
Abstract
Catalytic processes are an indispensable part of a large number of contemporary technologies that stimulate a constant research and development effort in the field. Computational methods represent a valuable tool to investigate crucial steps of catalytic cycles able to reveal the main characteristics of a catalyst and provide a basis for the design of materials with superior catalytic activity. This review is focused on the recent advances in density functional theory studies of the interactions of reactive species and intermediates with solid surfaces. As examples, we discuss the catalysts for the CO oxidation and electrocatalysis of H2 and O2 electrode reactions. We demonstrate how the theoretical modelling can contribute to the understanding of catalytic processes and help to design new catalysts and electrocatalysts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Catalysis plays a vital role in the modern world, affecting our lives in numerous ways. Heterogeneous catalysis by itself presents a basis of approx. 20 % of the total industrial production in the world [1]. In the near future, it is expected that the immense impact of the catalysis will increase even more, playing the main role in the development of sustainable energy solutions [2].
In general, catalysis relates to a possibility of a given material to affect the rate of chemical transformation through the control of the rate of chemical bond cleavage and formation. It also relates to the control of the yields of desired products. In heterogeneous catalysis, chemical processes are affected by the presence of a solid surface which mediates chemical transformations. Electrocatalysis can be considered as a special type of a heterogeneous catalysis where electrode potential appears as an additional kinetic parameter. Electrocatalysis can be considered operative in any case when the rate of an electrode reaction depends on the electrode material properties. In this sense, one can say that practically every electrochemical reaction is an electrocatalytic one [3]. Regardless of which type of catalytic process is considered, a catalyst reduces the activation energy of a chemical process without affecting the thermodynamics. In this way, a catalyst affects the reaction path of the desired chemical reaction to increase the chemical transformation rate and selectivity with minimal input of energy.
Some of the basic principles in catalysis have been defined almost one century ago, and are still used in current research. One of them is the Sabatier principle [4], which states that the strength of the interaction of reactants and intermediates with the catalyst has to be “optimal”. If too weak, reactants will not bind to the catalyst and the reaction will fail to take place. If the interaction is too strong, the active sites on the catalyst surface get blocked by reactants, intermediates or product, which in this case act as catalytic poisons. This principle is intuitively clear, but leaves researchers with the task to find what the “optimal” binding is for each catalytic reaction of interest. Another important concept, widely utilized by research community, is the Brønsted–Evans–Polanyi (BEP) principle [5–7] which states that, for a given family of chemical reactions, the difference in activation energy between two reactions is proportional to the difference of their reaction enthalpies. For the BEP principle to be operative, the pre-exponential factor of the Arrhenius equation and the position of the transition state along the reaction coordinate should be similar for all reactions in the considered family of chemical reactions.
Although the basic physical/chemical principles underlying the catalytic processes are the same as at the time when they were recognized, our understanding of the physics and chemistry of materials significantly deepened since then. Recent tremendous development of new experimental and theoretical techniques enabled us to resolve chemical processes at the atomic level. In this way, the main task of the catalysis science has shifted from the development of new active catalysts for different processes to the understanding of how to design catalyst structures to control catalytic activity and selectivity [8, 9].
The role of theory in catalysis science
With the increase of computer power, development of new concepts and algorithms, the theory and computations have become very important in the contemporary catalysis research. In this sense, three aspects can be outlined. Theoretical tools can (i) enable the understanding of known catalytic systems, (ii) provide qualitative and quantitative insights into experimental measurements and (iii) suggest new systems, which could be promising catalysts [8]. Although the arrays of experimental techniques have been upgraded with a number of powerful methods enabling the atomic scale resolution and providing the insight into chemical processes at the atomic level, certain spatial and temporal domains remain out of the reach of experimental techniques. In this sense, theoretical methods became irreplaceable in catalysis science.
With the main task set to the understanding of how to design a new catalytic material, the trial-and-error approach in the catalyst development, has to be abandoned and a paradigm should be shifted to a rational catalyst design. This demands the understanding of catalytic process at the molecular level, which identifies the crucial properties of a catalyst and suggests how to compose new catalysts or optimize already existing ones. It should be emphasized that catalysis is all about increasing the rate of chemical transformations and directing it through a desired pathway so that selectivity is maximized. This means that a precise estimate of thermodynamic (such as, reaction enthalpy and free energy) and kinetic parameters (such as, activation energies) is absolutely essential. To illustrate the importance of accuracy, a simple calculation can be performed. As the kinetics of elementary steps can be described by Arrhenius equation, an error in the activation energy of only 0.06 eV changes the reaction rate by the factor of 10 at the room temperature (the error in the reaction rate, however, vanishes as the temperature increases). Electronic structure theory made its way into catalysis science with two methods having the largest impact: (i) high-level ab initio molecular orbital (MO) theory and (ii) density functional theory (DFT). DFT is more approximate than ab initio MO theory (having in mind the treatment of electron exchange correlation), but also more attractive because it enables the treatment of systems complex enough to be linked with the real world. Moreover, its accuracy is high enough to explain and predict reactivity trends. This work will focus on the role of the quantum physics calculations in heterogeneous catalysis. However, we notice that the impact of computations in catalysis science does not stop at the level of the electronic structure theory, but also includes diverse methods to investigate chemical processes at different temporal and spatial domains. Furthermore, it should be emphasized that the computational and experimental approaches in catalysis should not be considered separately, as these two complement each other and only by their combination it is possible to achieve a complete understanding of a catalytic process. Considering the electronic structure theory, its role in catalysis research is in modeling and understanding of catalytic processes at the electronic/atomistic level and identification of general trends and underlying principles specific for various classes of catalytic processes.
In the following text, first we discuss formation of a chemical bond between various types of adsorbates and surfaces (having in mind its electronic structure), which is a crucial step in heterogeneous catalysis, in order to rationalize the mechanisms of bond activation (“Essential step: the formation of a chemical bond at the surface” section). Then we proceed with the introduction of the concept of catalytic activity descriptor (“A few quantities instead of many: The concept of the catalytic activity descriptor” section), which enables the link between the fundamental properties of a catalyst and its macroscopic kinetic behavior. Finally, for the cases of the catalytic CO oxidation and the electrocatalysis of H2 and O2 electrode reactions, we demonstrate how the electronic structure calculations can be used to understand catalytic processes and model new catalytic systems.
Essential step: the formation of a chemical bond at the surface
An essential step in every heterogeneous catalytic process is the formation of a chemical bond between the catalyst surface and reactant/intermediate. For a molecular species in the gas phase, the electron density is optimized to a maximal bonding. When a molecule approaches the surface, the electron density gets redistributed to maximize bonding in the new system composed of the surface and adsorbate. This means that the chemical bonding in the adsorbate molecule gets compromised and some bonds might get weakened: this is actually how solid catalyst activates chemical bonds and enables the adsorbate to undergo a chemical change. The way electron density redistributes in this process is determined by the electronic structure of the surface and adsorbate.
Transition metals play a very important part in heterogeneous catalysis and for them the theory of chemical bonding is highly elaborated [10]. The concept of Hammer and Nørskov [11] can account for the trends in adsorption strength from one metal to another based on the position of their d-band. Within this picture, the interaction of the electronic states of an adsorbate with metal electronic states can be described as a two-step process. First, the electronic state of the adsorbate gets widened by the interaction with the wide half-filled s-states (the same for all transition metals). Then, the adsorbate states interact with the narrow d-states giving rise to bonding and anti-bonding states. Their relative occupancies, determined by the positions of the adsorbate state and metal d-states, determine the strength of the interaction. According to Pettersson and Nilsson [10], the d-band model requires a radical state on the adsorbed atom to form a covalent bond with the substrate. On the other hand, the electron-pairing concept for molecular systems requires re-hybridization in order to form a radical state at an interacting atom or molecule to make the formation of a chemical bond feasible. In some cases, such a re-hybridization is too expensive and only a weak interaction with the surface can be achieved (Fig. 1).

