all the other tungsten-doped samples, as well. By mechanically mixing the γ-alumina support with STA, the resulting mixture exhibit the typical spectrum of the slicotungstic acid (Si–O, W=O and W–O–W vibrations occurring at 1,016, 977, 920 and 883 cm−1, respectively) overlapped by the alumina background (Fig. S2d). However, when the STA is introduced not by mechanical mixing, but by wet impregnation from an aqueous solution, the IR spectrum of the resulting material changes dramatically, compared to the mechanical mixture. The bands belonging to STA deteriorate significantly, and a new absorption band at 823 cm−1 turns up (Fig. S2c). This absorption band is expected to belong to W=O vibrations, similar to that of occurring in tungstates [14, 15]. Upon calcination, a further deterioration of the signals originating from STA, accompanied by the increase of the intensity of the W=O signal can be observed (Fig. S2b). This means that the original Keggin structure of the STA decomposes readily leaving mono-oxo wolframyl species on the alumina surface even at ambient temperature. This transformation is further promoted by calcination.
The as-prepared catalyst did not exhibit remarkable absorption bands above the wavenumbers of 1,000 cm−1. However, the spent 15H4STA/Al2O3 catalyst showed a complex absorption spectrum in the interval of 1,000–3,500 cm−1 (Fig. S3c), representing the absorption of the deposited tar, responsible for the catalyst deactivation. The IR spectrum of the tar is a superposition of the absorption spectrum of the cyclohexylamine (bands at 2,922, 2,854, 1,604, 1,448 and 1,465 cm−1) with several adjunct overlapping absorption bands, mainly in the regions 1,250–1,400 and 1,500–1,700 cm−1, respectively. The four main adjunct absorption bands (1,631, 1,570, 1,386 and 1,338 cm−1) can be ascribed to different functional groups. The combination of bands 1,386 and 1,570 cm−1 can be assigned to nitro, nitroso or nitrite compounds; while the bands at 1,338 cm−1 (C–N), 1,570 or 1,631 (N–H), together with a broad band at ~3,050 cm−1 (aromatic C–H) can be ascribed to aromatic amines. Interestingly, the C=O stretching bands, expected to occur at 1,700–1,800 cm−1 are missing. Hence, the tar, responsible for the catalyst deactivation, is expected to be comprised of mainly aliphatic cyclic amines, with some smaller degree of aromatization and oxidation, the oxygen atom being preferentially bound to nitrogen rather than to a carbon atom.
The TPR pattern of the 15H4STA/Al2O3 catalyst exhibits one distinct reduction peak at 640 °C (Fig. 1c). Below this temperature region, no reduction peaks occur. At first glance it refers to a rather uniform nature of the tungsten oxide species in terms of their reducibility. Moreover, it indicates that at the temperature of 180 °C, at which the catalytic tests were carried out, tungsten does not change its oxidation state. The position of the reduction peak corresponds well with the value around 637 °C reported for tetrahedral dimeric WOx species [16]. Octahedrally coordinated WOx species (occurring in the STA as well) are more easily reducible than tetrahedrally coordinated ones, having reduction maxima in the temperature range of 327–527 °C [16]. The presence of octahedral tungsten species, hence, can be ruled out.

TPR-patterns of the catalysts 15H4STA/Al2O3 (c), and the 15H4STA/Al2O3 catalysts doped with copper in W:Cu ratios 10:1 (b) and 3:1 (a). Reducing agent: 10 % H2/He, gradient 10 K min−1
In bimetallic catalysts, the addition of further metal generally lowers the reduction temperature of tungsten. The addition of copper shifted the reduction maxima for tungsten from 640 to 620 °C and 585 °C for W:Cu ratios 10:1 and 3:1, respectively (Figs. 1a and b). However, these temperatures are still far above the reaction temperature employed in catalytic tests. The reduction peaks at lower temperatures are assigned to copper, although, a presence of small amounts of octahedrally coordinated tungsten species, represented by the broad peak at 250–400 °C for the W:Cu ratio 3:1, is conceivable.
Influence of the flow rate of the oxidant
The equilibrium of adsorbed cyclohexylamine and reaction products on the catalyst surface is expected to be affected by a changing flow rate of the oxidant at a given reaction temperature. Higher flow rate increases the evaporation rate of more volatile products and decreases the contact time of the gaseous components of the reaction mixture with the catalyst. Moreover, it increases the molar ratio of oxygen:cyclohexylamine, which, in turn, has an additional influence on the distribution of oxidation products.
