
Abstract
Orange carotenoid protein (OCP) is a water-soluble photoactive protein responsible for a photoprotective mechanism of nonphotochemical quenching in cyanobacteria. Under blue–green illumination, OCP converts from the stable orange into the signaling red quenching form; however, the latter form could also be obtained by chemical activation with high concentrations of sodium thiocyanate (NaSCN) or point mutations. In this work, we show that a single replacement of tryptophan-288, normally involved in protein-chromophore interactions, by alanine, results in formation of a new protein form, hereinafter referred to as purple carotenoid protein (PCP). Comparison of resonance Raman spectra of the native photoactivated red form, chemically activated OCP, and PCP reveals that carotenoid conformation is sensitive to the structure of the C-domain, implicating that the chromophore retains some interactions with this part of the protein in the active red form. Combination of differential scanning fluorimetry and picosecond time-resolved fluorescence anisotropy measurements allowed us to compare the stability of different OCP forms and to estimate relative differences in protein rotation rates. These results were corroborated by hydrodynamic analysis of proteins by dynamic light scattering and analytical size-exclusion chromatography, indicating that the light-induced conversion of the protein is accompanied by a significant increase in its size. On the whole, our data support the idea that the red form of OCP is a molten globule-like protein in which, however, interactions between the carotenoid and the C-terminal domain are preserved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carotenoids are natural pigments, mainly C40 terpenoids, which occur in the form of hydrocarbons or keto- or hydroxylated derivatives and can be found in bacteria, algae, fungi, and higher plants (Frank and Cogdell 1996). They contribute to light harvesting, as well as to protection mechanisms against conditions of excessive light functioning as antioxidants. The xanthophyll cycle involving zeaxanthin and violaxanthin plays an important role in light energy dissipation within the chlorophyll-containing antenna of algae and higher plants via non-photochemical quenching (NPQ) (Demmig-Adams and Adams 1996; Pascal et al. 2005). In cyanobacteria, additional light harvesting occurs in supramolecular water-soluble pigment-protein complexes—phycobilisomes (Liu et al. 2013), and the specialized mechanism of photoprotection has been evolutionary developed. A specific 35 kDa water-soluble carotenoid binding protein, the orange carotenoid protein (OCP), plays an essential role in this process (Boulay et al. 2008). Numerous experiments showed that OCP is a photoactive protein capable of reversibly changing its spectral and structural properties triggered by photon absorption and resulting in formation of an active red form which interacts with phycobilisomes and efficiently quenches excessive excitation energy (Berera et al. 2012; Chergui et al. 2013; Kirilovsky and Kerfeld 2013; Liu et al. 2014; Maksimov et al. 2014; Stadnichuk et al. 2012, 2013; Wilson et al. 2010, 2011, 2012; Zhang et al. 2014).
OCP consists of two structural domains (N- and C-), between which the carotenoid chromophore is bound (Leverenz et al. 2014; Wilson et al. 2012). X-ray crystallography provided valuable information about the structure of the stable orange (OCPO) form (Wilson et al. 2010) and the red carotenoid protein (RCP) (Leverenz et al. 2015) containing only the N-terminal portion of the protein, which suggested to be responsible for the characteristic spectral and structural features of the active signaling state. The echinenone (ECN) as chromophore is responsible for the OCP absorption in the visible spectral region and exhibits a red shift upon photoswitching to the photoactivated OCP form, which is referred to as a red form (OCPR) (Berera et al. 2012; Chabera et al. 2011; Chergui et al. 2013; Polívka et al. 2005). Photoconversion of OCP is also accompanied by characteristic changes of the carotenoid’s resonance Raman spectrum, namely, the intensity of ν 4 vibration (~980 cm−1), related to the hydrogen-out-of-plane (HOOP) mode and by the alteration of ν 1 mode (~1520 cm−1 C=C stretch vibrations of polyene chain) (Kish et al. 2015; Leverenz et al. 2014; Maksimov et al. 2015). However, the molecular mechanism of OCP photoconversion and the complete sequence of conformational rearrangements which leads to the formation of the OCP form capable of phycobilisome fluorescence quenching (and the structure thereof) are still under discussion.
Several alternative possibilities were reported in the literature allowing to obtain the active OCP form without illumination that spectrally and functionally mimics OCPR. First, it was shown that the OCP protein without the C-domain, referred to as the RCP, exhibits absorption spectrum similar to the OCPR (Leverenz et al. 2015). The facts that RCP can be obtained from OCPO by proteolysis upon removal of the C-domain and that the resultant RCP is spectrally and functionally similar to OCPR, including its ability to effectively quench the fluorescence of phycobilisomes, raised the idea that OCP exhibits structural and functional modularity (Leverenz et al. 2014). Second, a quenching red form of the OCP could be obtained in the absence of light via chemical activation by chaotropic agents, e.g., by addition of 1.5–2.0 M NaSCN (King et al. 2014). Finally, a red form can be obtained as a result of point mutations in the vicinity of the chromophore in OCP (Wilson et al. 2011).
The general considerations about the changes in OCPR conformation compared to OCPO include the following. The secondary structure of OCP is unaltered upon its photoactivation while an increase in the protein volume was observed (Gupta et al. 2015; Leverenz et al. 2014; Liu et al. 2016), giving rise to the idea of the separation of the N- and C- domains upon photon absorption by OCPO. It was also shown that the kinetics of OCP photocycle followed by the transient changes in the chromophore absorption is identical to the changes in the intrinsic and extrinsic fluorescence of OCP, indicating that the local rearrangements of OCP in the vicinity of its chromophore are complemented by the global changes of the tertiary structure (Maksimov et al. 2015). Recent studies and the data obtained from small angle X-ray scattering (SAXS) experiments confirmed that separation of the N and C terminal domains occurs in the OCPR form (Gupta et al. 2015). This result, combined with the 12 Å shift of the chromophore into the N-domain in RCP (Leverenz et al. 2015), led to the suggestion that the chromophore segregates from the C-domain on native OCP upon the photoconversion. At the same time, it is commonly believed that it is the chromophore that stabilizes the OCPO structure due to the hydrogen bonds formed between its rings and the neighboring amino acid groups; hence, substitution of the latter via point mutations may result in OCPO destabilization and formation of the OCPR-equivalent form. Despite the fact that site-directed mutagenesis experiments showed the essential role of tyrosine-201 and tryptophan-288 within the C-terminal domain for the photoactivity of OCP (Wilson et al. 2011), substitution of these residues to histidine or serine did not result in formation of the red form with its characteristic spectra.
It is well established that certain gene clusters of carotenoid synthesis from Erwinia species in combination with other carotenogenic genes can be used to produce complex C40 carotenoids-like β-carotene and other keto- or hydroxylated products thereof, including zeaxanthin and ECN, with high yield and excellent purity in non- carotenogenic E. coli. In the present work, we make use of the plasmids carrying the genes for ECN biosynthesis to accomplish the insertion of these carotenoids into Synechocystis OCP upon co-expression in E. coli. This strategy also allows for the generation of carotenoid-complemented OCP mutants (de Carbon et al. 2015) and circumvents the strenuous and time-consuming generation of mutant Synechocystis strains, thus eventually speeding up structure–function investigations on OCP. This approach allowed us to obtain a fully photoswitchable 6xHis-tagged OCP (further OCP) and a stable 6xHis-tagged W288A-OCP mutant, which is characterized by unusual red-shifted absorption spectrum, and thus purple color (further Purple Carotenoid Protein, PCP).