Schematic illustrations of the five types of chemical bond formation on metal surfaces. Reprinted from [10] © Springer a radical b diatomic molecules c unsaturated hydrocarbons d lone pair molecules e saturated hydrocarbons
The simplest case of a radical adsorption is when an unpaired electron interacts with metal states giving rise to bonding and anti-bonding states. Besides the covalent bond formation, the ionic contribution can be significant in some cases [10]. If an adsorbate has no unpaired electrons but unsaturated π-electron system (such as N2, CO, O2, unsaturated hydrocarbons), a partial mixing or an excitation process involving the π and π* orbitals [12] can create a virtual radical state necessary for the adsorbate to bind to the surface. The actual mechanism of the formation of virtual radical state depends on the π → π* excitation energy and this will ultimately determine the adsorption geometry (Fig. 1b and 1c) [10, 12]. The cases described in Fig. 1d and 1e are related to much weaker bonding. The formation of bonding and antibonding states in electron pair donor bonding mechanism (Fig. 1d) is due to the interaction of a lone pair with the positive metal ion core, which appears when rigid σ lone pair ‘‘digs a hole’’ in the metal sp density of states. This mechanism is operative, for example, in the case of a water adsorption on metal surfaces via negatively charged O atom. In the last case (Fig. 1e), the XH moiety interacts weakly with metal surfaces as only the X–H σ* states are available for bonding and these lie very high in energy, hindering strong hybridization between metal and adsorbate states.
Although oxide surfaces play very important roles in surface science and catalysis, a general and commonly accepted view of atomic and molecular adsorption on this type of materials is not available. This is possibly due to a great variability of this class of materials in terms of their crystal structure, electronic structure and magnetic properties, which makes the observation of general rules in atomic and molecular adsorption rather difficult. However, also in this case, it is clear that chemisorption properties are determined by the electronic structure of a particular combination of the substrate and the adsorbate and that the charge redistribution upon adsorption is responsible for the bond activation and catalytic effect. As an example, let us consider a molecular adsorption on the ideal (001) surface of MgO (Fig. 2) [13], one of the simplest oxide materials having tremendous importance in heterogeneous catalysis [14–17].

Projected density of states (PDOS) and integrated local density of states (ILDOS) for isolated (top) and adsorbed (bottom) B2 (left), C2 (middle) and F2 (right). Density of states of clean MgO (scaled) is included. Vertical dashed line denotes the highest occupied states of investigated molecules (top) or the highest occupied state of X2,ads + MgO(001) complex. Presented structures give ILDOS in the energy windows designated on Projected Density of States. Reprinted from [13] with the permission by Elsevier
For a diatomic molecule to dissociate on MgO(001), its adsorption parallel to the surface plane is required so that the intramolecular bond is cleaved, while the energy lost in this process is compensated by the creation of new surface bonds. The analysis of H2 adsorption and the homonuclear diatomic molecules of the second row elements [13], shows that the investigated molecules split into three groups: (i) molecules weakly interacting with MgO (H2, N2, O2), (ii) molecules strongly adsorbing on MgO(001) (B2, C2) and (iii) molecules spontaneously dissociating on MgO(001) (F2). The first group of molecules is characterized by very strong intramolecular bonds due to filled bonding states (H2, N2) or partially filled anti-bonding states (O2). In this case, the involvement of antibonding states would destabilize the molecules that cannot be compensated by the adsorption of dissociation fragments on MgO(001). The second group molecules (B2 and C2) have partially filled bonding orbitals, located in the same energy window as the MgO valence band, and these states participate in bonding with MgO(001). The homolytic dissociation of F2 is justified on the basis of its electronic structure: the empty σ* orbital, being located just below the MgO valence band, gets filled upon the interaction with MgO as a result of a charge transfer to F (with the high electronegativity of fluorine being the main driving force) [13] (Fig. 3).

The discussion given above exemplifies the power of electronic structure calculations which provide us with an in-depth view of the interaction of different types of atomic and molecular species with various substrates and give us the essential fundamental knowledge at the atomic level. However, the electronic structure calculations are the most demanding ones among different computational approaches used in catalysis research and the detailed analysis of reaction mechanisms for a particular catalytic reaction on many possible substrates can be extremely time-consuming and impractical. Fortunately, such elaborated calculations are not always needed.
A few quantities instead of many: the concept of the catalytic activity descriptor
Electronic structure calculations can be used to evaluate many kinetic and thermodynamic parameters of a given catalytic reaction and to develop a microkinetic model, which captures macroscopic kinetic behavior of the catalytic system. There are examples of such kind of studies available in the literature [18, 19], but it is obviously a demanding procedure which needs to be repeated for hundreds of thousands of times in order to describe a single reaction. However, the Sabatier principle and BEP relations indicate that it might be possible to link the macroscopic kinetic behavior to certain properties of the catalytic material or thermochemistry of considered catalytic reaction. Indeed, such an idea had appeared long before the electronic structure theory became a powerful tool in catalysis. One of the best known examples is Trasatti’s formulation of volcano curve for the hydrogen evolution reaction (HER) in acidic solutions: when the exchange current density (measure of activity, analogous to turnover frequency) is plotted versus the formation energy of the hydride of metal catalyst, volcano-shaped curve is obtained with Pt located at its apex [20]. This approach was re-examined and questioned ever since it was published and still is [21, 22], but it presents a beautiful idea to link a few properties of a catalyst with its macroscopic kinetic behavior. Upon the development of the electronic structure theory it became possible to evaluate many microscopic properties of catalytic materials as well to describe the kinetics of catalytic processes, providing us with the microscopic characteristics of a catalyst which can be linked to its catalytic performance. These quantities are called catalytic activity descriptors and can be used to rationalize the catalytic activity of a given material or establishment of a design concept in search for new catalysts [2].
The d-band model [11, 23] contributed tremendously to the elaboration of the concept of catalytic activity descriptor. As it correlates the adsorption energies and activation energies with a single parameter describing the electronic structure of transition metal surfaces [24] it is tempting to use it in the search for new catalytic materials. This model, however, has its limitations as its use gets rather complicated already in the case of multicomponent metallic surfaces [25–27]. On the other hand, BEP relations [7] and the scaling of adsorption energies of different adsorbates on solid surfaces [28, 29] helps to reduce the dimensionality of the data set required to describe the kinetics of a given catalytic reaction and to identify the catalytic activity descriptor(s). Their use usually results in some kind of a volcano relationship with the most active catalyst located at its apex. Once the proper descriptor(s) have been identified, the electronic structure calculations can be used to screen many possible systems and identify new possible catalysts. Of course, the final judgments are left to experimental verification, which would help to refine models and incorporate missing elements. There are many catalytic activity descriptors identified so far for various catalytic reactions. Without any introspection of the underlying physical principles, which qualify these quantities as catalytic activity descriptors, we provide an overview of these quantities in Table 1. Some of these examples will be discussed in more detail later on, where we address the use of the electronic structure calculations in understanding of selected catalytic processes and development of new catalysts.