The catalytic tests using different flow rates of the oxidant—containing 33 vol% of oxygen—were studied over pure γ-alumina as a catalyst. Theoretically, a flow of 19.7 ml min−1 of such oxidant is sufficient for 100 % yield of cyclohexanone oxime.
The average conversions and average yields of cyclohexanone oxime and of cyclohexanone were calculated from the averaged data during the first 7 h of time on stream (TOS). The maximal conversion of cyclohexylamine over γ-alumina, 9.0 %, as seen in Fig. 2, was observed at a flow rate of the oxidant of ca. 24 ml min−1. The deterioration of the yields of the reaction products at higher flow rates of the oxidant, above 24 ml min−1, corresponds to the decrease of the conversion of cyclohexylamine. The increase of the flow rate of the oxidant leads to preferred formation of cyclohexanone oxime: at the flow rate of the oxidant of 5 ml min−1, the ratio of yields of cycloheanone:cyclohexanone oxime is 1:0.6. However, by increasing the flow rate of the oxidant to 40 ml min−1, this value increases to 1:2.5. The observed rate of formation of carbonaceous deposits on the catalyst surface, measured after 7 h of TOS, increases from 1.3 to 4.6 % by increasing the flow rate of the oxidant: from 5 to 50 ml min−1. A decrease of the selectivity of cyclohexene formation, from 0.5 to 0.15 % was registered upon increasing the flow rate of the oxidant, from 5 to 50 ml min−1.

The effect of the flow rate of the oxidant (33 % O2 in N2) on the average conversions of cyclohexylamine (diamond), average yields of cyclohexanone oxime (open triangle) and cyclohexanone (open square) over pure γ-alumina catalyst. Reaction conditions: temperature 180 °C, LHSV of cyclohexylamine: 1.0 ml h g−1
Influence of the flow rate of cyclohexylamine
The effect of the flow rate of cyclohexylamine was tested using a GHSV of the oxidant (33 % O2 in nitrogen) of 24 ml min−1 and the LHSV of cyclohexylamine of 0.75, 1.00 and 1.25 ml h−1 g−1.
As expected, the conversion of cyclohexylamine decreases by its increasing flow rate as the contact time between cyclohexylamine and the γ-alumina catalyst decreases (Fig. S4). It is worth noticing that a lower cyclohexylamine flow rate brings about a higher concentration of oxygen in the reactant stream. The selectivity of cyclohexanone oxime formation increases by increasing cyclohexylamine flow rate, from 33 to 43 % in the third hour of TOS, but this increase does not compensate the loss of the conversion of cyclohexylamine upon increasing its flow rate from 0.75 to 1.25 ml h−1 g−1. Thus, the yield of cyclohexanone oxime is higher at lower cyclohexylamine feed rate (3.0 % for the lowest, and 1.7 % for the highest feed rate). The selectivity of cyclohexanone formation varies within the range of 40–55 % and slightly decreases upon increasing the flow rate of cyclohexylamine. The sum of selectivities of the oxidation products (cyclohexanone oxime, cyclohexanone and nitrocyclohexane) is 80 ± 5 % and it is not significantly influenced by the LHSV of cyclohexylamine in the range from 0.75 to 1.25 ml h−1 g−1. By increasing the LHSV of cyclohexylamine from 0.75 to 1.25 ml h−1 g−1, the formation of carbonaceous deposits increases from 2.1 to 3.3 % after the third hour of TOS.
The effect of the introduction of silicotungstic acid
A deposition of 15 % of Na2H2STA on the γ-alumina support, increased remarkably the catalytic activity, reaching as high as 33 % of cyclohexylamine conversion at the LHSV of cyclohexylamine 1 ml g−1 h−1, and GHSV of the oxidant (33 % O2 in nitrogen) of 24 ml min−1. The introduction of tungsten into the γ-alumina support led to an increase of the selectivity of the formation of cyclohexanone oxime (see “The effect of the introduction of silicotungstic acid” section).
Influence of the oxygen content in the oxidant stream
To study the effect of the oxygen content in the oxidant stream, a 15Na2H2STA/Al2O3 catalyst was used at the LHSV of cyclohexylamine of 1 ml g−1 h−1, and GHSV of the oxidant of 24 ml min−1, using a range of oxygen contents in the oxidant stream of 13–45 %.
At low oxygen contents (13 and 21 vol%), in the initial period of the experiments, no cyclohexanone oxime was found in the reaction products, it appeared in the product stream only after a 2.5 h and 1.5 h induction period, respectively (Fig. 3). At higher oxygen contents (>25 vol%), the formation of cyclohexanone oxime was observed since the beginning of the catalytic tests.