In this work, we performed a comparative study of three activated OCP forms, namely, photoconverted OCPR, chemically activated OCP form and PCP to find similarities and differences between them, aimed at the elucidation of the role of protein structure and protein-chromophore interactions in the formation of OCPR-like states. At first, we studied the environment of the chromophore in all three forms of OCP using absorption and Raman spectroscopy. Since the principal role of the carotenoid-protein interaction in the formation of the red form of OCP is undebatable, the differences in the spectral properties of carotenoid in three OCP forms could be related to the structural differences of the latter. Second, we studied the temperature-induced unfolding of the three OCPR-like forms using differential scanning fluorimetry (DSF), aimed at the comparison of its stability, which is critical for understanding the organization of signaling state of photoactive proteins. Finally, as OCPR is characterized by the expanded volume compared to OCPO (Gupta et al. 2015; Leverenz et al. 2014), we analyzed the hydrodynamic properties of all the three forms using time-resolved anisotropy measurements, size-exclusion chromatography (SEC), and dynamic light scattering (DLS) measurements. With this combination of experimental approaches, we aim to elucidate distinguishing features in the family of the OCPR-like forms of OCP.
Materials
cDNA constructs and cloning
For the expression of the OCP apoprotein from Synechocystis sp. PCC 6803, we designed a plasmid harboring the cDNA of OCP according to the published amino acid sequence (PubMed/UniProt entry P74102) optimized for codon usage in E. coli by artificial gene synthesis (GeneOptimizer® algorithm (Graf et al. 2000), GeneArt, Life Technologies, sequence available upon request). The cDNA was excised by BamHI and NotI and ligated in-frame into a modified pQE81L vector (Qiagen, Hilden, Germany) harboring an engineered NotI restriction site and carrying an ampicillin resistance gene. This resulted in a protein sequence with an N-terminal 6xHis tag (N-terminal sequence MRGSHHHHHHTDPAT) preceding the first methionine of OCP. Generation of OCP mutants was performed using the QuikChange® site-directed mutagenesis kit (Stratagene) according to manufacturer’s instructions. The complete open reading frames of all constructs were verified by sequencing (Eurofins MWG Operon, Germany).
In order to accomplish carotenoid cofactor insertion into the OCP protein already upon expression in E. coli, NEBturbo cells (New England Biolabs) were transformed with a 1:1 mixture of the pQE81L-Amp plasmid carrying the OCP sequence and either the plasmid pACCAR16ΔcrtX (harboring the contiguous gene cluster consisting of crtY, crtI, crtB, and the crtE gene from Erwinia uredovora and a chloramphenicol resistance cassette) for the accomplishment of β-carotene biosynthesis in E. coli, or the plasmid pACCAR25ΔcrtX (harboring the contiguous gene cluster consisting of crtY, crtI, crtB, crtZ, and the crtE gene from Erwinia uredovora and a chloramphenicol resistance cassette) for the accomplishment of zeaxanthin biosynthesis, as described previously (Misawa et al. 1995). The CrtE gene product (a geranylgeranyl-pyrophosphate synthase) forms geranylgeranyl-pyrophosphate from isopentyl-pyrophosphate and farnesyl-pyrophosphate, these latter two precursors are natively formed in E. coli. The CrtB gene product (a phytoene synthase) forms phytoene from two geranylgeranyl-pyrophosphate molecules. The CrtI gene product (a phytoene desaturase) forms lycopene from phytoene and the CrtY gene product (a lycopene cyclase) forms β-carotene from lycopene. The CrtZ gene product (a β-carotene hydroxylase) converts β-carotene in two hydroxylation steps at the 3 and 3′ position of the β rings to zeaxanthin. Synthesis of the ECN cofactor was accomplished by means of the pACCAR25ΔcrtXZcrtO plasmid, on which the crtZ gene was inactivated by cloning the crtO gene encoding a β-carotene ketolase (Breitenbach et al. 2013; Fernández-González et al. 1997) into the HpaI cloning site of the crtZ gene. After transformation, cells were plated on LB agar plates supplemented with 50 µg/ml ampicillin and 34 µg/ml chloramphenicol and grown overnight at 37 °C. Starter cultures of 10 ml lysogeny broth (LB) medium with the aforementioned antibiotics were inoculated from single colonies and grown overnight at 37 °C at 200 rpm in an orbital shaker (infors HT, Bottmingen, Switzerland).
Protein expression and purification
For protein expression, 2 × 500 mL LB-medium with 34 µg/mL chloramphenicol and 50 µg/mL ampicillin were inoculated from a starter culture to OD 0.1 and grown at 37 °C in an orbital shaker at 200 rpm to an OD of 0.8. After induction with 500 µM isopropyl β-d-1-thiogalactopyranoside (IPTG), cells were grown for 48 h at 25 °C. Cells were harvested by centrifugation yielding reddish-colored pellets indicating carotenoid synthesis. Cell pellets were resuspended in phosphate buffer with 100 mg lysozyme and Complete® (Roche) protease inhibitor and lysed by 3–4 freeze/thaw cycles on dry ice with absolute ethanol. After removal of the cell debris by centrifugation (12,000×g, 4 °C), the clarified and intensively colored supernatant was purified by affinity chromatography with an ÄKTA FPLC purification system (GE Healthcare) using 5 ml HiTrap Talon crude Co2+-loaded affinity columns (GE Healthcare). Upon elution by an imidazole gradient from 20 mM up to 500 mM, an intensively orange/red-colored protein solution was obtained. After concentrating with Amicon Centrifugal Filter Units (Merck Millipore), the protein solution was desalted with PD-10 desalting columns (GE Healthcare) and stored at -80 °C in phosphate buffered solution until use.
Methods
Absorption measurements
Absorption spectra were recorded using a Perkin Elmer Lambda-25 spectrophotometer as described in (Maksimov et al. 2015). In all experiments, a M455L3 (Thorlabs, USA) 900 mW light-emitting diode (LED) with maximum emission at 455 nm was used for blue–green illumination of the samples (further—actinic light for OCPO → OCPR photoconversion). Absorption spectra were measured at 5 °C in order to reduce the rate of OCPR → OCPO conversion while measuring optical density of the red form, obtained after 3 min of exposition to 2000 µmol photons m−2s−1 of actinic light.
Steady-state fluorescence measurements. Differential scanning fluorimetry (DSF)
The steady-state fluorescence measurements were performed using FluoroMax-4 spectrofluorimeter (Horiba Jobin–Yvon, Japan-France). Samples were diluted to OD ~ 0.1 units at 280 nm to avoid inner filter effects and reabsorption. Fluorescence emission of the hydrophobic probe Nile Red (Sigma Aldrich) was measured in OCP solution during the temperature-induced unfolding as described in (Niesen et al. 2007). The excitation wavelength was set to 560 nm, and the emission was recorded in the 590–730 nm region. Temperature of the sample was stabilized by a Peltier-controlled cuvette holder Qpod 2e (Quantum Northwest, USA).