Theoretical insights into the mechanisms of catalytic processes and design of new catalytic materials
During the catalytic cycle, even for simplest reactions, a number of chemical bonds break and form, while at the end, the catalyst remains unchanged. It is rather difficult to capture each step using experimental techniques, irrespective of how sophisticated they are. In this sense, the electronic structure calculations, especially methods based on DFT, demonstrate their power. We proceed with some specific examples of the CO oxidation reaction, as well as two cases of electrocatalytic reactions: hydrogen evolution and oxygen reduction reactions.
CO oxidation
The CO oxidation is one of the simplest catalytic reactions, which is described by the following overall equation:
It is used as a benchmark for the understanding of heterogeneous catalysis. In electrocatalysis, it presents a basis for the understanding of the oxidation of small molecules of organic fuels (methanol, ethanol, formic acid, etc.). From a technological aspect, the reaction is of high importance in the control of car-exhaust emission, air purification and sensors. Three major types of catalysts for the CO oxidation can be distinguished: platinum group metallic catalysts (whose surface might be oxidized), bulk oxide catalysts (such as CeO2), and oxide-supported metal clusters.
CO oxidation on densely packed transition metal surfaces
Although being a relatively simple reaction, the CO oxidation might proceed via different mechanisms, depending on the nature and the state of the catalyst surface. Three possible mechanisms have been proposed: (i) Langmuir–Hinshelwood (L–H) [47] (Fig. 4a), (ii) Eley–Rideal (E–L) [48, 49] (Fig. 4b) and (iii) Mars-van Krevelen mechanism (MvK) [50, 51] (Fig. 4c), with the first one being widely accepted. Briefly, L–H mechanism assumes that CO adsorbs on the catalyst surface while O2 follows dissociative adsorption path to form two O adatoms (Oads). Then, adsorbed CO and Oads combine to give CO2 which desorbs from the surface (Fig. 4a). Within E–L mechanism CO reacts with Oads from the gas phase, that is, without the adsorption, to produce CO2 (Fig. 4b). According to MvK mechanism, assumed for some oxides and oxidized surfaces, CO is oxidized by lattice oxygen atom (Fig. 4c). Upon formation of CO2, surface vacancy is formed which heals upon the exposure to O2 from the gas phase.

Possible mechanisms of CO oxidation on platinum group metal surface. a Langmuir–Hinshelwood mechanism, b Eley–Rideal mechanism, c Mars-van Krevelen mechanism
Some of the first attempts to provide a generalized atomic picture of the CO oxidation on close-packed transition metal surfaces can be found in the work by Zhang et al. [52]. They compared the CO oxidation on Pt (being the most active CO catalyst) and Ru (very poor catalyst at low O2 and CO pressures, active only at high O2 partial pressures) assuming Langmuir–Hinshelwood mechanism. They concluded that the lower activation energy of the CO oxidation on Pt(111) (1 eV) compared to Ru(0001) (1.4 eV) can be correlated with the lower chemisorption energy of atomic oxygen (Oads) and CO. This means that at the transition state CO and Oads should come closer to each other on Ru(001) than on Pt(111), that hinders the kinetics of the CO oxidation at low oxidizing reaction conditions. However, as shown by Over and Muhler [53] Ru is superior to Pt, Rh and Pd at strongly oxidizing conditions due to the transformation of Ru into RuO2 under experimental conditions. Experimental measurements and DFT calculations suggested that in this case the crucial step was the recombination of adsorbed CO molecules with bridging O atoms from the oxide surface, with the activation energy of only 0.6 eV. Upon CO2 desorption, the surface gets regenerated when exposed to oxygen that heals the vacancy on the RuO2 surface. Gong et al. [54] offered a systematic DFT study of the CO oxidation on Ru(0001), Rh(111), Pd(111), Os(0001), Ir(111), Pt(111), and their corresponding metal oxides. As a general conclusion, it was outlined that (i) metal oxides are in general superior CO oxidation catalysts compared to their parental metals and (ii) reaction barrier increases as the chemisorption energy of the initial state (COads + Oads) increases, irrespective of the nature of the catalyst. The authors claimed that the geometric effects make oxides more active than metals, while they found that the barrier for the reaction increases as the OC–O bond distance in the transition state decreases (Fig. 5) [54]. According to the authors, the relation between the reaction barrier and the geometry of the transition state is due to the energy costs necessary to achieve the transition state and involve the destabilization of surface–COads and surface–Oads bond. If the two surface bonds are strong, COads and Oads have to be brought close together for the reaction to take place, which includes large energy requirements and raises the reaction barrier.

Reaction barrier as a function of the OC–O distance in the TS. The solid line is a first order exponential decay curve with R2 being 0.96. Reprinted with permission from [54]. Copyright (2014) American Chemical Society
More recently, Jiang et al. [55] used the DFT calculations to show the dependence of the CO oxidation activity of (111) close-packed surfaces, (211) stepped surfaces, (532) kinked surfaces, 55 atom cuboctahedral clusters, and 12 atom cluster on the coordination number of atoms at the active sites (Fig. 6). Using a microkinetic model, which assumes Langmuir–Hinshelwood mechanism, the CO oxidation activity is found to be a quasicontinuous function of the openness of the surface. As the main effect behind the structure variation, the strength of the adsorbate–metal bond at different structures was suggested, and this effect was ascribed to the variations in the electronic structure of the investigated systems [55]. This specific study suggests possible directions of the search for new CO oxidation catalysts connecting the electronic structure and surface morphology with the macroscopic kinetic behavior of the catalyst.

Contour plot of the Sabatier activity A S = k B T ln(r S/k B Th −1) in eV at (top) low temperature (T = 273, pO2 = 0.21 bar, pCO = 0.01 bar) and (bottom) high temperature (T = 600 K, pO2 = 0.33 bar, pCO = 0.67 bar), as a function of the CO and O adsorption energies on the a (111) surfaces (filled circle, black), b (211) surfaces (filled square, red), c (532) surfaces (filled diamond, green), and d M12 clusters (filled triangle, blue). The values for several elemental metals are shown. The activity is calculated under typical experimental conditions for gold nanoparticles. Reprinted with permission from [55]. Copyright (2009) American Chemical Society. For the detailed description of the microkinetic model and the activity estimate procedure, the reader is referred to the original work, Ref. [55]. (Color figure online)
CO oxidation on bulk oxides
Direct CO oxidation is catalyzed by some oxide materials able to release a lattice O atom to form CO2. In analogy to oxidized transition metal surfaces, the mechanism of this process is of Mars-van Krevelen type, as confirmed for the Co3O4 (110) surface by means of DFT calculations [56]. The authors outlined the importance of the effective electron sink which picks up electrons upon CO2 formation, before the surface gets re-oxidized by O2. For this particular material, the electron sink is the reaction Co3+ + e− → Co2+ which is essential for the O abstraction from the catalyst surface.