Time dependence of the selectivities of the formation of cyclohexanone oxime over 15Na2H2STA/Al2O3 catalyst as a function of the oxygen content (13–45 %) in the oxidant stream. Reaction conditions: temperature 180 °C, LHSV of cyclohexylamine: 1.0 ml h g−1, GHSV of the oxidant: 24 ml min−1
The observed selectivity of cyclohexanone formation is considerably high at the beginning of the catalytic tests, up to 85 % for the most oxygen-lean condition, utilizing 13 % of oxygen in nitrogen as an oxidant. Contrary to the trend observed for the selectivities of cyclohexanone oxime formation, the selectivities of the formation of cyclohexanone are monotonously decreasing for all the oxygen contentrations used (Fig. S5).
The conversion vs. TOS curves for cyclohexylamine usually exhibit maxima (Fig. S6). Generally, the lower the oxygen content of the oxidant, the later these curves reach maxima.
The highest conversion of cyclohexylamine (33 %) was observed at the concentration of oxygen 33 vol% after 3 h of TOS.
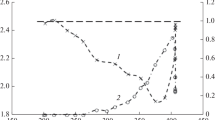
At higher concentrations of oxygen, one would expect higher conversions of cyclohexylamine, potentially overoxidation of cyclohexanone oxime e.g. to nitrocyclohexanone. Out of accord with this hypothesis, at oxygen concentrations above 33 % the conversion of cyclohexylamine significantly decreases, while the selectivities of cyclohexanone oxime formation—except during the induction period in the cases where lower oxygen concentrations were utilized—were not distinctively diverging; instead, for oxygen concentrations >25 %, the selectivity curves were almost identical (compare to Fig. 4).

Influence of oxygen concentration in the oxidant stream on the time dependence of the yields of cyclohexanone oxime (a) and cyclohexanone (b) over 15Na2H2STA/Al2O3 catalyst. Reaction conditions: temperature 180 °C, LHSV of cyclohexylamine: 1.0 ml h g−1, GHSV of the oxidant: 24 ml min−1
A build-up of carbonaceous deposits, measured after 1 h TOS was found to be proportional to the oxygen content of the oxidant, being 0.2 % for the lowest oxygen content (13 %) and 0.9 % for the highest oxygen content (45 %). These data correspond to the visual observation of the spent catalysts: at higher oxygen contents, the darkening of the used catalysts was more pronounced. The faster the formation of the carbonaceous deposits, the sooner the conversions of cyclohexanone reach a maximal value, after which a gradual loss of the conversion takes place.
Comparing the selectivities toward cyclohexanone oxime (Fig. 3) and toward cyclohexanone (Fig. S5), the complementarity of the formation of these products is obvious. The sum of the selectivities of the formation of cyclohexanone and cyclohexanone oxime is relatively constant, regardless of the concentration of oxygen in the oxidant stream, varying in a relatively narrow interval, between 80 and 90 % (Fig. S7). This sum slightly increases by increasing concentration of oxygen in the oxidant stream. In addition, the oxygen-lean conditions favor the formation of byproducts as N-dicyclohexylamine, N-cyclohexylaniline, cyclohexanol, cyclohexene, cyclohexane and benzene, what explains the higher deficits of the sums of selectivities in oxygen-lean conditions. The above-mentioned byproducts are supposedly formed via hydrogenation-dehydrogenation pathways [12].
Concerning the yields of the two main oxidation products, apparently, at a given TOS, the trend of yields of cyclohexanone oxime follow the observed trends of conversions (Fig. 4a). The maximal cyclohexanone oxime yield, 22.5 %, was observed for oxygen concentration of 33 % in the oxidant stream in 3–4 h of TOS. The cyclohexanone yields increase by the concentration of oxygen at a given TOS, and decrease by time on stream (Fig. 4b).
Using other commercial types of γ-alumina supports, the yields of the two main reaction products, cyclohexanone and cyclohexanone oxime, were practically unaffected.
Extraction of the adsorbed reaction products
The boiling points of cyclohexylamine, 134 °C, and of cyclohexanone, 156 °C, are below the temperature of the catalyst bed, 180 °C. However, the boiling points of cyclohexanone oxime, 207–210 °C, and of the Schiff base, 251 °C, are higher; which brings about their condensation on the catalyst´s surface. For nitrogen-containing compounds, such as cyclohexylamine, cyclohexanone oxime and the Schiff base, one should expect their adsorption on the acidic surface of the catalyst as well. Thus, the desorption of the cyclohexanone oxime and the Schiff base seems to be the most restricted. On the other hand, the weakly adsorbed and volatile cyclohexanone is not expected to be retained on the catalyst’s surface at the temperature of 180 °C.
