
Abstract
Mutations in the CER9 gene of Arabidopsis thaliana L. contribute to the amplification of the cuticular wax and consequently mitigation of water loss, thereby fortifying drought resilience. Recently, genes homologous to CER9, termed SUD1 genes, have been annotated in bread wheat (Triticum aestivum L.). However, no research has been done on these genes in T. aestivum. Hence, our study aimed to employ CRISPR/Cas technology to knock out the CER9/SUD1 gene orthologs in bread wheat. For this, five guide RNAs were meticulously chosen and merged into a singular vector. Delivery of the CRISPR/Cas components was arranged through Agrobacterium tumefaciens, utilized for transforming immature embryos of two agricultural spring bread wheat varieties: Taya and Sigma. Among the 13 transgenic plants procured, four manifested positivity for the reporter gene GFP and Cas9 gene. Notably, substantial deletions ranging from 284 to 398 bp within the CER9/SUD1 gene were discerned in these plants. Additionally, two of the edited plants exhibited an absence of CER9/SUD1 transcripts, while the other two displayed a noteworthy 5.4-fold reduction in CER9/SUD1 gene expression compared to the wild type. Intriguingly, the genome-edited plants of the T1 generation showcased enhanced growth parameters compared to the wild type under both standard and drought conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drought’s acceleration, propelled by diminished precipitation, global warming, and the depletion of surface and subterranean water reservoirs, now poses an increasingly severe menace to global agriculture, profoundly constricting crop productivity. Anticipations lean toward genetic modifications capable of limiting transpirational water losses, a potential way to bolster plant resilience against drought (Lu et al. 2012). The cuticle, enfolding the majority of a plant’s aboveground structures, assumes a pivotal role in preserving moisture within plant tissues, furnishing them with the capacity to thrive amidst water-scarce environments. Comprising intracuticular wax ensconced within the cutin polymer matrix and epicuticular wax situated on the external surface of the cutin, the cuticle acts as a barrier. This multifaceted layer regulates plant water loss, chiefly linked to non-stomatal transpiration, while also exerting influence on transpiration via stomatal pores (Lu et al. 2012; Lai et al. 2007). Several eceriferum (cer) mutations affecting the structure and composition of cuticular wax have been identified in Arabidopsis thaliana L. The most interesting mutation is observed in the CER9 gene, which leads to an increased content of primary alcohols in the cuticular wax of stems (Jenks et al. 1995). These CER9 mutants have exhibited noteworthy shifts in wax profiles, notably elevating the presence of very long-chain fatty acids—tetracosanoic acid (C24) and hexacosanoic acid (C26)—which are typically detected in minute quantities in the wild type (Jenks et al. 1995; Zhao et al. 2014). Albeit maintaining the ester chain length distribution akin to the wild type, CER9 mutants manifest higher alcohol content (Lai et al. 2007). The wax quantity in CER9 mutants has shown a substantial increase, by 27% in one variant and by 57% in another, relative to the wild type (Lu et al. 2012). Soil-based experiments have unveiled heightened drought tolerance in these mutants (Lu et al. 2012). It was revealed that the CER9 gene encodes E3 ubiquitin ligase, which is involved in cuticle biosynthesis and maintaining the water status of plants (Zhao et al. 2014). This gene, also known as SUD1 in A. thaliana, is situated at the AT4G34100 locus (Doblas et al. 2013). Thus, the knockout of the CER9/SUD1 gene may fortify the waxy layer of the cuticle, rendering the CER9/SUD1 gene a convenient target in increasing drought tolerance in crops (Goodwin et al. 2005). While experimentation with CER9/SUD1 genes has centered on A. thaliana, the presence of orthologs in bread wheat is also conceivable. Unfortunately, orthologs of the CER9/SUD1 gene in other plant species have not been investigated. Thus, among the primary objectives was the exploration and identification of CER9/SUD1 gene homologs within the bread wheat genome. Advancements in CRISPR/Cas genome editing for wheat have facilitated this study’s pursuit of knocking out the ortholog of the CER9/SUD1 gene within the bread wheat genome (Kuluev et al. 2022). Thus, the goal of this study was to knock out the ortholog of the CER9/SUD1 gene in the genome of bread wheat using CRISPR/Cas technology. A distinctive feature of this study was the use of production varieties of bread wheat grown in Russia rather than model varieties. It was assumed that the edited plants would be characterized by increased drought tolerance compared to the original variety.