Another material that possesses a rather high CO oxidation activity, is CeO2. Some dopants may affect the CO oxidation activity by influencing the formation energy of oxygen vacancy, as shown by DFT calculations [30, 57–59]. The later quantity affects the step of the deduction of an O atom from the lattice needed for the formation of CO2 via Mars-van Klevelen mechanism, and subsequent healing of the formed vacancy by exposure to gaseous O2. Hence, the formation energy of an oxygen vacancy, which can be evaluated using the electronic structure theory calculations, can be used as a catalytic descriptor for the CO oxidation reaction on this type of materials, as suggested by Aryanpour et al. [30]. According to these authors, the activity of doped CeO2 towards the direct CO oxidation increases as oxygen formation energy decreases, but according to Shapovalov and Metiu [57], the formation energy of the vacancy should be “proper” in value. This means that the formation energy of an oxygen vacancy should be low enough to enable the release of lattice O to form CO2, but high enough to make the vacancy annihilation due to O2 possible. In other words, by tuning the formation energy of oxygen vacancy by doping CeO2, one can optimize the catalyst to maximize the activity while preserving the catalyst stability. The structural sensitivity of CeO2 towards CO oxidation was investigated showing that the lattice oxygen of the (110) surface is more reactive than that of the (111) surface [60]. For the same CeO2 surface terminations, it was shown that the activities of lattice O atoms can be reduced due to the presence of the neighboring O vacancy. The origin of this effect is the presence of the localized 4f orbitals of Ce, as the vacancy-induced electron localization hinders any further vacancy formation necessary for the CO oxidation on this material [61]. This reinforces the conclusion regarding the necessity of an effective regeneration mechanism of the CeO2 surface to have effective CO oxidation catalyst.
Oxide-supported metal clusters
In practice, by dispersing the active catalyst component over a suitably chosen support one enhances its active surface area and the number of active sites that results in an increased catalytic activity. Sometimes the support itself can contribute to the catalytic activity and, in such cases, the interface between the support and the catalyst particle becomes crucially important. If the support contributes to the catalytic activity, it is called active. A detailed DFT calculation of the CO oxidation on gold surfaces, performed at the GGA-PBE level by Liu et al. [62] suggested that the oxidation of CO on Au nanoparticles dispersed over an inactive support, occurs on Au steps via a two-stage mechanism: CO + O2 → CO2 + O, followed by CO + O→CO2. In a case of an active support, the CO oxidation also follows the two-step mechanism with reactions occurring at the interface (Fig. 7).

Direct CO oxidation mechanism on Au supported by inactive support (oxide, i) and active support (oxide, ii). Reprinted (adapted) with permission from [62]. Copyright (2002) American Chemical Society
The already mentioned oxide-supported nanodispersed Au, exceptionally active towards the direct CO oxidation [63, 64], is the best known member of this family of the CO oxidation catalysts. Although the macroscopic gold is noble and not susceptible to oxidation, nanosized Au is a highly reactive and very potent catalyst. To observe this effect the reader is referred to Fig. 6 (the first row): when moving from the densely packed (111) surface to the 12-atoms cluster, Au evolves from inactive to the most active material towards CO oxidation. However, the size of the supported Au cluster should be large enough to enable the co-adsorption of reactants (CO and O2). DFT calculations suggested that a monatomic dispersion of Au and Au dimers supported by MgO should not be active towards the CO oxidation, while a catalytic effect is expected to set in starting with MgO-supported trimers [14]. Namely, Au atoms supported by MgO cannot co-adsorb CO and O2, while the dimer gets blocked by CO as the binding is too strong. It is also interesting to consider the effects of dimensionality of supported Au clusters on the adsorption of CO and O2. The DFT calculations performed by Amft et al. [65] suggested that a flat and a three-dimensional Au13 clusters supported by MgO equally bind CO, but O2 binds significantly only to the three-dimensional Au13 isomer. Although the properties of the support material are important [66], we should notice that non-supported Au clusters are also active towards the CO oxidation [67], starting with the negatively charged Au dimer [68]. For an isolated Au10 cluster, Lopez and Nørskov [67] suggested that its reactivity is associated with the special reaction geometries available at small particle sizes, combined with an enhanced ability of low coordinated gold atoms to interact with molecules from their surroundings. When considering supported Au, Lopez et al. [69] claim that the most important effect, determining the CO oxidation activity of small supported Au clusters, is the availability of many low-coordinated gold atoms, while the effects of the support are considered to play less important role. When a very small particle interacts with the support, practically every atom of the particle has its own individuality and certain sites at the particle surface can be highly catalytically active. Molina and Hammer [70] suggested that most reactive sites at Au/MgO appeared where gold shelters the MgO support, in this way creating a cavity where several low-coordinated Au atoms and Mg2+ cations from the substrate can interact simultaneously with an adsorbate. A more recent work by the same authors [71] suggested that irrespective of the Au particle size and morphology, the CO oxidation reaction proceeds via the CO adsorption and subsequent O2 trapping, which results in a metastable CO·O2 reaction intermediate. The complex CO·O2 intermediate then dissociates into CO2 and adsorbed atomic oxygen. The atomic oxygen can react directly with gaseous CO to produce CO2. This mechanism agrees with the one proposed by Liu et al. [62]. Recently, Stamatakis et al. [72] studied Au/MgO catalyst poisoning using first-principle kinetic Monte Carlo simulations and showed that the catalyst performance depended critically on the presence of vacancies in the MgO support. As a model system they employed a MgO-supported Au6 cluster. It was shown that when the support had a low amount of defects or Mg vacancies, the catalyst got poisoned by CO, as O2 did not bind to the clusters. If Au6 interacts with O vacancy, a catalyst deactivation takes place through the formation of carbonate.
Other oxide supports, such as TiO2 [73, 74] and CeO2 [75], have also been investigated in much detail. In contrast to MgO, these supports are (more) active ones and they contribute significantly to the reaction kinetics. For TiO2, DFT calculations suggested that CO oxidation occurred at the interface between Au and TiO2 with very small reaction barrier, while the O2 adsorption at Au/TiO2 interface was the key step in the overall reaction [73]. It was suggested that TiO2 promotes CO oxidation through an enhanced electron transfer from Au to the antibonding states of O2, which induced an ionic bonding between adsorbed O2, Au, and Ti cation, and activated O2 towards CO oxidation [73]. In the case of CeO2, the activity of Au/CeO2 depends on the surface termination of the support. Song and Hensen modeled Au nanorods supported on CeO2(110) and CeO2(111) surfaces using DFT calculations, showing that Mars-van Krevelen mechanism (Fig. 4c) was consistent with the experiments regarding the involvement of lattice O [75]. They suggested that Au/CeO2(111) should be active towards CO oxidation only if the support is defective, which is connected with the high formation energy of O vacancy on CeO2(111). The authors predicted also the deactivation of Au/(defective CeO2(111)) catalyst under the oxidizing conditions, due to the healing of surface vacancies [75].
There were also reports, based on the first principle calculations, saying that the presence of water can enhance the CO oxidation rate on oxide supported Au clusters. For the case of the Au8 cluster supported by MgO it was suggested that the complex formed upon H2O adsorption opened a catalytic reaction pathway towards CO oxidation [76]. This behavior seems to be affected by the particle size as more recent calculations for the Au1–4/MgO(001) systems did not reveal any beneficial role of water towards CO oxidation [77]. In the case of a more reactive TiO2 support, DFT calculations suggested that water could dissociate on TiO2, and the presence of OH formed on the support enhanced the O2 adsorption [74]. It is suggested that this effect is due to the charge transfer from TiO2 to O2 in the presence of adsorbed OH, while O2 adsorption energy is correlated to the charge transfer in a linear manner [74].
