
Abstract
Iron (Fe) is an essential mineral element, and its deficiency in soil largely affects crop productivity. In plants, the molecular mechanisms underlying the genetic regulation of Fe-deficiency responses pinpointing microRNA (miRNA)-mediated regulation of Fe-deficiency response and its regulatory network are largely unaddressed. In the present study, we performed a small RNA-targeted whole-genome transcriptome analysis from hexaploid wheat and identified small RNAs (sRNAs) responding to Fe deficiency. Detailed analysis identified 105 differentially expressed miRNAs corresponding to Fe-deficiency response, and nine miRNAs were found to be novel in this study. Interestingly, tissue-specific regulation of Fe-deficiency response also participates through miRNA-mediated regulation. We identified 17 shoot-specific miRNAs and 18 root-specific miRNAs with altered expression. We validated the tissue specificity of these miRNAs by stem-loop quantitative RT-PCR that confirmed a temporal regulation. Furthermore, an attempt was made to predict their targets to speculate their participation in Fe-deficiency response. The miRNA target prediction analysis suggested a few major target genes, such as multicopper oxidases, E3 ubiquitin ligases, GRAS family, and WRKY transcription factors; those are previously known to play key roles in Fe homeostasis. The first preliminary information generated here will classify the repository of wheat miRNAs (with few novel miRNAs) for their role in Fe-deficiency response. Our work provides insights into miRNA-mediated regulatory pathways during Fe deficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Iron (Fe) is an essential micronutrient. Being the principal component of chlorophyll, Fe–S clusters of enzymes and cofactors, it participates in various biochemical processes, including photosynthesis and respiration (Briat et al. 2015). Despite being the fourth most abundant element in the Earth’s crust, its bioavailability to plants is restricted owing to its presence as sparingly soluble Fe3+ form in aerobic and high-pH soil environment (Buckhout and Schmidt 2013; Morrissey and Guerinot 2009). Fe deficiency in plants causes interveinal chlorosis and drastically impacts vegetative growth and crop yield (Abadía et al. 2011). Plants have devised distinct uptake mechanisms and efficient modes of Fe translocation for its tissue-specific distribution via specific transporters and chelators (Kobayashi et al. 2019). Dicots and non-graminaceous monocots utilise the strategy-I mode of Fe uptake by reduction of Fe3+ to Fe2+ at the root surface, followed by internalization of soluble Fe by membrane-localized iron-regulated transporter-1 (IRT1) (Hell and Stephan 2003). Graminaceous plants like maize and wheat, on the other hand, chelate Fe3+ by excreting phytosiderophores, i.e. chelation-based strategy, also known as strategy-II. The resulting complexes are then taken up by yellow stripe-like (YSL) transporter in the plasma membrane of the roots. Molecular components regulating Fe homeostasis in plants comprise distinct families of transcription factors (TFs) like basic helix-loop-helix (bHLH), WRKY and members of no apical meristem (NAC), IDE-binding factors (IDEF1 and IDEF2) (Colangelo and Guerinot 2004; Heim et al. 2003; Long et al. 2010; Ogo et al. 2007; Yan et al. 2016; Zhang et al. 2017; Zheng et al. 2010). Some of these TFs are regulated at post-translational levels by E3-ubiquitin ligases such as BRUTUS (BTS) in Arabidopsis and haemerythrin motif-containing zinc-finger protein (HRZ1 and HRZ2) in rice to tackle the deleterious consequences of activation of Fe uptake and transport machinery during Fe deficiency (Kobayashi et al. 2013; Selote et al. 2015).