Materials and Methods
Finding the Target Gene and gRNA Design
The study aimed to ensure comprehensive knockout of the CER9/SUD1 gene across diverse wheat variants, accounting for potential gene variant disparities. The first step was to search for orthologs of the CER9/SUD1 gene in the bread wheat genome. There are 19 genes and 30 transcript variants published in GenBank under the name SUD1 in Triticum aestivum. Using the sequence of AtCER9 gene (NM_119571) and BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi), a search for homologous sequences in the genes of T. aestivum was carried out. Subsequently, nine homologous genes sharing nucleotide sequence similarities with the AtCER9 gene (XM_044569287.1; XM_044569288; XM_048693585; XM_037601843; XM_048693586; XM_037601844; XM_020295452; XM_044580090; XM_020295453) were selected for further analysis. All these sequences were aligned using the MEGA11 (Molecular Evolutionary Genetics Analysis version 11), and conserved DNA regions were identified. Next, conserved regions of the target gene were analyzed via the online gRNA design program CRISPOR (http://crispor.gi.ucsc.edu/). Secondary structures of designed gRNAs were analyzed using the RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). The ideal secondary structure of an gRNA molecule should include two hairpins and a maximum of eight complementary nucleotides in the spacer region (Fig. S1). Suitable gRNA sequences for the CER9/SUD1 gene were checked in the Ensembl database (https://www.ensembl.org/) to detect potential sites of off-target editing. If such regions were not found, or they fell into non-coding regions (introns, tandem repeats), the gRNA sequence was considered suitable for use.
Assembly of CRISPR/Cas Vector for Genome Editing
The selected gRNA nucleotide sequences were saved as FASTA files and uploaded to the online platform http://crispr-multiplex.cbs.umn.edu (Cermak et al. 2017). PCR was carried out using high-fidelity SuperFi DNA polymerase (Invitrogen, USA). PCR conditions: 1 cycle: 98 °C, 1 min; 30 cycles: 98 °C, 10 s; 60 °C, 15 s; 72 °C, 15 s; 1 cycle: 72 °C, 2 min. The amplicons were introduced into the pMOD_b2103 plasmid using the Golden Gate Assembly and cloned into Escherichia coli strain XL1-blue. Plasmids for knockout of the CER9/SUD1 gene were created using a multipurpose toolkit developed by Daniel Voytas according to the instructions (Cermak et al. 2017). The gRNAs amplified using SuperFi II DNA polymerase (Invitrogen, USA) were introduced into the pMOD_b2103 plasmid using the Golden Gate Assembly and cloned into Escherichia coli strain XL1-blue. Next, single colonies were selected, the insertion of gRNAs was checked by PCR with primers TC320/TC089R (Table S1), and the resulting amplicon was sequenced. Plasmid DNA was isolated from bacterial colonies using a commercial kit (Biolabmix, Russia), and a modular construct was assembled in the backbone pTRANS_230 using the pMOD_A0503 and pMOD_C3003 modules by the Golden Gate reaction. The created vectors were cloned into E. coli strain DH10B; single colonies were selected and analyzed by PCR and sequencing with primers TC430/NB463 (Cermak et al. 2017). The sequences of the primers are presented in Table S1.
Bacterial Transformation
Chemical transformation of E. coli was carried out according to a standard protocol using heat shock at 42 °C. After transformation, bacteria were sown on LB medium containing a combination of two selective antibiotics: kanamycin (for the target vector, 50 mg/l) and tetracycline (for strain XL1-blue, 100 mg/l) or streptomycin (for strain DH10B, 100 mg/l). Transformation of Agrobacterium tumefaciens strain EHA105 was carried out by electroporation using the MicroPulser (Bio-Rad, USA). Next, the bacteria were inoculated on an LB medium containing the selective antibiotics kanamycin (100 mg/l) and rifampicin (100 mg/l).
Agrobacterium-Mediated Transformation of Immature Bread Wheat Embryos
Before the start of the research, an analysis of wheat transformation methods was carried out, which is described in detail in our review article (Kuluev et al. 2022). An analysis of the literature has shown that an effective method of genetic transformation of bread wheat is Agrobacterium-mediated transformation combined with organogenesis using immature embryos as explants (Hensel 2020). For the Agrobacterium-mediated transformation of wheat, we used the Taya variety provided by the P.P. Lukyanenko National Grain Center (Krasnodar, Russia) and the Sigma variety provided by the Omsk Agricultural Scientific Center (Omsk, Russia). These varieties are cultivated in Russia and are not model objects of biotechnology.