Electrocatalysis of hydrogen and oxygen cathode reactions
In contrast to heterogeneous catalytic reactions taking place at the solid/gas interface, the electrochemical interface is much more complicated. To treat it explicitly, one has to take into account the presence of the solvents, as well as an additional parameter—the electrode potential, being the main driving force of any electrochemical process. At the electrochemical interface, the electric field is enormous, reaching up to 109 V m−1. This could present a tremendous problem for the first principle modeling of electrochemical reactions, as there is no proper scheme to account for the electrode potential [78]. However, it appears that the treatment of the electrochemical reactions from the aspect of the electrocatalysis does not require any inclusion of the electrode potential, while the presence of a solvent is usually disregarded, or sometimes included implicitly through the correction of the adsorption energetics of reaction intermediates [79]. In this way, electrocatalytic reactions could be treated theoretically as reactions at solid/gas interface.
Here we proceed with some specific examples of theoretical studies in the search for new catalysts for HER and oxygen reduction reaction (ORR). The first one is particularly important for H2 production via electrochemical water splitting, while the second one is the cathode reaction in fuel cells. ORR is of particular interest, as there is no catalyst which can provide reversible ORR kinetics, and a large overvoltage at the fuel cell cathode is the main source of energy losses in this energy conversion system.
Hydrogen evolution reaction (HER)
The hydrogen evolution reaction is one of the most important electrochemical reactions and it proceeds through apparently simple mechanism. In acidic solutions the mechanism is described as:
-
i. \({\text{H}}^{ + } +^{*}+ e^{ - } \to {\text{H}}_{\text{ads}} \, \left( {\text{Volmer step}} \right)\)
-
iia. \({\text{H}}_{\text{ads}} + {\text{H}}_{\text{ads}} \to {\text{H}}_{ 2} + 2^{*}\,\left( {\text{Tafel step}} \right)\)
-
iib. \({\text{H}}_{\text{ads}} + {\text{H}}^{ + } + e^{ - } \to {\text{H}}_{ 2} +^{*}\left( {\text{Heyrovsky step}} \right)\)
In the mechanism given above, the star (*) represents the adsorption site. The rationalization of HER activity in terms of volcano curve [20] (Fig. 3) appears intuitive but it has been criticized. The criticism is based on the fact that there are different states of the electrocatalyst surface around the potential of hydrogen evolution: some metals are covered by oxide (W, Mo, etc.), some are covered by underpotential deposited H (HUPD, strongly bound hydrogen; metals behaving this way are Pt, Pd, Ir, etc.), while some are reduced, i.e., in metallic state [80]. The variations in the behavior of different catalysts under the HER conditions and an elusive nature of the real reaction intermediates for HER (usually called weakly bonded H or overpotential deposited H, HOPD [3]) did not stop the research community in using H adsorption energies as a catalytic activity descriptor. A general prescription in this search for new HER electrocatalysts is deduced form the volcano-curve: H binding strength should be optimal, which translates into the condition that ΔG for the H adsorption should be around 0.
Considering H adsorption energetics on transition metal surfaces, obtained by DFT calculations on GGA-PBE level, Nørskov et al. [44] provided a microkinetic model for HER, reinforcing the conclusion that ΔG = 0 for hydrogen adsorption, calculated with respect to H2 molecule in the gas phase, splits volcano curve into two branches with Pt displaying the highest activity. Although Pt has the highest catalytic activity out of all monometallic systems it is not found at the very apex of the volcano curve (Fig. 3), but is very close to it. This gives a possibility to find more active HER catalysts, which could bring an activity improvement of up to the factor of 5 as compared to Pt. Alternatively, due to the high price of Pt, one can search for new Pt-free or low-Pt content catalysts, which optimize the price-to-performance ratio.
The work of Greeley et al. [45] demonstrated an extensive DFT-based high-throughput screening of more than 700 surface alloys and overlayer structures comprised by the combination of 16 transition and p-metals. With the main search criterion reducing to |ΔGH| → 0, possible candidates were also checked for undesirable segregation processes and dissolution under the HER conditions. The search indicated that there were three most stable and active materials: Pt (as expected), and BiPt on Pt and RhRe on Re surface alloys. The PtBi surface alloy was then prepared experimentally upon annealing of Pt with irreversible adsorbed Bi (Pt–Biir), obtained by Bi underpotential deposition, and confirmed to have a higher HER activity than pure Pt (Fig. 8).

Voltammograms for the HER after each stage of Bi–Pt surface alloy synthesis. 1 Pt film 2 immediately after Bi UPD 3 after second anneal to form the BiPt surface alloy. The inset represents a control sample—Pt film without Bi after first and second annealings. Reproduced with permission from Ref. [45] ©2006 Nature Publishing Group
The number of DFT calculations presented in ref [45] is tremendous, so Björketun et al. [81] have used empirical and semi-empirical filters to reduce the number of systems requiring explicit DFT treatment (Fig. 9).

Flowchart illustrating the systematic search for affordable, binary surface alloys and/or overlayers active towards HER. Starting from a large set of metals (0), viable metals are selected by filtering with the ‘‘corrosion’’ (1) and ‘‘cost’’ (2) filters. From the remaining set of metals, potentially stable and active substrate-overlayer combinations are then selected, based on estimated segregation energies, giving thermodynamically stable substrate-overlayer combinations (3) and trends in d-band shifts for pseudomorphically deposited metal overlayers (4). Finally, the activities and stabilities of the most promising surface alloys are evaluated by explicit DFT calculations (5, 6). Reproduced from Ref. [81] with permission of The Royal Society of Chemistry
Applied filters reduced the number of possible candidates from more than 1,500–36. Then DFT calculations qualified 20 candidates for HER catalysts. The authors have chosen “Cu-overlayer–W-substrate” system to be produced experimentally. Though faced with significant difficulties during the production of the catalyst, the improved HER activity of the Cu-W system, as compared to pure W and Cu, was confirmed. In contrast to the work of Greeley et al. [45], this catalyst does not involve Pt and its activity is far from that of Pt, but the price should also be considered before making the final judgment.
In recent years, transition metal carbides (TMC), particularly WC, have become very interesting from the point of view of electrocatalysis [82–85]. It was suggested that WC has a Pt-like behavior and that the deposition of Pt onto the WC surface should not induce large ligand and strain effects [83]. This led to the conclusion that Pt monolayers deposited onto WC (PtML/WC) should behave just as massive Pt in terms of their HER activity. Esposito et al. [84] calculated the hydrogen binding energies for Pt(111), WC(0001) and PtML/WC(0001) surfaces using the DFT approach, and predicted quite similar behavior for Pt(111) and PtML/WC surfaces, although the actual HER activity of the monolayer was slightly lower than that of pure massive Pt [84]. It was reported that metal monolayer/TMC substrate systems obey a commonly observed volcano relationship between their HER activity and hydrogen binding energy [82]. The recently published DFT calculations for Pt and Pd mono- and bilayers on WC [86] suggested that, in fact, the electronic structure of a Pt monolayer on WC(001), compared to clean Pt(111) surface (Fig. 10), was significantly altered, predominantly due to the ligand effect [86]. Moving into the film, the ligand effect vanished quickly, already in the second Pt layer. The calculated hydrogen binding energies and the measured HER exchange current densities, collected from different literature sources, were then used to obtain a volcano-type curve, with Pt displaying a higher HER activity than the other systems considered in the study. It was also suggested that the improvement of HER activity for WC supported nanosized Pt did not have an electronic origin but is rather due to H spillover onto a partially oxidized WC support [86].