Approximation of the experimental data, i.e., the dependence of normalized NR fluorescence F in proteins on temperature T, was performed using a Boltzmann-type equation:
This allowed us to determine melting temperatures T* and the transition range \(\Delta T\). After that, the effective enthalpy of transition (\(\Delta E\)) was calculated according to the van’t Hoff criterion as
where \(\Delta T\) is determined by the range of the most significant fluorescence changes. \(\Delta E\) represents the difference between the energies of the two states.
Picosecond time-resolved fluorescence and anisotropy measurements
In this work, we used the fluorescent Nile Red (NR) dye as a probe to follow the structural transitions of protein as described elsewhere (Maksimov et al. 2015). We also measured anisotropy kinetics of the NR fluorescence to estimate the correlation time of bound dye, which is related to size and shape of the host protein.
Fluorescence decay kinetics with picosecond time resolution were collected by time- and wavelength-correlated single photon counting setup (Becker&Hickl, Germany). Excitation was performed with a 635 nm laser diode BHL-700 (Becker&Hickl, Germany), driven at a repetition rate up to 50 MHz. Notch filter NF633-25 (Thorlabs, USA) was used to block excitation light. Fluorescence decay curves were approximated by a sum of exponential decay functions with the SPCImage (Becker and Hickl, Germany) software package. To compare different decay curves, we calculated the average decay time according to the following expression:
where τ i and a i are the lifetime and the amplitude (normalized to unity: \(\sum\nolimits_{i}^{n} {a_{i} = 1}\)) of the i-th fluorescence decay component, respectively. To obtain the time-integrated fluorescence spectra, the number of photons in each spectral channel was summed up.
To measure fluorescence anisotropy kinetics, we used a filter-based system with a set of two ultrabroadband wire-grid polarizers WP25M-UB (Thorlabs, USA). Fluorescence decay kinetics I(t) were measured at different positions of emission polarizer—in parallel (∥) and perpendicular (⊥) orientation to excitation polarizer, and the anisotropy kinetics r(t) was calculated as
In some experiments, we observed a complex polyphasic kinetics of anisotropy (see Fig. 3), which is characteristic for so-called associated decay (Chib et al. 2014; Lakowicz 2013). In order to quantify such anisotropy decay, one may assume that each fluorophore type obtains a single lifetime (τ m ) and a single correlation time (θ m ). The anisotropy decay for this mixture is then given by
where α m and r 0m are the amplitude and anisotropy of the m-th component at t = 0.
In our experiments, we used the hydrophobic dye Nile Red (NR), which has a low fluorescence lifetime in aqueous solutions and a higher lifetime when bound to the hydrophobic sites of proteins. If we consider that our system has only these two states, the kinetics of anisotropy could be approximated by the following equation:
where the indexes 1 and 2 correspond to free and bound NR molecules, respectively. In the first state NR movement (rotation) is fast, while in the second state it is limited by the size of binding site and rotation of the whole protein. To increase the quality of approximation of the experimental data by Eq. 6, we reduced the number of free parameters values by (1) setting α 1 + α 2 = 1 and (2) using the lifetimes τ 1 and τ 2 obtained directly from the fluorescence decay and approximated with the biexponential function.
Raman spectroscopy measurements
Raman spectra were obtained under continuous illumination at 532 nm. The laser beam was focused on a 0.1 mm glass capillary connected to a peristaltic pump (Heidolph, Germany). Raman-scattered light was collected and subsequently imaged using a microscope-based system (NTMDT, Ntegra Spectra, Russia). OCPO Raman spectra were collected under rapid-flow conditions (2 mL/min) in order to minimize the effect of OCPO → OCPR photoconversion in the registered volume of the sample. Turning off the peristaltic pump led to an immediate conversion of the illuminated sample into the red form.
Dynamic light scattering
Measurements of the protein size distribution were performed using the Nano-ZS device (Malvern, UK). Scattered light (632.8 nm of a helium–neon laser) was detected at an angle of 173° (in a backscattering geometry).
Analytical size-exclusion chromatography
Protein samples (100 µl) with different protein contents were subjected to SEC on a Superdex 200 Increase 10/300 column (GE Healthcare) equilibrated with a filtrated and degassed 20 mM Tris–HCl buffer, pH 7.6, containing 200 mM NaCl, 0.1 mM EDTA and 3 mM ME. The column was operated at 25 °C at 1.5 ml/min flow rate using a chromatographic ProStar325 UV/Vis system (Varian Inc.). The elution profiles were monitored by absorbance at 540 nm with or without the constant illumination by a blue LED (445 nm; 900 mW) and the maxima of the peaks were used to determine corresponding elution volumes (V e). To estimate the Stokes radii of the species, the column was experimentally calibrated with the following standards: blue dextran (V e = 8.49 ml), bovine serum albumin trimer (V e = 11.52 ml; R s = 66.9Å), bovine serum albumin dimer (V e = 12.54 ml; R s = 53.2 Å), bovine serum albumin monomer (V e = 14.47 ml; R s = 35.5 Å), ovalbumin (V e = 15.69 ml; R s = 30 Å), maltose-binding protein (V e = 15.91 ml; R s = 28.5 Å), α-lactalbumin (V e = 17.61 ml; R s = 15.7 Å). The Stokes radii were calculated using the experimentally determined elution volumes (V i), from the calibration curve R s vs [–log(K av)]0.5, where K av = (V i—V 0)/(V t—V 0), V t being a total column volume (24 ml).
All experiments were performed in vitro in aqueous solutions of isolated OCP. Each experiment was repeated at least three times.
Results and discussion
Absorption and resonance Raman spectroscopy measurements
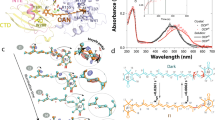
Figure 1a shows steady-state absorption spectra of OCPO (6xHis-OCP, hereinafter OCP, orange curve) with characteristic vibronic structure, and its three signaling forms. The absorption spectrum of OCPR (red curve), that was obtained by photoconversion, is red-shifted compared to that of OCPO and lacks vibronic structure, that is often interpreted as an increased heterogeneity of the chromophore. The absorption spectrum of another red form, obtained by chemical activation as described in (King et al. 2014) via 1.5 M NaSCN treatment, is slightly red-shifted compared to OCPR (black curve). The most distinct shift of absorption, which confers purple color to the sample, was observed for the 6xHis-W288A OCP mutant (PCP, blue curve). Specifically, PCP also exhibits a profound shoulder on the red flange, with the corresponding maximum of the transition band at around 565 nm. We observed no transient changes of PCP absorption under actinic illumination. It should be noted that according to Wilson et al. (2011) the W288H substitution does not change the color of the sample, which stays orange but loses photoactivity and ability to induce NPQ. Together, the differences between these mutants emphasize an essential role of Trp-288 in photoactivity and structural organization of OCP.