Initially, the lines of agrobacteria were grown for 16–20 h in a liquid LB medium with the addition of selective antibiotics in an orbital shaker at 27 °C at 180–200 rpm. Then, 10 ml of Agrobacterium suspension was transferred into 100 ml of LB medium, and the overnight culture was grown for 14–16 h until OD600 = 0.6–0.8. An overnight culture of agrobacteria was centrifuged at 3000 g for 10 min and resuspended in 10 ml of minA medium with the addition of 100 mM acetosyringone. The bacteria were grown for another 4 h under the same conditions and used for plant transformation.
Immature wheat grains were selected from plants that were cultivated in the open ground during the period of milky-wax maturity (75–80 days after seed sowing). The grains were isolated from the central part of the spikelet and then surface sterilized with 70% ethyl alcohol for 2 min and a 10% solution of commercial bleach “Belizna” (Sterlitamak, Russia) for 15 min, after which the grains were washed with sterile distilled water four times. The embryos were mechanically removed from the grains and placed in a liquid MS medium (Table S2). Ten milliliters of bacterial suspension was added to 10 ml of liquid MS medium for transformation (Table S3) with immature embryos and cultured for 3 h with shaking on an orbital shaker (120 rpm, room temperature). After this, the suspension of agrobacteria was poured through sterile filter paper, and the explants remaining on the paper were slightly dried and carefully transferred to a solid MS medium for co-cultivation (Table S4) in the dark at 22 °C for 2 days. After this, the explants were placed on the same MS medium containing the antibiotic cefotaxime to eliminate agrobacteria, and the Petri dishes were left in the dark at 25 °C for 2 days. The composition of the co-culture medium is given in Table S4. The procedures of transformation, co-cultivation, callus induction, regeneration, and rooting were carried out on nutrient media recommended in the study of Sparks and Jones (2009).
Callus Formation, Regeneration, Rooting, and Acclimatization
After the co-cultivation step, the explants were transferred into callus induction media (Table S5). The calli were then transferred to a regeneration medium (Table S6) containing glufosinate ammonium (glufosinate, PanReac, USA) (7.5 mg/l) to screen out non-transformed plants at the stage of induction of shoot regeneration. Regenerated shoots were placed in Magenta vessels with a root-inducing medium. Rooted plants were transferred into growing vessels containing a mixture of perlite and soil (1:1 ratio), covered with thin plastic film (the first 2 weeks), and placed in growth chambers at 25 °C, 60% humidity, 10,000 lx illumination, and 16-h light period. The composition of all culture media is given in the Supplementary information (Tables S2-S6).
PCR Analysis of Transgenic Plants, Sequencing
DNA for PCR analysis was isolated from wheat that was successfully acclimated to soil conditions. To control the insertion of T-DNA, PCR analysis was performed in the presence of the Cas (primers Cas2F/Cas3R) and GFP (primers GFPF/GFPR). To analyze the changes that occurred in the target gene, PCR was performed, and the edited region of the gene was sequenced with primers Seq_cer 1F and Seq_cer 3R (Table S1).
The amplicons were purified using a diaGene reagent kit (Dia-M, Russia). When sequencing fragments of the CER9/SUD1 gene, the final volume of the reaction mixture was 10 µl and contained 1 µl of primer, 1 µl of purified DNA template, 7.5 µl of sterile deionized water, and 0.5 µl of BigDye v3.1 (Thermo Fisher Scientific, CШA). The order of the sequencing reaction cycles was as follows: denaturation at 96 °C for 10 s, primer annealing at 53 °C for 5 s, and elongation at 60 °C for 4 min for all 30 cycles. Fluorescently labeled amplicons were analyzed using a genetic analyzer Nanophore 05 (Syntol, Russia). Sequencing of the studied regions of each selected line was carried out from both ends using forward and reverse primers (Table S1) in two technical (each amplicon was sequenced twice) replicates. Then, for each plant, by aligning the resulting nucleotide sequences, one consensus sequence was compiled using the program MegAlign (DNASTAR, CШA). Also, using this program, a computer analysis of nucleotide sequences was performed; nucleotide numbering was carried out using the reference gene XM_044569287.1 from GenBank.
Real-Time Quantitative Reverse Transcription PCR
Total RNA was isolated with TRIzol reagent from leaves, and the first strand of cDNA was synthesized using oligo(dT) primer and M-MuLV reverse transcriptase (Evrogen, Russia). Quantification of mRNA for CER9/SUD1 gene converted to cDNA was performed via real-time PCR in the presence of SYBR Green I intercalate dye in Rotor-Gene™ 6000 thermocycler (Corbett Research, Australia). Primers CER9RTF/CER9RTR were used for RT-PCR of the CER9/SUD1 gene (Table S1). Calculations were performed using mRNA of RLI (RNase L Inhibitor-like protein) as standard, its expression level taken as 100%. RT-PCR of the RLI gene was performed using primer pair RLIF/RLIR (Table S1). RT-PCR conditions: 1 cycle: 94 °C, 1 min; 30 cycles: 94 °C, 20 s; 53 °C, 20 s; 72 °C, 20 s; 1 cycle: 72 °C, 2 min. For each transgenic variant, leaves from three different T1 plants were used (n = 3). The significance of differences was assessed using Duncan’s test.