PDOS of Pt atoms in WC-supported mono- and bilayers. The d-band centers are indicated by thick vertical lines, while exact numbers are given within the layers. Energy scales are referred to Fermi level. The term “expanded” is used to denote that Pt(111) single crystal surface is artificially laterally expanded in such a way that their lattice constants fit the one of WC(0001) surface. Adapted from [86]
The same class of HER electrocatalysts was investigated in Ref. [87] where the activity of new metal overlayer/WC support systems was estimated. The estimation was based on the volcano curve constructed using the hydrogen binding energies to the metallic surface and overlayers for which the HER exchange current densities were known. Two metal overlayers on WC(0001), RhML/WC(0001) and CuML/WC(0001), were estimated to have HER activity as high as platinum (and higher than PtML/WC) (Fig. 11). These systems were also predicted to be stable under the HER conditions.

Volcano curve correlating hydrogen–metal binding energy (E H–M) with corresponding experimentally determined HER activities of studied surface (expressed by log j 0, j 0 in A cm−2) consolidating clean metal surfaces (open circle) and WC-based HER electrocatalysts (open square). Predictions made for CuML/WC(0001) and RhML/WC(0001) are indicated by diamonds (filled diamond). Error bars indicate the scattering of experimentally determined log j 0 values. Reproduced from [87]
For these two systems, the experimental verification of the predicted HER activity was not performed yet, although one of them, RhML/WC(0001), was prepared previously [88]. However, it is interesting to recall that the study of Björketun et al. [81] also pointed to a beneficial W-Cu interaction for achieving HER catalytic effects, although W and Cu separately are exceptionally poor HER catalysts. To see whether this is something realistic and promising or just a “false alarm”, more detailed work is needed.
Oxygen reduction reaction (ORR)
The oxygen reduction reaction is characterized by very sluggish kinetics for all known materials. In acidic solutions, the complete reduction of O2 to H2O is described by the overall reaction:
with standard electrode potential of 1.23 V. For the best known electrocatalysts, the overpotential of at least 0.15 V is required for ORR to proceed at a measurable rate. Depending on the nature of the electrode material, ORR takes different pathways employing 2 or 4 electrons to reduce O2 to hydrogen peroxide or water. The 4-electron pathway can have different mechanisms, known as dissociative and associative. The best known elemental catalyst for ORR is platinum [3]. For a detailed description of ORR related to fuel cell applications the reader is referred to the work of Markovic and Ross [3].
As oxygen is a very strong oxidizing agent, ORR commences at rather high potentials, where most of the metals are at least partially covered by oxide layer or adsorbed OH, formed due to water discharge under the ORR conditions. Although ORR has been investigated for decades, the main breakthrough occurred just recently [89–91]. This became possible due to a concerted advancement in single crystal preparation (so that proper model systems can be realized experimentally), surface science techniques (so that model systems can be characterized properly) and electronic structure theory calculations (so that materials properties and reaction mechanisms can be evaluated and connected with experimental measurements). The work was devoted to Pt alloyed with 3d transition metals [89–91], showing that the ORR activity was determined by (i) the surface coverage of spectator species (adsorbed OH) and (ii) adsorption energetics of reaction intermediates (which determines reaction barriers). Both factors are affected by the electronic structure of the surface layer and its behavior captured by the d-band model [11, 23] and scaling relationships [28]. It should be noted that ORR in acidic medium is not only complex from the aspect of its mechanism; its complexity is raised further due to the possible changes of the catalyst surface structure, especially under harsh ORR conditions (high acidity, high anodic potentials). Some to the possible processes are surface segregation and metal dissolution which can also be treated theoretically [92, 93]. However, this only gives additional task for the computations to consider surface models which account for these processes, where expected, while the general strategy remains the same.
The work of Nørskov et al. [43] provided a simple approach to treat ORR theoretically, enabling the rationalization of ORR activity trends for a series of transition metals. The authors developed a microkinetic model, which used the input parameters obtained from DFT calculations while the electrode potential was included implicitly. As a reaction barrier, the authors used the largest reaction free energy among different steps included in the considered ORR mechanisms, which usually turned out to be electron/proton-transfer to adsorbed O or OH. This approach made it possible to construct an activity map that connects the ORR activity with O adsorption energy and OH adsorption energy (Fig. 12). As O and OH adsorption energies scale mutually [28], it turns out that both O and OH adsorption energies can be used as ORR activity descriptors. As shown by Calle-Vallejo et al. [94], this leads to a general prescription in the search for new ORR electrocatalysts using a descriptor based approach: the new catalyst should bind OH, by ~0.1 eV, and/or O, by ~0.2 eV, weaker than platinum. In terms of the electronic structure of Pt-based catalysts, the d-band model suggests that ORR electrocatalysts should have their d-band center positioned at lower values, compared to the one of Pt. In addition, Wang and Balbuena [95] have investigated systematically the interaction of single transition metal atoms and metal trimers with ORR reaction intermediates using DFT calculations. This approach is, strictly speaking, related to a gas phase reaction of small metal clusters, so the reactivity cannot be linked to the d-band structure (as there are no electronic bands). However, quite general conclusions have been derived, pointing how different ORR elementary steps depend on the metal electronic structure and showing that efficient ORR catalysis requires the formation of bimetallic catalysts combining metals with vacant and occupied d orbitals. The first one enhances the kinetics of the rate determining step, while the second one improves the removal of adsorbed O and OH [95].

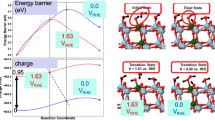
Relying on the descriptor-based search, new materials have been identified. Following the general scheme for the estimation of the ORR activity [43], the Pt3Sc and Pt3Y alloy catalyst have been singled out as possible ORR catalysts by Greeley et al. [96], and the predictions obtained by calculation have been confirmed experimentally. As suggested by the calculated free energy profile for ORR on Pt, Pt3Sc and Pt3Y (Fig. 13), the modification of the electronic structure of Pt upon alloying result in the change of the rate determining step. On Pt3Sc and Pt3Y OH removal is not the rate determining step but the first electron/proton-transfer to O2 is. Compared to pure Pt used as a benchmark catalyst, the activity of polycrystalline Pt3Sc and Pt3Y electrodes is improved by a factor of 1.5–1.8 and 6–10, respectively, in the potential range 0.9–0.87 V.

Calculated free energy diagram for the oxygen reduction reaction at 0.9 V with respect to the reversible hydrogen electrode (RHE) under standard conditions for Pt(111) (solid line) and for Pt overlayers on the Pt3Sc(111) and Pt3Y(111) surfaces (dashed). The free energy changes for the formation of OOH (ΔG 1) and the removal of OH (ΔG 2) are indicated. Reproduced from [96] © 2009 Nature Publishing Group
Thus, the descriptor-based approach allows one to screen many possible candidates, some of them being quite unexpected from the point of view of the traditional approach. In particular, its application led to the discovery of Pt5La [97] and Pt5Gd ORR catalysts [98]. Both materials show significantly improved ORR kinetics compared to pure Pt, which is explained by the dissolution of La and Gd, resulting in a Pt-enriched active surface layer. Due to a compressive strain effect, the Pt d-bands get stabilized, which results in weaker bonding of ORR intermediates, and thus enhanced ORR kinetics.
Similarly, the alloys of Pt with p-metals were not traditionally viewed as ORR catalysts. However, it was recently reported that the incorporation of In into Pt improved the ORR kinetics in alkaline media [99]. This combination of metals was selected on the basis of the electronic structure calculations for a series of Pt surface alloys [100]. More detailed DFT calculations on PtIn surface, subsurface and bulk alloys confirmed that the incorporation of In into Pt reduced the adsorption energies of O atoms, which was beneficial for ORR kinetics. Hence, a new ORR catalyst with the nominal composition denoted as Pt9In was produced, and its activity toward ORR was found to be improved by a factor of 2.5, as compared to pure Pt. The experimental results were found to agree with the predictions made by DFT calculations on the basis of O adsorption energies (Fig. 14).