Hence, a question emerges, whether in those cases, when in the initial period of the catalytic tests there were no cyclohexanone oxime detected in the liquid reaction products, the apparent absence of the cyclohexanone oxime was or not caused by its adsorption on the catalyst surface.
To monitor the amount of the cyclohexanone oxime adsorbed on the catalyst’s surface, the catalytic tests were periodically interrupted and the catalyst extracted twice by 10 ml of methanol. The molar ratio of cyclohexanone:cyclohexanone oxime was then analyzed separately in the condensed liquid reaction products and in the methanolic extract (Table 1).
Comparing these ratios in the liquid products and in the methanolic extract, a pronounced disbalance was found after 2 h of TOS. Using 33 % O2 in nitrogen as an oxidant, after 2 h of TOS, a cyclohexanone:cyclohexanone oxime ratio of 1:0.21 was found in the liquid reaction products. At the same time, this ratio in the methanolic extract was 1:2.3. This disbalance, however, is attenuated in the course of time on stream. After 6 h of TOS the cyclohexanone:cyclohexanone oxime ratio was 1:2.4 in the liquid reaction products and 1:3.6 in the methanolic extract. This indicates that in the initial period of the catalytic test, cyclohexanone oxime, formed from cyclohexylamine, accumulates on the catalyst´s surface. Thus, during the induction period, the appearance of the cyclohexanone oxime in the liquid products is hindered by its adsorption on the catalyst surface.
The extraction of the carbonaceous deposits by methanol (after 6 h of TOS) led to a partial restoration of the activity of the catalysts in terms of cyclohexanone oxime yields. The methanol-extracted catalysts in the second reaction cycle yielded ~70 % of the initial amount of cyclohexanone oxime. The performance of the methanol-extracted catalysts in the consecutive reaction cycles decreased likewise. The carbonaceous deposits, insoluble in methanol can be, however, decomposed completely by purging the catalysts wit air at a temperature of 500 °C for 1 h; such a regeneration restored completely the original performance, and no apparent decrease in the catalytic activity was observed even after 5 reaction-regeneration cycles.
The effect of the extra Schiff base
N-Cyclohexylidene-cyclohexylamine, the Schiff base of cyclohexanone and cyclohexylamine, has, in comparison with other reaction products, the lowest volatility, and, thus is expected to condensate on the catalyst’s acidic surface. The iminic double bond in the molecule of the Schiff base, in addition, is sensitive to radical polymerization. The radical formed on the Schiff base can react with other Schiff base molecule to form a polymer, or by oxygen to form peroxy radical, an initiator of further oxidation or polymerization. The IR spectra of polymers—carbonaceous deposits were discussed recently [10].
Accordingly, the formation of the oligomers or polymers can block the catalyst´s surface. On the other hand, though, the water, formed as a reaction product of the oxidation, can cleave the Schiff base, thus setting free the catalyst’s surface.
To test the role of the Schiff base in the inhibition of the oxidation process over a 15Na2H2STA/Al2O3 catalyst, 1 g of the Schiff base was injected directly to the catalyst bed at standard reaction conditions using 33 % O2 in nitrogen as an oxidant. Two sets of experiments were carried out: (i) one with the introduction of the Schiff base prior to the introduction of the amine, and (ii) one with the introduction of the Schiff base past the introduction of the amine.
When the Schiff base was introduced prior the saturation of the catalyst by cyclohexylamine, the formation of cyclohexanone oxime was blocked completely during the first 6 h of TOS. In the liquid reaction products, only N-cyclohexylaniline and N-dicyclohexylamine were found along with the unreacted cyclohexylamine. N-cyclohexylaniline is a dehydrogenated derivative of the Schiff base, N-dicyclohexylamine is a hydrogenated one. Their yields, however, did not exceed 1.5 % in sum. Since cyclohexene was not found among the reaction products, the N-dicyclohexylamine in this case is presumably not formed by acid catalyzed alkylation of cyclohexylamine by cyclohexene, but instead, by hydrogenation of the Schiff base.
When the catalyst’s bed was saturated by cyclohexylamine prior to the introduction of the Schiff base, the formation of cyclohexanone oxime was not significantly deteriorated. In the first 4 h of TOS, the production of cyclohexanone oxime was similar to that of observed without the addition of the Schiff base. Supposedly, in this case the Schiff base, bearing lower basicity compared to the cyclohexylamine, cannot completely make up for the cyclohexyl amine on the catalyst surface. Hence, the added Schiff base was simply washed out by cyclohexyl amine (The fact that the Schiff base was eluted was confirmed by the analysis of the effluent stream).