Morphometric Analysis of Edited Plants
Seeds from the T0 generation’s edited plants were sown into 1-l growing vessels filled with Terra vita universal soil (Nevatorf, Russia), encompassing ten seeds per vessel. The experiment unfolded in two variants: the control group received bi-weekly watering with 150 ml of water, while the experimental group, subjected to the same water volume, was watered only once a week. Plant height, shoot freshness, and dry weight measurements transpired precisely 2 months post-seed sowing. The research findings were graphically illustrated via histograms presenting average values, with the bars delineating the standard error of the mean. Statistical assessment of differences across all experiments was conducted using the Mann–Whitney U test.
Results
Design of gRNAs and Assembly of a Module Plasmid Based on pTRANS_230 Backbone
Nine homologous nucleotide sequences of the CER9/SUD1 gene were taken from GenBank and were aligned using the MEGA11 program with the Muscle method. The alignment results showed homogeneity of the open reading frame (ORF) sequences of all nine selected variants of the CER9/SUD1 gene. The length of the nucleotide sequence encoding the CER9/SUD1 protein is 3300 bp. The identified conserved gene regions were analyzed via the CRISPOR system (http://crispor.tefor.net/) to design gRNAs. The 2185 bp input sequence contained 250 potential gRNA sequences ranked by specificity according to Hsu et al. (2013).
Post-gRNA design, verification of their existence in the complete genome version of the gene from the Ensembl database ensued. Alignment with chosen spacers transpired in MEGA11, disclosing the presence of three homeologs of the CER9/SUD1 gene on chromosomes 7A (TraesCS7A02G524600), 7B (TraesCS7B02G442100), and 7D (TraesCS7D02G513500) in Ensembl. Impressively, all five generated gRNAs proved suitable for targeting these three identified homeologs of the CER9/SUD1 gene. Rigorous analysis of off-target gRNA spacer attachments was conducted using Ensembl, with the exclusion of all gRNAs found to be complementary to off-target genes.
A pivotal facet of gRNA design involved meticulous scrutiny of their secondary structures, acknowledging their substantial impact on the efficiency of the CRISPR/Cas system. Supplementary Information (Fig. S1) outlines an exemplary suitable secondary structure. Consequently, five gRNA sequences, detailed in Table 1, were meticulously selected. The corresponding protospacer locations are delineated in Fig. S2.
To increase the possibility of successful knockout of the CER9/SUD1 gene, a vector for multiplex targeting of all five selected gRNAs was created (Table 1). The primary objective aimed at inducing diverse deletions within the target gene, ultimately resulting in its complete knockout and the absence of the gene’s protein product. The intricate process of assembling the multiplex vector unfolded within the plasmid pMOD_B2103. Initially, the utilization of the restriction enzyme BanI was imperative to cleave the plasmid pMOD_B2103 in the first PCR reaction (refer to Fig. S3), crucial for preventing the formation of amplicons of varying lengths (as depicted in Fig. S4). The plasmid pMOD_B2103 map depicting integrated guide RNAs is presented in Supplementary Information (Fig. S5). Following each step of the Golden Gate reaction, sequencing protocols were rigorously executed to scrutinize the inserts’ integrity and their orientation. This process yielded 17 colonies of E. coli strain DH10B, each harboring the target vector (as illustrated in Fig. S6). For the subsequent phase involving the delivery of the vector (refer to Fig. S7) into plant cells, the Agrobacterium-mediated transformation method was selected. This process would leverage strain EHA105 to facilitate efficient and delivery into the plant cells.
Creating Transgenic Wheat Plants
For the Taya variety, 390 immature embryos were isolated, of which 163 formed callus tissue (Table 2). For the Sigma variety, 236 immature embryos were obtained, of which 89 formed calli (Table 2).