The ORR volcano-curve according to the theoretical (dashed line) and experimental results in Pt3M (M = Ti, Fe, Co and Ni) systems compiled from Ref. [91]. Activity was estimated at 0.9 V versus RHE. For the analyzed Pt–In model systems presented by ball models, the calculated oxygen adsorption energies are assigned by vertical arrows. Their positions, relying to the presented experimental volcano plot, allow expecting the activity enhancement factor for ORR to range 1.5–3.5 (within the grey rectangle). Reproduced from Ref. [99] ©2013 with permission from Elsevier
Summary and outlook
Recent developments of experimental and computational techniques in surface science and catalysis enabled us to understand the basic principles of catalysts operation. However, this also imposes a new task to understand how to design catalyst structures to control their catalytic activity and selectivity. This work was intended to demonstrate what kind of information we can obtain from the electronic structure calculations and how this approach can help in designing new materials. At this point, we are able to obtain an atomic-level insight into different catalytic processes, in some cases we are also able to search for new catalysts, guided by general principles and approximate models, which reduce the dimensionality of the problem. In this sense, the descriptor concept has proven its value. Further advancement of the electronic structure theory as well as the creation of large materials properties databases, along with the algorithms enabling us to search through them, will contribute to the efficient search for new catalysts. As the catalytic activity essentially reflects the electronic structure of the catalyst, fundamental studies should be directed to the rationalization of structure–activity relationships, that is, to go beyond correlations. It must not be disregarded that the complete understanding requires other computational approaches to assess different temporal and spatial domains of catalytic reactions.
At the end, we note that for designing new catalytic structures we have a limited number of building blocks. When noble gases and radioactive elements are removed from the periodic table of elements there are approximately 70 elements left to be combined in the ways Nature allows us to. This leaves us with still a huge number of possible combinations to search through, imposing the need for solid and rational underlying principles to guide our quest.
References
Maxwell IE (1996) Stud Surf Sci Catal 101:1–9
Nørskov JK, Abild-Pedersena F, Studt F, Bligaard T (2011) Proc Natl Acad Sci 118:937–943
Marković NM, Ross PN (2002) Progr Surf Sci 45:117–229
Sabatier P (1911) Ber Deutch Chem Soc 44:1984–2001
Brønsted JN (1928) Chem Rev 5:231–338
Evans MG, Polanyi NP (1938) Trans Faraday Soc 34:11–24
Bligaard T, Nørskov JK, Dahl S, Matthiesen J, Christensen CH, Sehested J (2004) J Catal 224:206–217
Opportunities for Catalysis in the 21st Century, Basic Energy Sciences Advisory Committee Subpanel Workshop Report (2002). http://science.energy.gov/~/media/bes/besac/pdf/Catalysis_report.pdf. Accesses 15 Sept 2014
Putanov P (2010) New breakthroughs in catalysis and the quiet scientific and technological revolution. In: Putanov P (ed) Catalysis in scientific and educational programs and in public development of Serbia. Serbian Academy of Sciences and Arts, Branch in Novi Sad, Novi Sad pp 13–25
Pettersson LGM, Nilsson A (2014) Top Catal 57:2–13
Hammer B, Nørskov JK (1995) Nature 376:238–240
Nilsson A, Pettersson LGM (2008) In: Nilsson A, Pettersson LGM, Nørskov JK (eds) Chemical bonding at surfaces and interfaces. Elsevier, Amsterdam
Pasti IA, Baljozović M, Skorodumova NV (2014) Surf Sci. doi:10.1016/j.susc.2014.09.012
Amft M, Skorodumova NV (2010) Phys Rev B 81:195443
Yoon B, Häkkinen H, Landman U, Wörz AS, Antonietti JM, Abbet S, Judai K, Heiz U (2005) Science 307:403–407
Frusteri F, Freni S, Spadaro L, Chiodo V, Bonura G, Donato S, Cavallaro S (2004) Catal Commun 5:611–615
Frusteri F, Freni S, Chiodo V, Spadaro L, Di Blasi O, Bonura G, Cavallaro S (2004) Appl Catal A Gen 270:1–7
Hansen EW, Neurock M (2000) J Catal 196:241–252
Linic S, Barteau MA (2003) J Catal 214:200–212
Trasatti S (1972) J Electroanal Chem 39:163–184
Jerkiewicz G (1998) Prog Surf Sci 57:137–186
Quaino P, Juarez F, Santos E, Schmickler W (2014) Beilstein J Nanotechnol 5:846–854
Hammer B, Nørskov JK (1995) Surf Sci 343:211–220
Mavrikakis M, Hammer B, Nørskov JK (1998) Phys Rev Lett 81:2819–2822
Pallassana V, Neurock M, Hansen LB, Nørskov JK (2000) J Chem Phys 112:5435–5439
Tang H, Trout BL (2005) J Phys Chem B 109:17630–17634
Pašti IA, Gavrilov NM, Mentus SV (2014) Electrochim Acta 130:453–463
Abild-Pedersen F, Greeley J, Studt F, Rossmeisl J, Munter TR, Moses PG, Skúlason E, Bligaard T, Nørskov JK (2007) Phys Rev Lett 99:016105
Mom RV, Cheng J, Koper MTM, Sprik M (2014) J Phys Chem C 118:4095–4102
Aryanpour M, Khetan A, Pitsch H (2013) ACS Catal 3:1253–1262
Fajin JLC, Cordeiro M, Illas F, Gomes JRB (2010) J Catal 276:92–100
Vojvodic A, Hellman A, Ruberto C, Lundqvist BI (2009) Phys Rev Lett 103:146103
Cheng J, Hu P (2008) J Am Chem Soc 130:10868–10869
Andersson MP, Bligaard T, Kustov A, Larsen KE, Greeley J, Johannessen T, Christensen CH, Nørskov JK (2006) J Catal 239:501–506
Studt F, Abild-Pedersen F, Bligaard T, Sørensen RZ, Christensen CH, Nørskov JK (2008) Science 320:1320–1322
Jacobsen CJH, Dahl S, Clausen BS, Bahn S, Logadottir A, Nørskov JK (2001) J Am Chem Soc 123:8404–8405
Grabow LS, Studt F, Abild-Pedersen F, Petzold V, Kleis J, Bligaard T, Nørskov JK (2011) Angew Chem Int Ed 50:4601–4605
Sauer J (2008) In: Morokuma K, Musaev DG (eds) Computational modeling for homogeneous and enzymatic catalysis. Wiley, Weinheim
Sauer J, Döbler J (2004) Dalton Trans 19:3116–3121