Absorption (a) and resonance Raman (b and c) spectra of different OCP forms. Absorption was measured at 5 °C in order to reduce the rate of OCPR → OCPO conversion while measuring optical density of the red form (red curve), obtained after 3 min of exposition to 2000 µmol photons m−2s−1 of actinic light. Raman spectra were recorded upon excitation by 532 nm laser. Raman spectra are shifted along the y-axis for better comparison. c Close-up view of the ν 1 band position for different activated OCP species
Considering the hypothesis that the chromophore ECN leaves the C-terminal domain upon photoconversion to the red form, we expected exactly the same Raman spectra for the photoconverted sample, NaSCN-treated sample and PCP. Indeed, the obtained spectra share a lot of common properties, especially regarding the low intensity of the HOOP mode (~980 cm−1 on Fig. 1b), indicating that in all samples the chromophore is in a planar configuration. At the same time, we found that our samples exhibited different positions of the ν 1 mode peak (Fig. 1c). It was previously shown that the ν 1 position changes from 1525 to 1516 cm−1, during OCP photoconversion (Kish et al. 2015; Leverenz et al. 2014), which is in a good agreement with our data. In (Kish et al. 2015), it was shown that such changes may arise from cis-cis to trans–trans isomerization along C6–C7/C6′–C7′ bonds resulting in an increase of conjugation length; in other words, the observed changes may be due to rotation of both β-ionylidene rings of the ECN chromophore. Similar results were previously obtained by (Wei-Long Liu et al. 2008) for β-carotene from DFT calculations. Plotting the ν 1 band position of PCP at 1510 nm against the corresponding 0-0 transition energy of 17,700 cm−1 (565 nm) places the W288A mutant along the data points of lycopene (longest conjugated system from Kish et al. 2015), so one indeed might infer that the PCP spectral characteristics arise from the longest possible system of conjugated bonds with trans–trans configuration of the β-ionylidene rings. Due to the fact that OCPR → OCPO conversion occurs even under actinic light sample may contain insignificant amounts of the orange form. However, photoconversion was absent for NaSCN-treated sample or PCP, so we assume that the observed spectral differences are due to protein-chromophore interactions. This finding raises the question why ECN conformation could be sensitive to a single amino acid substitution in the C-terminal domain if ECN in OCPR is completely translocated to the N-terminal domain, as proposed by the RCP structure according to (Leverenz et al. 2015). This problem will be addressed in the “Discussion and conclusions” section below.
As it was mentioned, the inability to form stable orange form could be induced by point mutations of residues, essential for the formation of hydrogen bonds. However, as binding of a chromophore to a protein occurs only upon a release of free energy, such mutations may cause the reduction of chromophore content in such samples. Indeed, Vis (450 nm) to UV (280 nm) absorption ratio of our PCP sample was much lower (~0.2) comparing to the wild type (~1.8). In the next sections, we provide the results of the investigation of protein-chromophore interactions on the stability of the system and the size of the complex.
Differential scanning fluorimetry (DSF) of OCP
The temperature-induced unfolding of different OCP species was followed by fluorescence changes of nonspecifically binding hydrophobic dye Nile Red (Fig. 2a), which was successfully used previously to study the OCP photocycle (Maksimov et al. 2015). This measurement protocol is referred to as DSF and is described in (Niesen et al. 2007). Temperature dependency of fluorescence intensity of NR in DMSO was used as a baseline and subtracted from experimental data shown in Fig. 2a, and after that the curves were normalized to obtain Fig. 2b.

a Normalized fluorescence intensity of the hydrophobic dye Nile Red complexed with OCP in orange and red forms (orange squares and red circles), OCP in the presence of 1.5 M NaSCN (black squares), Apo-W288A OCP (blue triangles), and BSA (open squares) during their temperature-induced unfolding. Temperature dependency of fluorescence intensity of NR in DMSO was used as a baseline and subtracted from experimental data to obtain (b) temperature-induced transients of OCP unfolding, which were approximated by Eq. (1). Protein concentration and NR to protein ratio were the same in all samples
As shown in Fig. 2a, the studied samples were stable up to ca 45 °C, as fluorescence of NR in proteins coincided with that for NR in DMSO in this temperature range. Temperature-induced changes of NR fluorescence were less pronounced in Apo-W288A OCP (the OCP W288A apoprotein) and almost absent in the NaSCN-treated OCP sample. This fact was presumably due to the high initial background fluorescence of the hydrophobic probe, the effect known to occur in the DSF experiments (Niesen et al. 2007). This indicates that hydrophobic sites of Apo-W288A were occupied by the NR even before the melting. In case of the NaSCN-treated sample, we assume that its tertiary structure was already changed or destabilized by the chaotropic agent resulting in the absence of pronounced temperature-induced melting.
The normalized dependences of NR fluorescence in the presence of OCPO, OCPR, and PCP shown in Fig. 2b are s-shaped, and the half-transition temperature corresponds to protein melting. To validate our experimental setup, we studied the unfolding of bovine serum albumin (BSA, Sigma). We found that melting of BSA occurs in the region of 47–75 °C, and fitting of the corresponding curve with Eq. (1) provided the melting temperature equal to 61.4 °C, that is in a good agreement with the literature data (Flora et al. 1998).
We note that photoactivation of OCP led to a significant enhancement of NR fluorescence during the temperature-induced unfolding (see Fig. 2a), that is probably associated with an increase in the number of hydrophobic sites available for non-specific binding of NR. Such an exposure of hydrophobic sites to the solvent is characteristic of molten globule-like proteins (Shakhnovich and Finkelstein 1989).
From Fig. 2b, it is clear that Apo-W288A OCP is less thermodynamically stable compared to other samples, thus indicating that the chromophore stabilizes the protein, probably, by providing the connection between C and N-terminal domains.
It should be noted that even at 95 °C OCPO sample remained orange without visible aggregates or sediment; however, upon cooling the sample was not photoactive under any intensities of actinic light (data not shown).
Time-resolved measurements of the Nile Red fluorescence anisotropy
In addition to the steady-state fluorescence of NR and DSF measurements, we analyzed temperature-induced unfolding of different OCP species in a time-resolved regime to access the quantum yield and anisotropy of NR. First of all, it should be noted that NR in Apo-W288A OCP is characterized by a significantly higher fluorescence lifetime (~2 ns, see Fig. 3a), compared to any other OCP species with chromophore (less than 1 ns). This fact could be explained by (1) excitation energy transfer (EET) from the NR molecule to ECN, or (2) a presence of several unspecific binding sites (for example, on the surface of the protein and in the cavity between C and N domains). However, as ECN is absent in Apo-W288A and bi-exponential signals were recorded here as well, we attribute the bi-exponential fluorescence decay to multiple sites of NR binding with different solvent accessibilities. Moreover, the amplitude of the fast component increased with the increase of temperature upon the protein unfolding (Fig. 3a). Thus, analysis of NR fluorescence requires additional explanation.