After 7–10 days of cultivation in an induction medium supplemented with 5 mg/l glufosinate, some of the callus cultures began to turn yellow or brown. At this stage of cultivation, 142 samples of callus of the Taya variety and 82 samples of callus of the Sigma variety were isolated. Next, callus cultures were transplanted into a regeneration medium containing 7.5 mg/l glufosinate. After 2 weeks of cultivation on a medium with 7.5 mg/l glufosinate, the calli were transferred to a regeneration medium without a selective agent. At this stage, 85 calli of the Taya variety and 64 calli of the Sigma variety were selected. Within 2–3 weeks after transferring from the co-culture medium, some callus cultures began to produce green points of plant regeneration. However, some calli were highly hydrated, some of which turned yellow or brown. After 4 weeks of cultivation on media for the callus induction and regeneration, 42 green calli with shoot regeneration points were obtained among the Taya variety and 21 among the Sigma variety (Fig. 1a, b). Regenerated shoots 1.5–2 cm high were transferred into Magenta vessels containing regeneration medium without hormones (Fig. 1c, d).

Regeneration of wheat from immature embryos at various stages: a green calli, which subsequently gave rise to seedlings. b Seedlings developing on maternal calli in a regenerative medium. c, d Growth and rooting of shoots. e, f Seedlings acclimatized to the soil conditions and used to obtain T1 seeds. Bars = 1 cm
After 4 weeks of cultivation, ten rooted plants of the Taya variety and eight of the Sigma variety were obtained (Table 2). It was possible to acclimatize seven plants of the Taya variety and six plants of the Sigma variety to soil conditions (Fig. 1e, f). Total DNA was isolated from these 13 plants for subsequent PCR analysis to identify the Cas and GFP genes.
PCR Analysis of Transgenic Plants for the Presence of Cas and GFP Genes and Changes in the Size of the CER9/SUD1 Gene
Thirteen DNA samples were analyzed for the presence of the Cas gene, which is integrated into the plant genome along with the T-DNA of the pTRANS_230 vector. It turned out that all wheat plants of the Taya and Sigma varieties acclimatized to soil conditions contain the Cas gene (Fig. 2a). Since the T-DNA of the vector also contains the GFP gene, PCR analysis was also carried out for the presence of this reporter gene. It was shown that all seven plants of the Taya variety and six plants of the Sigma variety contain the GFP gene in their genome (Fig. 2b).

a Results of PCR analysis for the presence of the Cas9 gene using primers Cas3F and Cas2R (PCR product length is 1218 bp). 1–13: DNA samples of wheat seedlings (1–7: Taya variety, 8–13: Sigma variety), 14–16: positive PCR control (transgenic wheat plants with the Cas gene), 17: negative PCR control, 18: 1 kb DNA Ladder (Evrogen, Russia). b Results of PCR analysis for the presence of the GFP gene using primers NB463 and CFPF (PCR product length is 772 bp). 1–7: DNA samples of wheat seedlings of the Taya variety, 8–10: positive PCR control (transgenic wheat plants with the GFP gene), 11: 1 kb DNA Ladder (Evrogen, Russia). 12–17: DNA samples of wheat seedlings of the Sigma variety, 18: negative control. c Results of PCR analysis of the edited region of the CER9/SUD1 gene using primers Seq_cer1F and Seq_cer3R (PCR product length is 1139 bp). 1: 1 kb DNA Ladder, 2–14: DNA samples of various wheat seedlings (2–8: Taya variety, correspond to sample numbers 1–7; 9–14: Sigma variety, correspond to sample numbers 1–6), 15: positive PCR control, 16: negative PCR control
To check the presence of mutations in the CER9/SUD1 gene, we performed PCR of a region of this gene in all 13 isolated DNA samples. PCR with the primers Seq_cer1F and Seq_cer3R (amplicon size 1139 bp, Table S1) showed the presence of the DNA fragment in all 13 analyzed samples (Fig. 2c). Samples 1, 5, and 6 showed smaller sizes of amplicons than the wild type (Fig. 2c, lanes 2, 6, and 7). Sample 5 had an amplicon size smaller by approximately 300 bp, while samples 1 and 6 had a smaller amplicon size of approximately 400 bp. In addition, sample 6 also exhibited an amplicon of the same size as the wild type. In the Sigma variety, amplicons of different sizes were detected in samples 2, 5, and 6, corresponding to lanes 10, 13, and 14 on gel electrophoresis (Fig. 2c). In addition, samples 2, 5, and 6 exhibited amplicons approximately the same size as the wild type. In these three samples, the altered amplicon was smaller than in the wild type by approximately 400 bp (Fig. 2c). Thus, the results obtained indicate the possible presence of deletions in the CER9/SUD1 gene in the analyzed plant samples. To prove the presence of deletions, amplicons differing in size from samples 1 and 5 of the Taya variety and 2 and 5 of the Sigma variety were eluted from the gel for further analysis of nucleotide sequences.