Suntivich J, May KJ, Gasteiger HA, Goodenough JB, Shao-Horn Y (2011) Science 334:1383–1385
Vojvodic A, Nørskov JK (2011) Science 334:1355–1356
Suntivich J, Gasteiger HA, Yabuuchi N, Nakanishi H, Goodenough JB, Shao-Horn Y (2011) Nat Chem 3:546–550
Nørskov JK, Rossmeisl J, Logadottir A, Lindqvist L, Kitchin JR, Bligaard T, Jónsson H (2004) J Phys Chem B 108:17886–17892
Nørskov JK, Bligaard T, Logadottir A, Kitchin JR, Chen JG, Pandelov S, Stimming U (2005) J Electrochem Soc 152:J23–J26
Greeley J, Jaramillo TF, Bonde J, Chorkendorff I, Norskov JK (2005) Nat Mater 5:909–913
Lee YL, Kleis J, Rossmeisl J, Shao-Horn Y, Morgan D (2011) Energy Environ Sci 4:3966–3970
Engel T, Ertl G (1979) Adv Catal 28:1–78
Eley DD, Rideal EK (1946) Nature 146:401–402
Stampfl C, Scheffler M (1997) Phys Rev Lett 78:1500–1503
Hendriksen BLM, Frenken JWM (2002) Phys Rev Lett 89:046101
Hendriksen BLM, Bobaru SC, Frenken JWM (2004) Surf Sci 552:229–242
Zhang C, Hu P, Alavi A (1999) J Am Chem Soc 121:7931–7932
Over H, Muhler M (2003) Prog Surf Sci 72:3–17
Gong XQ, Liu ZP, Raval R, Hu P (2004) J Am Chem Soc 126:8–9
Jiang T, Mowbray DJ, Dobrin S, Falsig H, Hvolbæk B, Bligaard T, Nørskov JK (2009) J Phys Chem C 113:10548–10553
Broqvist P, Panas I, Persson H (2002) J Catal 210:198–206
Shapovalov V, Metiu H (2007) J Catal 245:205–214
Andersson DA, Simak SI, Skorodumova NV, Abrikosov IA, Johansson B (2007) Phys Rev B 76:174119
Andersson DA, Simak SI, Skorodumova NV, Abrikosov IA, Johansson B (2007) Appl Phys Lett 90:031909
Chen F, Liu D, Zhang J, Hu P, Gong XQ, Lu G (2012) Phys Chem Chem Phys 14:16573–16580
Song YL, Yin LL, Zhang J, Hu P, Gong XQ, Lu G (2013) Surf Sci 618:140–147
Liu ZP, Hu P, Alavi A (2002) J Am Chem Soc 124:14770–14779
Haruta M, Kobayashi T, Sano H, Yamada N (1987) Chem Lett 16:405–408
Haruta M (1997) Catal Today 36:153–166
Amft M, Johansson B, Skorodumova NV (2012) J Chem Phys 136:024312
Kung MC, Davis RJ, Kung HH (2007) J Phys Chem C 111:11767–11775
Lopez N, Nørskov JK (2002) J Am Chem Soc 124:11262–11263
Häkkinen H, Landman U (2001) J Am Chem Soc 123:9704–9705
Lopez N, Janssens TVW, Clausen BS, Xu Y, Mavrikakis M, Bligaard T, Nørskov JK (2004) J Catal 223:232–235
Molina LM, Hammer B (2003) Phys Rev Lett 90:206102
Molina LM, Hammer B (2004) Phys Rev B 69:155424
Stamatakis M, Christiansen MA, Vlachos DG, Mpourmpakis G (2012) Nano Lett 12:3621–3626
Liu ZP, Gong XQ, Kohanoff J, Sanchez C, Hu P (2003) Phys Rev Lett 91:266102
Liu LM, McAllister B, Ye HQ, Hu P (2006) J Am Chem Soc 128:4017–4022
Song W, Hensen EJM (2013) Catal Sci Technol 3:3020–3029
Bongiorno A, Landman U (2005) Phys Rev Lett 95:106102
Amft M, Skorodumova NV (2001) arXiv:1108.4669v1 [cond-mat.mes-hall]
Santos E, Quaino P, Schmickler W (2012) Phys Chem Chem Phys 14:11224–11233
Greeley J, Rossmeisl J, Hellmann A, Nørskov JK (2009) Z Phys Chem 221:1209–1220
Conway BE (1995) Prog Surf Sci 49:331–452
Björketun ME, Bondarenko AS, Abrams BL, Chorkendorff I, Rossmeisl J (2010) Phys Chem Chem Phys 12:10536–10541
Esposito DV, Hunt ST, Kimmel YC, Chen JG (2012) J Am Chem Soc 134:3025–3033
Esposito DV, Chen JG (2011) Energy Environ Sci 4:3900–3912
Esposito DV, Hunt ST, Stottlemyer AL, Dobson KD, McCandless BE, Birkmire RW, Chen JG (2010) Angew Chem Int Ed 49:9859–9862
Nikolic VM, Perovic IM, Gavrilov NM, Pašti IA, Saponjic AB, Vulic PJ, Karic SD, Babic BM, Marceta Kaninski MP (2014) Int J Hydrogen Energy 39:11175–11185
Vasić DD, Pašti IA, Mentus SV (2013) Int J Hydrog Energy 38:5009–5018
Vasić Anićijević DD, Nikolić VM, Marčeta-Kaninski MP, Pašti IA (2013) Int J Hydrog Energy 38:16071–16079
Kelly TG, Stottlemyer AL, Ren H, Chen JG (2011) J Phys Chem C 115:6644–6650
Stamenkovic VR, Fowler B, Mun BS, Wang G, Ross PN, Lucas CA, Marković NM (2007) Science 315:493–497
Stamenkovic VR, Mun BS, Arenz M, Mayrhofer KJJ, Lucas CA, Wang G, Ross PN, Markovic NM (2007) Nat Mater 6:241–247
Stamenković V, Mun BS, Mayrhofer KJ, Ross PN, Marković NM, Rossmeisl J, Greeley J, Nørskov JK (2006) Angew Chem Int Ed 45:2897–2901
Ramírez-Caballero GE, Ma Y, Callejas-Tovara R, Balbuena PB (2010) Phys Chem Chem Phys 12:2209–2218
Balbuena PB, Callejas-Tovar R, Hirunsit P, Martínez de la Hoz JM, Ma Y, Ramírez-Caballero GE (2012) Top Catal 55:332–335
Calle-Vallejo F, Koper MTM, Bandarenka AS (2013) Chem Soc Rev 42:5210–5230
Wang Y, Balbuena PB (2005) J Phys Chem B 109:18902-18706
Greeley J, Stephens IEL, Bondarenko AS, Johansson TP, Hansen HA, Jaramillo TF, Rossmeisl J, Chorkendorff I, Nørskov JK (2009) Nat Chem 1:552–556
Stephens IEL, Bondarenko AS, Grønbjerg U, Rossmeisl J, Chorkendorff I (2012) Energy Environ Sci 5:6744–6762
Escudero-Escribano M, Verdaguer-Casadevall A, Malacrida P, Grønbjerg U, Knudsen BP, Jepsen AK, Rossmeisl J, Stephens IEL, Chorkendorff I (2012) J Am Chem Soc 134:16476–16479
Pašti IA, Gavrilov NM, Baljozović M, Mitrić M, Mentus SV (2013) Electrochim Acta 114:706–712
Pašti I, Mentus S (2009) Mater Chem Phys 116:94–101
Acknowledgments
I.A.P. and S.V.M. acknowledge the support provided by the Serbian Ministry of Education, Science and Technological Development through the contract no. III45014. I.A.P. and N.V.S. acknowledge the support provided by the Swedish Research Council through the project “Catalysis by metal clusters supported by complex oxide substrates”. S.V.M. also acknowledges the support provided by the Serbian Academy of Sciences and Arts through the project “Electrocatalysis in the contemporary processes of energy conversion”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pašti, I.A., Skorodumova, N.V. & Mentus, S.V. Theoretical studies in catalysis and electrocatalysis: from fundamental knowledge to catalyst design. Reac Kinet Mech Cat 115, 5–32 (2015). https://doi.org/10.1007/s11144-014-0808-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-014-0808-x