a Examples of fluorescence decay kinetics and b fluorescence anisotropy decay of Nile Red mixed with Apo-W288A at 26 and 65 °C. c Temperature dependency of correlation time θ 2 of fluorescence anisotropy relaxation of Nile Red bound to OCPO (black squares) and Apo-W288A (red circles)
Typical anisotropy decay kinetics of the Nile Red fluorescence in OCP solution are presented in Fig. 3b. Such complex polyphasic kinetics of anisotropy is characteristic for the situation of a so-called associated decay (Lakowicz 2013). If a system contains several types of fluorophores which exhibit different lifetimes and different correlation times (e.g., free and protein-bound), the overall measured anisotropy will depend on the ratio of their populations. In other words, if one fraction of molecules shows fast anisotropy relaxation (e.g., on the surface of a protein), the overall anisotropy will rapidly decrease, but as their fluorescence intensity decreases exponentially according to their fluorescence lifetime (which is about 450 ps for the Nile Red, see Fig. 3a), anisotropy may increase again after some time due to the presence of another fraction of fluorophores (bound to the hydrophobic sites of the protein) with considerably higher fluorescence lifetimes (~2.4 ns for the Nile Red) and longer correlation times. Such a situation may occur if fluorophores have distinct lifetimes in different environments and only one fraction of them is bound to hydrophobic site(s) of the protein, while the other remains accessible to the solvent. Analysis of the Nile Red fluorescence decay in different OCP preparations and in DMSO shows that the lifetime of the dye is strongly temperature dependent resulting in a decrease of the overall fluorescence intensity in the temperature range of 5–35 °C (see Fig. 2), while the distribution of components changes dramatically upon temperature-induced unfolding of the protein in the 45–80 °C region. In this particular range, the NR fluorescence anisotropy kinetics of all studied samples show a characteristic “inflection” due to the high yield of the fast component with a short correlation time θ 1, indicating that during unfolding new NR molecules bind to the surface of the protein and thus are solvent accessible. Increase of the fast component yield in 45–80 °C region follows the overall changes of fluorescence intensities presented in the Fig. 2b. However, the behavior of the component with a long correlation time θ 2 is much more interesting as it follows the movements of the whole protein (Fig. 3c). The correlation time θ (for the rotation of a spherical protein) could be calculated according to Perrin equation (Lakowicz 2013):
where η is the viscosity of the solvent, M is molecular weight of the protein, ν and h are the partial specific volume of the protein and its degree of hydration (all in ml/g), respectively. For the 35 kDa spherical protein, θ is equal to 7.9 ns at zero hydration and 12.1 ns at 0.4, respectively (at 32 °C and water viscosity η about 0.76 cP), which is significantly higher compared to the observed θ 2 values; however, such twofold discrepancy is well known for many proteins (Yguerabide et al. 1970), and may indicate segmental flexibility of the protein structural domains. This, in turn, does not negate the fact that protein hydration degree may affect the rotation rates. On the other hand, according to X-ray and SAXS data (Gupta et al. 2015), the shape of OCP is non-spherical and could be better described by a prolate ellipsoid of revolution. In this case, increase of the protein size by a third along the major axis of symmetry will result in a 10 % increase of its correlation time θ (Lakowicz 2013), which is not sufficient to describe observed differences in θ 2 values of OCP in orange form, NaSCN-treated OCP and PCP. Therefore, we suggest that differences in θ 2 are simultaneously caused by changes of protein hydration and protein shape.
The temperature dependency of correlation time θ 2 in general follows the changes of fluorescence intensities as shown in Fig. 2, which shows that this approach is also suitable for the determination of protein stability. However, determination of correlation time allows to obtain more specific information about the initial and the final states of the protein which undergoes temperature-induced unfolding. According to our results (Fig. 3c), θ 2 of NR in Apo-W288A OCP increases ca 5 times in the temperature range from 50 to 70 °C, which clearly indicates the formation of protein aggregates (the following decrease of θ 2 is probably due to sedimentation of aggregates). In contrast, unfolding of OCP causes twofold increase in θ 2, that could be the evidence of nonspecific dimerization. Thus, time-resolved anisotropy measurements reveal the mechanism of changes underlying the process of Nile Red binding to the protein destabilized by high temperatures.
Hydrodynamic properties of OCP species as revealed by dynamic light scattering (DLS) and size-exclusion chromatography (SEC)
Since introduction of alanine instead of tryptophan-288 impairs the ECN binding by the mutated protein the resulting preparation inevitably contained a mixture of Apo and ECN-containing protein. However, due to characteristic absorption in the visible region, the hydrodynamic properties of the carotenoid-bound protein could be assessed by SEC without separation of the forms. Moreover, the shape and the maximum position of the peak on the elution profile of the PCP were substantially the same regardless of whether the profile was followed by 280 nm (total protein) or 540 nm (carotenoid-bound protein only) (data not shown). The position of the OCPO peak remained unchanged upon more than tenfold change of the protein concentration (Fig. 4), with the corresponding hydrodynamic radius of 26.4 Å, in agreement with what was expected from one crystallographic OCP subunit (PDB ID 3MG1). Of note, higher protein concentrations of OCPO resulted in appearance of a small peak corresponding to larger particles. We may speculate that it corresponded to a small fraction of nonspecific oligomers (probably, dimers) present in the sample. In contrast to OCPO, PCP had a pronounced concentration-dependent behavior showing a significant decrease of its elution volume (from 15.5 to 14.9 ml that corresponds to the increase of the hydrodynamic radius from 30.5 to 34.7 Å) with increasing protein concentration in the sample loaded on the column (Fig. 4, lower panel). Remarkably, the PCP peak position at the highest concentration used was comparable to that of the tentative nonspecific oligomers of OCPO (Fig. 4, lower and middle panels); and an equivalent mixture of OCPO and PCP gave two peaks corresponding to non-interacting individual proteins with the elution profile being indistinguishable from their algebraic sum (Fig. 4, upper panel, blue dashed line). It is likely, that these observations are explained by a concentration-dependent self-association of PCP; however, our attempts to supplement the sample with 0.1 % deoxycholate to prevent protein oligomerization, the approach which has successfully been used to promote dissociation of 14-3-3 protein dimers (Denison et al. 2014), did not give desired results (data not shown). Nonetheless, even at smaller protein concentration, the estimated radius of PCP was higher than that of OCPO (30.5 instead of 26.4 Å). Taking into account that illumination of the OCPO sample during pre-incubation and SEC run with blue light resulted in the peak shift and the increase of its hydrodynamic radius from 26.4 to 29.8 Å (Fig. 4, upper panel and inset), in agreement with the previous results (Gupta et al. 2015; Leverenz et al. 2014), we suggest that at least at low protein concentration PCP well imitates the photoactivated OCPR form, whose larger radius implies significant structural changes and indirectly supports the idea of its molten-globule-like behavior.