Sequencing of the CER9/SUD1 Gene Fragment
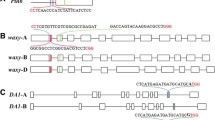
Four selected amplicons of the CER9/SUD1 gene region were sequenced on both sides using primers Seq_cer1F and Seq_cer3R. As a control, similar regions were sequenced from both ends in wild-type varieties Taya and Sigma. The obtained nucleotide sequences of samples Taya 1, Taya 5, Sigma 2, Sigma 5, Taya WT, and Sigma WT were aligned using the MegAlign (DNASTAR, USA). The analysis showed the presence of extensive deletions in the CER9/SUD1 gene in selected wheat samples (Fig. 3). The size of the deletion of sample Taya 1 was 394 bp, for Taya 5, 284 bp; for Sigma 2, 375 bp; and for Sigma 5, 398 bp. Based on the data, it was concluded that in these four samples, successful editing of the target gene occurred, and in the case of samples Taya 1 and Taya 5, possibly in a homozygous version (Fig. 2c), which should lead to a complete knockout of the CER9/SUD1 gene. In samples Sigma 2 and Sigma 5, one copy of the CER9/SUD1 gene was probably edited to form heterozygotes. The four samples of bread wheat were successfully grown to obtain seeds, which were used for morphometric analysis in the T1 generation under normal and drought conditions. Other samples of bread wheat, in which the size of the DNA fragment did not change, were also brought to the seed stage, but have not yet been used for morphometric analysis and subsequent propagation.

Comparative analysis of nucleotide sequences of the CER9/SUD1 gene region flanked by primers Seq_cer1F and Seq_cer3R in wild-type Taya and Sigma varieties and edited using CRISPR/Cas (Taya 1 and 5, Sigma 2 and 5). A region of 1139 bp in size (594–1732 bp) was sequenced; a region of 1098 bp in size (614–1712 bp) was used for alignment. The figure shows a fragment of the analyzed region measuring 960 bp. Numbering is given according to XM_044569287.1
Drought Tolerance of Edited Bread Wheat Plants
Seeds from plants of the T0 generation obtained after genome editing and acclimatization were sown on the soil to obtain seeds of the T1 generation. It was decided to measure the height of plants grown under normal conditions (Fig. 4a) and drought conditions (Fig. 4b). Under normal conditions, Taya 5, Sigma 2, and Sigma 5 plants exceeded the wild type in stem height (Fig. 4c). Under drought conditions, in Taya 1, Sigma 2, and Sigma 5 plants, stems were higher than in wild type (Fig. 4d). The edited plants showed no decreasing in growth compared to the wild type under normal or drought conditions.

Growing edited wheat plants of the T1 generation under normal (a) and drought (b) conditions. c Plant height under normal conditions. d Plant height under drought conditions. Taya WT and Sigma WT are wild-type wheats of the corresponding varieties. Taya 1 and 5, edited plants of the Taya variety. Sigma 2 and 5, edited plants of the Sigma variety. Data are presented as the mean ± SEM (n = 10; *p ≤ 0.05)
The edited plants of Taya 1, Taya 5, Sigma 2, and Sigma 5 were characterized by higher shoot fresh weight under both normal (Fig. 5a) and drought conditions (Fig. 5b). Also, all the edited plants analyzed differed significantly from the wild type in higher dry weight under normal (Fig. 5c) and drought conditions (Fig. 5d).

Fresh (a, b) and dry (c, d) shoot weight of T1-edited generation of bread wheat plants under normal (a, c) and drought action (b, d). Taya WT and Sigma WT are wild-type wheats of the corresponding varieties. Taya 1 and 5, edited plants of the Taya variety. Sigma 2 and 5, edited plants of the Sigma variety. Data are presented as the mean ± SEM (n = 10; *p ≤ 0.05)
Expression Analysis of CER9/SUD1 Gene in Edited Plants
The edited T1 plants selected after PCR were used to determine the level of CER9/SUD1 gene transcripts in their leaves. A rather high level of expression of the gene was detected in the leaves of Taya WT and Sigma WT (Fig. 6). In the Taya 1 and Taya 5 plants, CER9/SUD1 gene transcripts were not detected. At the same time, expression of the gene was detected in Sigma 2 and Sigma 5 plants (Fig. 6). It should be noted that these two plants showed a decrease in transcript content by an average of 5.4 times compared to the wild type.