The influence of water in the reactant stream
Water is one of the possible by-products of various oxidation reactions, which are mostly irreversible. Therefore, the presence of water cannot directly influence the formation rates of cyclohexanone oxime and carbonyl compounds by shifting the reaction equilibrium. Instead, it can act only indirectly, e.g. by changing the catalyst´s surface or the partial pressure of the reactants in the reaction mixture. Water on the catalyst´s surface typically converts Lewis acid sites to Brønsted ones, which were found to deteriorate the selectivity in terms of the cyclohexanone oxime formation [9, 10]. The hydroxyl groups, created by the hydration of Lewis acid sites can be reversibly dehydrated upon calcination, thus restoring the original Lewis acidity [17] Another effect of water, formed as a reaction product, is that it shifts the equilibrium of Schiff base formation back to cyclohexylamine and cyclohexanone (Scheme S1) [6]. Thus, water decomposes the Schiff base on the catalyst´s surface and—as the Schiff base readily undergoes condensation reactions [18]—prolongs the lifetime of the catalyst.
The influence of the presence of water on the oxidation of cyclohexylamine was tested using γ-alumina and 15Na2H2STA/Al2O3 as catalysts. Cyclohexylamine with water contents of: 0, 15, 30 and 50 wt% was fed by a rate of 1 ml h−1 g−1 at atmospheric pressure and a catalyst bed temperature of 180 °C.
The presence of water in cyclohexylamine significantly decreases not only the conversions of cyclohexylamine, but also the selectivity of cyclohexanone oxime formation (Figs. 5 and 6). As water is known to transform Lewis acid sites both on alumina and alumina-supported silicotungstic acid to Brønsted ones, this finding is consistent with the theoretical expectation.

Time dependence of the conversion of the cyclohexylamine as a function of the water content (0–50 %) in the reactant stream using a γ-alumina catalyst. Reaction conditions: temperature 180 °C, LHSV of cyclohexylamine: 1.0 ml h g−1, GHSV of the oxidant: 24 ml min−1

Time dependence of the conversion of the cyclohexylamine as a function of the water content (0–50 %) in the reactant stream using a 15Na2H2STA/Al2O3 catalyst. Reaction conditions: temperature 180 °C, LHSV of cyclohexylamine: 1.0 ml h g−1, GHSV of the oxidant: 24 ml min−1
Over pure γ-alumina, the conversion of cyclohexylamine did not exceed 5.8 % at the water content of 15 wt% (Fig. 5). By increasing the water content, the conversion of cyclohexylamine decreases; the lowest conversions were observed at a concentration of water in the reactant stream of 50 wt%. The same trend was observed also in the selectivity of cyclohexanone oxime formation, i.e. by increasing the water content from 0 to 50 %, the maximal selectivity of cyclohexanone oxime formation decreases from 55 to 20 % (Fig. S8). By decreasing the selectivity of the formation of cyclohexanone oxime—caused by the presence of water—, a formation of N-cyclohexylformamide became more pronounced, yielding up to 1 %.
Using 15Na2H2STA/Al2O3 catalyst, the conversions of cyclohexylamine were generally higher in comparison with the results obtained over pure γ-alumina catalyst (Fig. 6). The increasing water content has a negative effect on the oxidation of cyclohexylamine in terms of conversion and selectivity, similarly as it was observed in the case of pure γ-alumina catalyst. Adding 15 wt% of water to the reactant stream, the selectivity of cyclohexanone oxime formation reaches 55 %, which is more by 20 % compared to that of observed over pure γ-alumina catalyst (Fig. S9). In addition, the presence of water decelerated the relative decrease of the conversion of cyclohexylamine.
The influence of the neutralization of the alumina catalyst
To test the effect of acidic sites on cyclohexylamine oxidation, parent γ-alumina and neutralized γ-alumina catalysts were compared. γ-alumina, as described above, led to a 56 % selectivity to cyclohexanone oxime at 9.0 % of cyclohexylamine conversion.
Upon complete neutralization of the alumina by NaOH, no cyclohexanone oxime formation was detected in the catalytic tests. Only N-cyclohexylidene–cyclohexylamine (a Schiff base) was found in the reaction products, in a yields <1 %. Thus, the acidity of alumina is essential for the oxidation of cyclohexylamine.
As the impregnation of alumina by NaOH does not eliminate the presence of eventual transition metal impurities in alumina, the above mentioned results also mean that the oxidation of cyclohexylamine to cyclohexanone oxime over alumina is not a result of a catalytic effect of the presence of transitional metal impurities in alumina, but instead, it is the result of the catalytic activity of acidic sites of alumina itself.