Analytical SEC of OCP and PCP species performed at different protein concentrations loaded on a Superdex 200 Increase 10/300 column. Initial preparations (see insets) gave similar absorbance at 540 nm, and were diluted with a column buffer (corresponding dilutions are color-coded in the middle and bottom panels) and loaded on the column (profiles are shifted vertically for clarity). OCPO sample was optionally subjected to a constant illumination with a blue light (inset on the upper panel), yielding a decrease of the elution volume (marked by arrow). Alternatively, the mixture of OCPO and PCP was analyzed (dark-blue dashed line). Thin vertical dotted lines indicate peak positions for the highest protein concentration of OCPO (V e ~ 16.0 ml) or PCP (V e ~ 14.9 ml) loaded
These conclusions were additionally confirmed by qualitatively similar results obtained using DLS. In such experiments, we found that hydrodynamic radius of Apo-W288A was equal to 48.7 Å, while the corresponding parameter for the OCP in the orange form was equal to 30.4 Å, and the red form obtained by 1.5 M NaSCN treatment was characterized by 37.7 Å radius, which is in a good agreement with both the results of SEC and correlation times of NR, as observed by time-resolved anisotropy measurements (Table 1). Comparison of SEC and DLS data also indicates that a relative increase of OCP hydrodynamic radius after the chemical activation is comparable to the changes induced by photoactivation.
Discussion and conclusions
In this work, we performed a comparative study of the three OCPR-like forms at different levels of structural organization. First of all, we conducted the comparison of the protein-chromophore interactions based on its optical properties, namely the shape and the maximum position of absorption spectra maximum and the position of the ν 1 vibrational mode in the Raman spectra. The absorption spectra of all the three forms OCPR, NaSCN-treated OCP, and PCP represented structureless bands and were red-shifted compared to the spectrum of OCPO in a following manner: OCPR < NaSCN-treated OCP < PCP. In line with this, the position of the ν 1 vibrational mode in the Raman spectra was shifted compared to that of OCPO (1526 cm−1) in the order OCPR (1515 cm−1) < NaSCN-treated OCP (1513 cm−1) < PCP (1510 cm−1). These observations could be explained by different conjugation lengths for the chromophore in the three forms of OCP: activation of OCPO results in the weakening of the protein-carotenoid interactions and the rotation of the end-ring of chromophore that may lead to the increased conjugation length of ECN, which is more energetically favorable compared to that the orange form. This suggestion could be parallel to the case of another carotenoid binding protein, crustacyanin containing astaxanthin, whose color of which is also affected by the protein structure. The chemical basis of the so-called bathochromic shift in the lobster carapace is now well established due to X-ray crystallography and other spectral techniques (Bartalucci et al. 2007; Cianci et al. 2002; Salares et al. 1979). In the native state of crustacyanin, astaxanthin retains an energetically unfavorable conformation with the longest conjugation length, resulting in an extremely red (~650 nm) absorption and a low value of ν 1 band shift (~1494 cm−1, Salares et al. 1979). Denaturation of the protein causes disruption of hydrogen bonds between the carbonyl oxygen atom at the end rings of carotenoid and specific residues, leading to the relaxation of carotenoid conformation (including rotation of the end rings) and a reduction of conjugation length, thus resulting in the ν 1 band shift (increase up to 1520 cm−1) and the blue shift of the absorption.
Our results allow comparison of the chromophore environment of different red forms of OCP. Figure 5 demonstrates the correlation between the ν 1 frequency on the S0 → S2 transition frequency for OCPO, OCPR, NaSCN-treated OCP, PCP, and RCP. Based on the suggestion that the increase in conjugation length is due to the loosening of protein-chromophore interaction, the rigidity of the microenvironment of the ECN end-ring in the area of C-domain decreases as follows: OCPO > RCP ≈ OCPR > NaSCN-treated OCP > PCP. Surprisingly, the conjugation length is not the largest one for RCP, which lacks the C-domain, and, presumably, the chromophore is more released in OCPR, NaSCN-treated OCP, and PCP. We clearly see that the ECN conformation is sensitive to a single amino acid substitution in the C-terminal domain, which causes a significant shift in both absorption and Raman spectra. This suggests that ECN is possibly not completely translocated in the N-terminal domain as in the RCP structure according to (Leverenz et al. 2015), because further increase of the conjugation length exists.

Correlation between the position of the S 0 → S 2 electronic transition and the ν 1 band frequency for different OCP forms. Parameters for RCP were taken from (Leverenz et al. 2014)
Our results also suggest that the inability to form orange form could be induced by point mutations of tryptophan-288, which interfere with essential prerequisites for the formation of hydrogen bonds to the chromophore, and that the PCP and Apo-forms are the most unstable due to the lack of essential protein–chromophore interaction. This suggestion was further supported by our results on the thermodynamic stability and size of OCPO, OCPR, NaSCN-treated OCP, and PCP. For this purpose, we studied the temperature-induced unfolding process using the DSF. This method, based on the changes of dye fluorescence intensity, caused by the changes in its binding to protein at different temperatures, is capable to characterize protein stability based on its melting (half-transition) temperature (Niesen et al. 2007). Unfolding of small globular unidomain proteins is a cooperative process, which is characterized by the steep s-shaped dependence of protein parameters on the denaturing factors (Shakhnovich and Finkelstein 1989). The s-shaped dependence means that the molecule properties change from the native state to those in unfolded state, and the narrow region of transition is due to the cooperativity of the process (i.e., it involves numerous amino acid residues simultaneously). In our case, the behavior of DSF curves (Fig. 2b) and rotational correlation time on temperature (Fig. 3c) was s-shaped; moreover, melting (half-transition) temperatures obtained from the curves coincide, indicating that studied proteins undergo unspecific oligomerization (OCPO) and aggregation (PCP) upon the temperature-induced unfolding.
The parameters T*, \(\Delta T\) and \(\Delta E\) (melting temperature, transition range, and effective enthalpy of transition), obtained from the DSF curves shown in Fig. 2b using Eq. (1–2), are presented in Table 1. It should be noted that estimated \(\Delta E\) values for the OCPO unfolding are comparable with the activation energies for the OCPR → OCPO conversion rate (Maksimov et al. 2015), indicating that during photoactivation OCP undergoes significant rearrangements comparable to unfolding. Surprisingly, the parameters obtained for the OCPR form were close to that of OCPO, while we expected the red form to be significantly more unstable due to the fact that it is not the state with the minimum of free energy (as it exhibits a spontaneous transition to the stable orange form). This finding can be explained by a partial photoconversion of OCP in the DSF experiments. Indeed, the rate of OCPR → OCPO transition increases with temperature, and could be estimated as 1 s−1 at 50 °C, and the conversion efficiency is two times lower than at 25 °C (Maksimov et al. 2015). According to the DSF data, the Apo-W288A protein was significantly less stable than the other OCP form and exhibited the lowest melting temperature, presumably, due to the lack of carotenoid-induced stabilization of the protein structure.
OCPR is characterized by an almost identical (slightly lower) melting temperatures, compared to OCPO, and noticeably narrower region of temperatures \(\Delta T\) where melting occurs. Apo-W288A OCP was characterized by even more narrow \(\Delta T\). The observed differences could be explained by significantly higher hydration of Apo-W288A OCP, compared to OCP, which contains highly hydrophobic chromophore in a specifically organized environment. According to this data, we can assume that OCPR structure is less hydrophobic compared to OCPO, that is in agreement with our previous experiments revealing that quenching of NR fluorescence in OCPR may be due to the exposure of the NR binding site to the solvent (Maksimov et al. 2015).