Relative gene expression analysis of CER9/SUD1 gene with respect to the internal control used in this study. In Taya 1 and Taya 5 plants, transcripts of the CER9/SUD1 were not detected. Data are presented as the mean ± SEM. Letters indicate significantly different results after the Duncan test (n = 3)
Discussion
The most important physiological function of waxes is to limit non-stomatal water loss, so they are involved in increasing of drought tolerance of plants (Lai et al. 2007). Cuticular wax of leaves and stems plays an important protective role against various stress factors (Jenks et al. 1995). Mutation of the CER9 gene in A. thaliana, encoding the E3 ubiquitin ligase, causes an increase in the quantity of 18-carbon cutin monomers and very long-chain free fatty acids (C24, C26) in cuticular wax, both of which are associated with increased cuticular membrane thickness and drought tolerance (Zhao et al. 2014). Therefore, we hypothesized that the mutation of the CER9/SUD1 gene in wheat could also lead to the accumulation of wax and, accordingly, to a decrease in water loss through the cuticle. The genome of bread wheat is hexaploid and consists of three subgenomes (Kuluev et al. 2023), and for each gene in these subgenomes, there may be homeologs, which are also desirable to knock out during genome editing. Based on these considerations, we selected five gRNAs to knock out all putative three homeologs of the bread wheat CER9/SUD1 gene. Using a multipurpose toolkit for genome editing (Cermak et al. 2017), we were able to clone all five selected oligonucleotides. Thus, a vector to knock out the CER9/SUD1 gene in bread wheat was created during this investigation. According to our review article (Kuluev et al. 2022), the efficiency of Agrobacterium-mediated transformation of bread wheat may be higher than with a biolistic method. Therefore, A. tumefaciens was used for further genetic transformation. The Agrobacterium-mediated transformation method is used frequently for delivering CRISPR/Cas components into bread wheat cells (Singh et al. 2018; Zhang et al. 2018; Abe et al. 2019). However, most studies use model varieties of bread wheat for genome editing. The specifics of this research are the usage of Russian agricultural bread wheat varieties for genetic transformation. Taya and Sigma were selected among other industrially cultivated varieties through their ability to effectively regenerate and form the callus tissue (unpublished data). In our work, we used the standard methodology of Agrobacterium-mediated transformation of immature bread wheat embryos (Hensel 2020) and common nutrient media formulations (Sparks and Jones 2009) without any modifications. Since no model varieties of bread wheat were used in the study, the preliminary search for varieties capable of efficient regeneration was an important part of the whole work. From a large list of Russian varieties, Taya and Sigma were found to be such varieties. Thirteen transgenic plants were created and acclimated to soil conditions during the study. They were grown in a greenhouse until T1 seeds were obtained. Using PCR, it was proven that these 13 plants included the Cas and GFP genes (Fig. 2a, b). PCR of the edited region showed the presence of large deletions in six plants (Fig. 2c), four of them were studied in more detail; these plants were named Taya 1, Taya 5, Sigma 2, and Sigma 5, according to the name of the variety and sample number. Sequencing of this region confirmed the presence of large deletions in these four plants (Fig. 3). All deletions occurred close to PAM regions in the recognition region of our selected gRNAs. Seeds obtained from these plants were sown on the soil to obtain T1 generation plants. The edited plants did not differ significantly in phenotype from the wild type during normal and drought conditions (Fig. 4a, b). However, measuring the height of 2-month-old plants showed that the edited plants are higher both under normal (Taya 5, Sigma 2, and Sigma 5) and drought (Taya 1, Sigma 2, and Sigma 5) conditions (Fig. 4c, d). A more significant difference between wild-type and edited plants was found when comparing fresh and dry weights under both normal (Fig. 4a, c) and drought conditions (Fig. 4b, e). A more detailed morphometric analysis is planned to be held with plants of the T2 generation.
Thus, we selected and tested guide RNAs for knockout of the CER9/SUD1 genes in bread wheat. As a result of the transformation with an assembled vector, four plants with a knockout of CER9/SUD1 genes of the T0 generation were obtained, and 20 plants of the T1 generation were grown. It was shown that the edited plants differ from the wild type by higher and heavier by weight of shoots under normal and drought conditions.
Data Availability
No datasets were generated or analysed during the current study.