Another, weaker, base, tested for the neutralization of the acidity of γ-alumina, was ammonia. Ammonia at the reaction temperature of 180 °C occupies only the stronger acidic sites on the γ-alumina catalyst, whereas weak acidic sites desorb ammonia bellow 180 °C. A recent TPD-study [10, 13] showed that ca. 15 % of the total amount of acidic sites release ammonia bellow 180 °C, the rest is released above this temperature at atmospheric pressure. Hence, using an ammonia-saturated catalyst, cyclohexylamine, being a weak base, can occupy only those weakly acidic sites; the strong acidic sites are blocked by ammonia.
The blocking of the strong acidic sites affected the formation of cyclohexanone oxime and of cyclohexene. Cyclohexene is known to be a product of deamination of cyclohexylamine catalyzed by strong acidic sites [10]. As expected, the neutralization of the catalyst by ammonia ceased the formation of cyclohexene: the cyclohexene yield dropped from 0.4 % for parent alumina to zero for the ammonia-neutralized alumina catalyst. The cyclohexanone oxime formation was slightly suppressed upon neutralization by ammonia, to ca. 80 % of the original values (Fig. 7). The experiments with neutralized catalysts clearly show that in the formation of cyclohexanone oxime over γ-alumina catalyst there are the week acidic sites which play the most important role in the formation of cyclohexanone oxime.

Time dependence of cyclohexanone oxime yields over parent γ-alumina (full symbols) and ammonia-neutralized γ-alumina (open symbols) catalysts. Reaction conditions: temperature 180 °C, LHSV of cyclohexylamine: 1.0 ml h g−1, GHSV of the oxidant: 24 ml min−1
In a further experiment, 15 % of pure silicotungstic acid was used instead of Na2H2STA as a source of tungsten on the γ-alumina support. The partial neutralization of the silicotungstic acid improved both the selectivity to cyclohexanone oxime and the conversion of cyclohexylamine (Table 2). In addition, the partial neutralization of the strongest acidic sites for Na2H2STA apparently suppressed the formation of cyclohexene, benzene and cyclohexane.
Therefore, over the catalyst prepared from pure STA, dicyclohexylamine can be formed also from cyclohexene and cyclohexylamine by acid catalyzed reaction. However, benzene and cyclohexane is formed by a hydrogenation-dehydrogenation pathway from cyclohexene.
The influence of a selective poisoning of the catalyst by H2S
The formation of cyclohexanone oxime, as the desired product of the oxidation of cyclohexylamine is strongly correlated with the occurrence of weak Lewis acidic sites (see “The influence of water in the reactant stream” section).
Concerning the by-products of the oxidation of cyclohexylamine, one can distinguish two groups.
-
(i)
The deamination of cyclohexylamine to cyclohexene, as well as the formation of dicyclohexylamine from cyclohexene and cyclohexylamine are typical acid-catalyzed reactions, requiring strong acidic sites on the catalyst, the effect of which can be affected by the neutralization of the most acidic sites.
-
(ii)
Cyclohexane and benzene are formed from cyclohexene by a hydrogenation/dehydrogenation pathway, similarly as cyclohexylaniline is formed either form the Schiff base or from dicyclohexylamine. Hence, this group of by-products necessitates a presence of catalytic centers bearing hydrogenation/dehydrogenation activity.
Transitional metals as Pt, Cu, Ni catalyze the hydrogenation/dehydrogenation reactions at the reaction temperature of 180 °C even in trace amounts. (An intentional addition of copper to the tungsten-doped alumina catalysts—using either free sillicotungstic acid or its hemisodium salt as a tungsten source—, in molar ratios 10:1 and 3:1, led to a rapid deactivation of the catalysts, within 3 h of TOS, and the cyclohexanone oxime yields dropped below 5 %.) The activity of these metals in hydrogenation/dehydrogenation reactions is highly sensitive to the presence of hydrogen sulfide. However the hydrogenation/dehydrogenation performance of tungsten is supposed to be much less affected by hydrogen sulfide at 180 °C and atmospheric pressure [19].
To poison selectively the catalysts, 1 ml min−1 of gaseous H2S was fed to reactor along with the oxidant stream.
For the 15Na2H2STA/Al2O3 catalyst, the conversions of cyclohexylamine were deteriorated in the presence of H2S to the level of ½ of the original value (Fig. S10). However, the distribution of the reaction products, i.e. the selectivity toward particular products, is not significantly affected by the presence of H2S.
The above-mentioned results indicate that the hydrogenation/dehydrogenation pathway can be attributed directly to the 15Na2H2STA/Al2O3 catalyst, and it is not a result of a catalytic effect of other metal impurities.