Finally, we performed the investigation of OCP hydrodynamic radii using SEC and DLS, the results are presented in Table 1. Our results suggest that all the OCP forms (OCPO, OCPR, NaSCN-treated OCP, and PCP) are monomeric. This is in agreement with the data of (Gupta et al. 2015; Leverenz et al. 2014). However, by SEC we observed additional small peak in elution profiles of OCPO (Fig. 4), which is probably a feature of dimers, and this peak completely vanishes after illumination of the sample, which is in agreement with the results presented by (King et al. 2014), where it was shown by mass-spectroscopy that the OCPO can form homodimers, which are converted to monomers upon photoactivation. Estimations obtained by SEC, DLS, and fluorescence anisotropy reveal that protein size increases as follows: OCPO < OCPR ≤ NaSCN-treated OCP < PCP. This sequence is in line with characteristic changes in absorption and Raman (Fig. 5), indicating that destabilization of the protein–chromophore interactions leads to the increase of protein structure heterogeneity and its size, and vice versa denaturation of the protein results in a disruption of protein–chromophore interactions.
We also made a rough estimation of the relation between the hydrodynamic radii of the OCPO and OCPR based on the model introduced in (Gupta et al. 2015) on the basis of SAXS data. These models represent OCPR as an elongated ellipse with the N- and C-domains being completely separated in space, in contrast to their close contact in the native OCPO form. Assuming both forms to be prolate ellipsoids of revolution with different aspect ratios and taking the approximate axes sizes as 63 × 36 Å for OCPO and 90 × 27 Å OCPR, one can obtain the ratio α of the gyration radii for OCPR and OCPO to be close to α ≈ 1.22, that is higher than the same ratio obtained from SEC (α ≈ 1.11). At the same time, the obtained ratio is closer to the ones obtained for PCP and OCPO (α ≈ 1.26 (SEC)), suggesting that such an expanded structure with the maximum elongation of the protein’s globule is more characteristic for the PCP than for OCPR. This consideration is in line with our hypothesis that the PCP in which protein domain-linking is disrupted is the most unstable red form. In contrast, in the OCPR and NaSCN-treated OCP, the domains are not that expanded.
Thus, we assume that the overall heterogeneity and diversity of protein structures of the red forms support the idea that the red form is a molten globule-like protein in which, however, interactions between the chromophore and the C-terminal domain are preserved. Despite the fact that the study of apoproteins is beyond the scope of this work, we believe that the study of their structure (especially by SAXS and other structural methods) will provide valuable information about the mechanisms of conformational transitions in OCP and other photoactive proteins.
Abbreviations
- OCPO :
-
Orange carotenoid protein—orange form
- OCPR :
-
Orange carotenoid protein—red form
- PCP:
-
Purple Carotenoid Protein (W288A-OCP)
- RCP:
-
Red Carotenoid Protein
- ECN:
-
Echinenone
- NPQ:
-
Non-photochemical quenching
- τ :
-
Fluorescence lifetime
- φ fl :
-
Fluorescence quantum yield
- r :
-
Anisotropy
- θ :
-
Correlation time
- NR:
-
Nile red
- SEC:
-
Size-exclusion chromatography
- DSF:
-
Differential scanning fluorimetry
- DLS:
-
Dynamic light scattering
- HOOP:
-
Hydrogen-out-of-plane
References
Bartalucci G, Coppin J, Fisher S, Hall G, Helliwell JR, Helliwell M, Liaaen-Jensen S (2007) Unravelling the chemical basis of the bathochromic shift in the lobster carapace; new crystal structures of unbound astaxanthin, canthaxanthin and zeaxanthin. Acta Crystallogr B 63:328–337. doi:10.1107/S0108768106052633
Berera R, van Stokkum IH, Gwizdala M, Wilson A, Kirilovsky D, van Grondelle R (2012) The photophysics of the orange carotenoid protein, a light-powered molecular switch. J Phys Chem B 116:2568–2574. doi:10.1021/jp2108329
Boulay C, Abasova L, Six C, Vass I, Kirilovsky D (2008) Occurrence and function of the orange carotenoid protein in photoprotective mechanisms in various cyanobacteria. Biochim Biophys Acta 1777:1344–1354. doi:10.1016/j.bbabio.2008.07.002
Breitenbach J, Gerjets T, Sandmann G (2013) Catalytic properties and reaction mechanism of the CrtO carotenoid ketolase from the cyanobacterium Synechocystis sp. PCC 6803. Arch Biochem Biophys 529:86–91
Chabera P, Durchan M, Shih PM, Kerfeld CA, Polivka T (2011) Excited-state properties of the 16 kDa red carotenoid protein from Arthrospira maxima. Biochim Biophys Acta 1807:30–35. doi:10.1016/j.bbabio.2010.08.013
Chergui M et al (2013) Comparing the photophysics of the two forms of the Orange carotenoid protein using 2D electronic spectroscopy. EPJ Web of Conferences 41:08008. doi:10.1051/epjconf/20134108008
Chib R, Raut S, Sabnis S, Singhal P, Gryczynski Z, Gryczynski I (2014) Associated anisotropy decays of ethidium bromide interacting with DNA. Methods Appl Fluoresc 2(1):015003. doi:10.1088/2050-6120/2/1/015003
Cianci M, Rizkallah PJ, Olczak A, Raftery J, Chayen NE, Zagalsky PF, Helliwell JR (2002) The molecular basis of the coloration mechanism in lobster shell: beta-crustacyanin at 3.2-A resolution. Proc Natl Acad Sci USA 99:9795–9800. doi:10.1073/pnas.152088999
de Carbon CB, Thurotte A, Wilson A, Perreau F, Kirilovsky D (2015) Biosynthesis of soluble carotenoid holoproteins in Escherichia coli. Sci Rep 5:9085. doi:10.1038/srep09085
Demmig-Adams B, Adams WW (1996) The role of xanthophyll cycle carotenoids in the protection of photosynthesis. Trends Plant Sci 1:21–26
Denison FC, Gökirmak T, Ferl RJ (2014) Phosphorylation-related modification at the dimer interface of 14-3-3ω dramatically alters monomer interaction dynamics. Arch Biochem Biophys 541:1–12
Fernández-González B, Sandmann G, Vioque A (1997) A new type of asymmetrically acting β-carotene ketolase is required for the synthesis of echinenone in the cyanobacterium Synechocystis sp. PCC 6803. J Biol Chem 272:9728–9733
Flora K, Brennan JD, Baker GA, Doody MA, Bright FV (1998) Unfolding of acrylodan-labeled human serum albumin probed by steady-state and time-resolved fluorescence methods. Biophys J 75:1084–1096
Frank HA, Cogdell RJ (1996) Carotenoids in Photosynthesis. Photochem Photobiol 63:257–264. doi:10.1111/j.1751-1097.1996.tb03022.x
Graf M, Bojak A, Deml L, Bieler K, Wolf H, Wagner R (2000) Concerted action of multiple cis-acting sequences is required for Rev dependence of late human immunodeficiency virus type 1 gene expression. J Virol 74:10822–10826