References
Abe F, Haque E, Hisano H, Tanaka T, Kamiya Y, Mikami M, Kawaura K, Endo M, Onishi K, Hayashi T, Sato K (2019) Genome-edited triple-recessive mutation alters seed dormancy in wheat. Cell Rep 28:1362-1369.e4
Cermak T, Curtin SJ, Gil-Humanes J, Cegan R, Kono TJY, Konecna E, Belanto JJ, Starker CG, Mathre JW, Greenstein RL, Voytas DF (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29:1196–1217
Doblas VG, Amorim-Silva V, Pose D, Rosado A, Esteban A, Arro M, Azevedo H, Bombarely A, Borsani O, Valpuesta V, Ferrer A, Tavares RM, Botella MA (2013) The SUD1 gene encodes a putative E3 ubiquitin ligase and is a positive regulator of 3-hydroxy-3-methylglutaryl coenzyme a reductase activity in Arabidopsis. Plant Cell 25:728–743
Goodwin SM, Rashotte AM, Rahman M, Feldmann KA, Jenks MA (2005) Wax constituents on the inflorescence stems of double eceriferum mutants in Arabidopsis reveal complex gene interactions. Phytochemistry 66:771–780
Hensel G (2020) Genetic transformation of Triticeae cereals – summary of almost three-decade’s development. Biotechnol Adv 40:107484
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Jenks MA, Tuttle HA, Eigenbrode SD, Feldmann KA (1995) Leaf epicuticular waxes of the eceriferum mutants in Arabidopsis. Plant Physiol 108:369–377
Kuluev BR, Mikhailova EV, Kuluev AR, Galimova AA, Zaikina EA, Khlestkina EK (2022) Genome editing in species of the tribe Triticeae with the CRISPR/Cas system. Mol Biol 56:885–901
Kuluev AR, Kuluev BR, Chemeris AV (2023) The problem of the origin of subgenomes B, A, and D of bread wheat Triticum aestivum L.: old facts and new evidences. Biol Bull Rev 13:148–161
Lai C, Kunst L, Jetter R (2007) Composition of alkyl esters in the cuticular wax on inflorescence stems of Arabidopsis thaliana cer mutants. Plant J 50:189–196
Lu S, Zhao H, Des Marais DL, Parsons EP, Wen X, Xu X, Bangarusamy DK, Wang G, Rowland O, Juenger T, Bressan RA, Jenks MA (2012) Arabidopsis ECERIFERUM9 involvement in cuticle formation and maintenance of plant water status. Plant Physiol 159:930–944
Singh M, Kumar M, Albertsen MC, Young JK, Cigan AM (2018) Concurrent modifications in the three homeologs of Ms45 gene with CRISPR-Cas9 lead to rapid generation of male sterile bread wheat (Triticum aestivum L.). Plant Mol Biol 97:371–383
Sparks CA, Jones HD (2009) Biolistics transformation of wheat // Huw D. Jones and Peter R. Shewry (eds.), Methods in molecular biology, transgenic wheat, barley and oats. Springer Nature, pp 71–92
Zhang S, Zhang R, Song G, Gao J, Li W, Han X, Chen M, Li Y, Li G (2018) Targeted mutagenesis using the Agrobacterium tumefaciens-mediated CRISPR-Cas9 system in common wheat. BMC Plant Biol 18:302
Zhao H, Zhang H, Cui P, Ding F, Wang G, Li R, Jenks MA, Lu S, Xiong L (2014) The putative E3 ubiquitin ligase ECERIFERUM9 regulates abscisic acid biosynthesis and response during seed germination and postgermination growth in Arabidopsis. Plant Physiol 165:1255–1268
Acknowledgements
The authors extend their heartfelt appreciation for the wheat seeds utilized in this study, generously provided by the P.P. Lukyanenko National Grain Center (acknowledging Bespalova L.A.) and the Omsk Agrarian Scientific Center (with gratitude to Nikolaev P.N.).
Funding
The study of Khalit Musin, Elena Mikhaylova, Aizilya Galimova, Elvina Baimukhametova, Zarina Ibragimova, and Bulat Kuluev was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement no. 075–15-2021–1066, September 28, 2021). The study of Zaikina E.A., Kuluev A.R., Rakhmatullina I.F., and Berezhneva Z.A. was supported by state assignment no. 122030200143–8.
Author information
Authors and Affiliations
Contributions
KM, EM, EB: design of gRNA, assembly of CRISPR/Cas vector for genome editing, bacterial transformation. AG, ZI, IR: Agrobacterim-mediated transformation of immature bread wheat germs, creating of edited plants, PCR-analysis. AK: DNA sequencing. EZ: real-time quantitative reverse transcription PCR. ZB: morphometric analysis of edited plants. KB and KM: research design, data analysis, article writing. All authors took part in editing the article and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Musin, K., Mikhaylova, E., Galimova, A. et al. Knockout of the Bread Wheat CER9/SUD1 Gene Using CRISPR/Cas Technology. Plant Mol Biol Rep (2024). https://doi.org/10.1007/s11105-024-01495-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11105-024-01495-w