The suggested reaction pathways of the oxidation of cyclohexylamine
During the calcination at 500 °C, not only γ-alumina, but also silicotungstic acid is dehydrated. The elimination of the structural water from the γ-alumina support, as well as from the silicotungstic acid generates electron deficient Lewis acid sites, where the lone electron pair of oxygen or of the aminic nitrogen can be deployed (Fig. 8a–c).

Schematic description of the reaction mechanism of cyclohexylamine oxidation over γ-alumina-supported STA catalyst
Tungsten oxide is reportedly able to chemisorb oxygen, while the adsorption of compounds with lone electron pairs increases the conductivity of tungsten oxide species. The originally insolated electrons of tungsten oxide species shift to the conduction band resulting in the electrical conductivity of tungsten oxide. The formed electron vacancy may be filled by electrons of a chemisorbed molecule (amine, oxygen, etc.) [20]. In the chemisorbed amine, the adsorption on the wolframyl specie brings about the acidification of the aminic proton, which can therefore shift to the oxygen atom on the wolframyl specie, forming a W–OH moiety (Fig. 8d).
The chemisorbed molecule of cyclohexylamine, bound to a W–OH specie, upon losing a proton, exhibit a partial negative charge on the lone electron pair on the nitrogen atom. This pair of electrons can be easily oxidized, i.e. an electron can easily move via the conductivity band of tungsten to an electron-acceptor; in the case of chemisorbed oxygen this acceptor being the adsorbed oxygen in the vicinity, thus forming a superoxide radical while creating a cation radical of nitrogen (Fig. 8e, intermediate #1) [21, 22].
In the next step, the superoxide radical reacts with the cation radical giving a hydroperoxide intermediate (Fig. 8f). Depending on the relative strengths of the N–W and N–C bonds, the hydroperoxide decomposes giving either cyclohexanone oxime or cyclohexanol.
The cation radical in the intermediate #1 can, however, easily isomerize to give intermediate #2, which results in the formation of cyclohexanone [21–25].
Upon adsorption of cyclohexylamine on a strong acid site, not only the aminic hydrogen atoms, but also the hydrogen atoms on the β-carbon of cyclohexylamine are becoming acidic, and their release allowed. Releasing a proton from the β-carbon, a double bond is created between the α- and β-carbons of the cyclohexylamine molecule; and as a result, cyclohexylamine is deaminated (Fig. 9). Cyclohexene can be further activated on an acidic site (located e.g. on an aluminum atom), where it can react with a cyclohexylamine molecule to form dicyclohexylamine. The chemisorbed cyclohexene, activated on a strong acidic site can further undergo oxydehydrogenation, finally giving benzene.

A simplified reaction mechanism of N-dicyclohexylamine, cyclohexene and benzene formation over γ-alumina-supported STA catalyst (–R stands for a strong acidic site, located either on tungsten, aluminum, or represents a free proton)
Conclusions
The optimized reaction conditions for the formation of cyclohexanone oxime during oxidation of cyclohexylamine by molecular oxygen over alumina and alumina supported silicotungstic acid catalysts were found.
The highest yield of cyclohexanone oxime, 22.5 %, accompanied by a 12.5 % yield of cyclohexanone, was observed at the optimal oxygen content of the oxidant of 33 %. The optimal molar ratio of oxygen:cyclohexylamine was found to be very close to 1:1. In the oxygen-rich conditions the catalyst is rapidly deactivated by carbonaceous deposits. The optimal LHSV of cyclohexylamine was found 1 ml h−1 g−1. An increase of the LHSV of cyclohexylamine leads to a decrease in its conversion and increases the selectivity of cyclohexanone oxime formation. It leads to a faster deactivation of the catalyst as well.
The selectivity to cyclohexanone oxime generally increases by the first hours of TOS until mounting to a stable level. The conversion of cyclohexylamine, on the other hand, typically passes through a maximum value after ca 3 h on stream, after which it decreases. The apparent initial increase of the conversion of cyclohexylamine is caused by the adsorption of cyclohexanone oxime on catalyst surface, and its gradual release upon reaching the saturation of the surface, as it was confirmed by extraction of the adsorbed cyclohexanone oxime during the initial hours of TOS. The decrease of the conversion of cyclohexylamine after ca. 3–5 h of TOS is caused by the deactivation of the catalyst by carbonaceous deposits formed by oligomerization and polymerization of the Schiff base.
Cyclohexanone in the reaction products is present in two forms: as free cyclohexanone and in the form of N-cyclohexylidene–cyclohexylamine (a Schiff base formed between cyclohexanone and cyclohexylamine).