Gupta S et al (2015) Local and global structural drivers for the photoactivation of the orange carotenoid protein. Proc Natl Acad Sci USA 112:E5567–E5574. doi:10.1073/pnas.1512240112
King JD, Liu H, He G, Orf GS, Blankenship RE (2014) Chemical activation of the cyanobacterial orange carotenoid protein. FEBS Lett 588:4561–4565. doi:10.1016/j.febslet.2014.10.024
Kirilovsky D, Kerfeld CA (2013) The Orange Carotenoid Protein: a blue-green light photoactive protein. Photochem Photobiol Sci 12:1135–1143. doi:10.1039/c3pp25406b
Kish E, Pinto MM, Kirilovsky D, Spezia R, Robert B (2015) Echinenone vibrational properties: from solvents to the orange carotenoid protein. Biochim Biophys Acta 1847:1044–1054. doi:10.1016/j.bbabio.2015.05.010
Lakowicz JR (2013) Principles of fluorescence spectroscopy. Springer, Berlin
Leverenz RL, Jallet D, Li MD, Mathies RA, Kirilovsky D, Kerfeld CA (2014) Structural and functional modularity of the orange carotenoid protein: distinct roles for the N- and C-terminal domains in cyanobacterial photoprotection. Plant Cell 26:426–437. doi:10.1105/tpc.113.118588
Leverenz RL et al (2015) A 12 Å carotenoid translocation in a photoswitch associated with cyanobacterial photoprotection. Science 348:3
Liu WL, Wang ZG, Zheng ZR, Li AH, Su WH (2008) Effect of β-ring rotation on the structures and vibrational spectra of β-carotene: density functional theory analysis. J Phys Chem A 112(42):10580–10585
Liu H, Zhang H, Niedzwiedzki DM, Prado M, He G, Gross ML, Blankenship RE (2013) Phycobilisomes supply excitations to both photosystems in a megacomplex in cyanobacteria. Science 342:1104–1107
Liu H, Zhang H, King JD, Wolf NR, Prado M, Gross ML, Blankenship RE (2014) Mass spectrometry footprinting reveals the structural rearrangements of cyanobacterial orange carotenoid protein upon light activation. Biochim Biophys Acta 1837:1955–1963. doi:10.1016/j.bbabio.2014.09.004
Liu H et al (2016) Dramatic Domain Rearrangements of the Cyanobacterial Orange Carotenoid Protein upon Photoactivation. Biochemistry 55:1003–1009. doi:10.1021/acs.biochem.6b00013
Maksimov EG et al (2014) The time course of non-photochemical quenching in phycobilisomes of Synechocystis sp. PCC6803 as revealed by picosecond time-resolved fluorimetry. Biochim Biophys Acta 1837:1540–1547. doi:10.1016/j.bbabio.2014.01.010
Maksimov EG et al (2015) The Signaling State of Orange Carotenoid Protein. Biophys J 109:595–607. doi:10.1016/j.bpj.2015.06.052
Misawa N et al (1995) Structure and functional analysis of a marine bacterial carotenoid biosynthesis gene cluster and astaxanthin biosynthetic pathway proposed at the gene level. J Bacteriol 177:6575–6584
Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2(9):2212–2221. doi:10.1038/nprot.2007.321
Pascal AA et al (2005) Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 436:134–137
Polívka T, Kerfeld CA, Pascher T, Sundström V (2005) Spectroscopic Properties of the Carotenoid 3‘-Hydroxyechinenone in the orange carotenoid protein from the Cyanobacterium Arthrospira maxima. Biochemistry 44:3994–4003. doi:10.1021/bi047473t
Salares V, Young N, Bernstein H, Carey P (1979) Mechanisms of spectral shifts in lobster carotenoproteins The resonance raman spectra of ovoverdin and the crustacyanins. Biochim Biophys Acta (BBA) Protein Struct 576:176–191
Shakhnovich EI, Finkelstein AV (1989) Theory of cooperative transitions in protein molecules. I. Why denaturation of globular protein is a first-order phase transition. Biopolymers 28:1667–1680. doi:10.1002/bip.360281003
Stadnichuk IN, Yanyushin MF, Maksimov EG, Lukashev EP, Zharmukhamedov SK, Elanskaya IV, Paschenko VZ (2012) Site of non-photochemical quenching of the phycobilisome by orange carotenoid protein in the cyanobacterium Synechocystis sp. PCC 6803. Biochim Biophys Acta 1817:1436–1445. doi:10.1016/j.bbabio.2012.03.023
Stadnichuk IN et al (2013) Fluorescence quenching of the phycobilisome terminal emitter LCM from the cyanobacterium Synechocystis sp. PCC 6803 detected in vivo and in vitro. J Photochem Photobiol B 125:137–145. doi:10.1016/j.jphotobiol.2013.05.014
Wilson A et al (2010) Structural determinants underlying photoprotection in the photoactive orange carotenoid protein of cyanobacteria. J Biol Chem 285:18364–18375. doi:10.1074/jbc.M110.115709
Wilson A, Punginelli C, Couturier M, Perreau F, Kirilovsky D (2011) Essential role of two tyrosines and two tryptophans on the photoprotection activity of the Orange Carotenoid Protein. Biochim Biophys Acta 1807:293–301. doi:10.1016/j.bbabio.2010.12.009
Wilson A, Gwizdala M, Mezzetti A, Alexandre M, Kerfeld CA, Kirilovsky D (2012) The essential role of the N-terminal domain of the orange carotenoid protein in cyanobacterial photoprotection: importance of a positive charge for phycobilisome binding. Plant Cell 24:1972–1983. doi:10.1105/tpc.112.096909
Yguerabide J, Epstein HF, Stryer L (1970) Segmental flexibility in an antibody molecule. J Mol Biol 51:573–590
Zhang H, Liu H, Niedzwiedzki DM, Prado M, Jiang J, Gross ML, Blankenship RE (2014) Molecular mechanism of photoactivation and structural location of the cyanobacterial orange carotenoid protein. Biochemistry 53:13–19. doi:10.1021/bi401539w
Acknowledgments
V.Z.P. and E.G.M. thank the Russian Foundation for Basic Research (Projects Nos. 14-04-01536A and 15-04-01930A) for partial support of this work. E.A.S. and E.G.M. thank the Russian Scientific Foundation (Project 14-17-00451) for partial support of this work. E.G.M., G.V.T., and K.E.K. thank the Russian Ministry of Education and Science (Project MK-5949.2015.4) for partial support of this work. This work was supported, in part by a Dynasty Foundation Fellowship to E.G.M. The reported study was funded by RFBR and Moscow city Government according to the research Project No. 15-34-70007 «mol_a_mos» . T.F., F.-J.S, N.N.T., M.W., and M.M gratefully acknowledge support from the Stifterverband für die Deutsche Wissenschaft (Study reform Project “OPLChem”) and the German Research Foundation (DFG, Cluster of Excellence “Unifying Concepts in Catalysis”).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maksimov, E.G., Moldenhauer, M., Shirshin, E.A. et al. A comparative study of three signaling forms of the orange carotenoid protein. Photosynth Res 130, 389–401 (2016). https://doi.org/10.1007/s11120-016-0272-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-016-0272-8