The maximal yields of cyclohexanone were observed in the first hour of TOS. Cyclohexanone and cyclohexanone oxime are formed in parallel reaction pathways. Once cyclohexanone is formed, it cannot be further converted to cyclohexanone oxime under the studied reaction conditions. An introduction of cyclohexanone—either pure or in a form of a Schiff base—does not lead to an increase in cyclohexanone oxime yield, it leads instead to a deactivation of the catalyst.
An addition of water into the reactant stream decreases the conversion of cyclohexylamine, as well as the selectivity of the oxime formation, and increases the formation of N-cyclohexylformamide, and prologs the lifetime of catalyst. At the presence of water, the amount of trapped poly-condensation products was decreased on the catalyst by stripping the tar formed upon polycondensation of the Schiff base.
In addition to the matrix of reaction conditions (flow rates of cyclohexylamine and the oxidant, the oxygen content, desorption of the reaction products), there are the acidobasic properties of the catalyst, determining the formation of cyclohexanone oxime. A minor hydrogenation/dehydrogenation pathway, influencing the distribution of the reaction products, was identified as well.
Neither alumina, nor tungsten oxides on their own are not reducible under the reaction conditions studied. (Their reduction temperatures, identified by H2-TPR lies well above 180 °C.) The considerable redox activity of the STA/Al2O3 catalyst in the oxidation of cyclohexylamine to cyclohexanone oxime should therefore be attributed to weak Lewis acidic sites and a subsequent radical mechanism of oxidation. The conductivity band of tungsten accessible for adsorbed molecules further enhances the formation of cyclohexanone oxime.
References
Ichihashi H, Sato H (2001) Appl Catal A 221:359–366
Dahlhoff G, Niederer JPM, Hoelderich WF (2001) Catal Rev Sci Eng 43:381–441
Takagi K, Ishida T (1972) US Patent 3644526 (1972)
Yuan HX, Xia QH, Zhan HJ, Lu XH, Su KX (2006) Appl Catal A 304:178–184
Mantegazza MA, Padovan M (1994) US Patent 5320819
Kaszonyi A, Cvengrošová Z, Hronec M (2000) J Mol Catal A 160:393–402
Kabalka GW, Pagni RM (1997) Tetrahedron 53:7999–8065
Maggi R, Ballini R, Sartori G, Sartorio R (2004) Tetrahedron Lett 45:2297–2299
Armor JN, Carlson EJ, Riggitanol R, Yamanis J, Zambri PM (1983) J Catal 83:487–490
Rakottyay K, Kaszonyi A (2009) Appl Catal A 367:32–38
Gutiérrez-Alejandre A, Castillo P, Ramírez J, Ramis G, Busca B (2011) Appl Catal A 216:181–194
Jagadeeswaraiah K, Kumar ChR, Prasad PSS, Loridant S, Lingaiah N (2014) Appl Catal A 469:165–172
Rakottyay K, Kaszonyi A, Vajíček S (2010) Appl Catal A 378:33–41
Gotić M, Ivanda M, Popović S, Musić S (2000) Mater Sci Eng, B 77:193–201
Rajkumar T, Ranga Rao G (2008) Mater Chem Phys 112:853–857
Liu N, Ding S, Cui Y, Xue N, Peng L, Guo X, Ding W (2013) Chem Eng Res Des 91:573–580
Li H, Xu Y, Gao Ch, Zhao Y (2010) Catal Today 158:475–480
Meixiang G, Dong L, Liqiu M, Dulin Y (2013) Chin J Appl Chem 30:28–31
Mashkina AV (2003) Kinet Catal 44:277–282
Chen Z, Yang J (2007) Chin J Chem Phys 20:78–84
Suzuki K, Watanabe T, Murahashi SI (2013) J Org Chem 78:2301–2310
Schümperli MT, Hammond C, Hermans I (2012) ACS Catal. 2:1108–1117
Schümperli MT, Hammond C, Hermans I (2012) Phys Chem Chem Phys 14:11002–11007
K Suzuki, H Nagahara (2004) US Patent 20040077903 A1
Zotov RA, Molchanov VV, Volodin AM, Bedilo AF (2011) J Catal 278:71–77
Acknowledgments
A financial support from the Slovak Grant Agency VEGA 1/0556/13 is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Horváth, B., Kaszonyi, A., Rakottyay, K. et al. Towards the mechanism of the oxidation of cyclohexylamine by molecular oxygen over alumina-based catalysts. Reac Kinet Mech Cat 115, 231–250 (2015). https://doi.org/10.1007/s11144-015-0840-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-015-0840-5