Plant responses are regulated by a number of non-coding RNAs (ncRNAs) where these ncRNAs can be categorised into three major classes, i.e. small (18–30 nt), medium (31–200 nt) and long (> 201 nt) based on their nucleotide length (Yu et al. 2019). MicroRNAs (miRNAs) are a single-stranded non-coding endogenous class of small RNAs that regulate the target mRNAs by either causing their cleavage or translational repression (Chen 2009). In plants, miRNAs are known to play roles in diverse fundamental processes as controllers of vegetative and floral organ development, phytohormone signalling and regulation of various biotic and abiotic stress responses, including regulation of genes involved in nutrient uptake and transport under nutrient stress conditions (Gao et al. 2016; Guo et al. 2013; Jian et al. 2016; Li and Zhang 2016; Sunkar et al. 2012; Wang et al. 2014; Willmann and Poethig 2005; Zhang et al. 2013; Zhou et al. 2012). Previously, 24 miRNAs were found to have iron-deficiency responsive cis-elements (IDE1 and IDE2) in their promoter regions. Around 70% of them, i.e. 17 miRNAs, were responsive to Fe deficiency in Arabidopsis shoot and/or root (Kong and Yang 2010). In another study, 32 Fe-deficiency responsive miRNAs were identified using a microarray-based approach in leaves, roots and nodules of common bean (Phaseolus vulgaris) (Valdés-López et al. 2010). Differential expression pattern of miRNAs responsive to Fe deficiency in Arabidopsis rosette and shoot has also been analysed, pin-pointing eight miRNAs from seven different families (Waters et al. 2012). Recently, several seed iron concentration-related QTLs were found to be the targets of Fe-deficiency responsive miRNAs in rice recombinant inbred lines (RILs) (Agarwal et al. 2015). Seven of nine miRNAs identified in the study showed downregulation in response to Fe-deficient conditions. Furthermore, identifying 26 known and 55 novel Fe-deficiency responsive miRNAs in Citrus sinensis suggested a larger role being played by miRNAs during Fe deficiency (Jin et al. 2021).
Hexaploid wheat (Triticum aestivum L.) is the most widely grown cereal crop in many countries and accounts for a total of 20% of calorific intake by humans (FAOSTAT, http://www.fao.org/faostat/en/#data/FBS). Multiple miRNAs from wheat have already been characterized for their roles in various abiotic stresses, including nutrient starvation (nitrogen and phosphate), salinity and drought stress (Hou et al. 2020; Lu et al. 2011; Ravichandran et al. 2019). Wheat has a complex genetic architecture, and there is a dearth of knowledge about different means of genetic regulation of Fe homeostasis in this crop. Identifying distinct miRNAs and their targets during Fe-homeostasis will help develop regulatory networks. Previously, core components participating in strategy-II mode of Fe uptake and mobilization were identified using transcriptomics-based approaches in wheat (Kaur et al. 2019; Kaur et al. 2023; Wang et al. 2019, 2020). However, information on the role of miRNAs in the Fe-deficiency response in wheat is lacking. Therefore, we extended this work to get an insight into the miRNA-based regulation during Fe-deficient conditions. In the current study, we investigated the differentially expressed miRNAs in response to Fe deficiency to understand small RNA-mediated regulation of Fe-homeostasis. Our work identified a sub-set of the shoot and root-specific miRNAs targeting genes involved in Fe-mobilization in a tissue-specific manner.
Materials and Methods
Plant Material, Fe-deficiency Treatment and Sampling
Triticum aestivum cv. C306 (provided by Panjab Agricultural University, Ludhiana, India) was used for this experiment. Bread Wheat cv. C306 has good processing properties (including chapatti-making quality) (Bhatnagar et al. 2002; Singh et al. 2014). Wheat grains were stratified in the dark at 4 °C overnight and germinated for 4 days on Petri plates lined with Whatman filter paper. The endosperms were removed from the developing seedlings to cut out the nutrient supply from the seed. Subsequently, the seedlings were grown for 5 days in Hoagland’s nutrient solution and subjected to Fe-deficiency treatment. For Fe starvation (− Fe), 2 µM Fe (III) EDTA was used as the Fe source. For control plants (+ Fe), Hoagland’s nutrient solution was used, keeping other nutrients unchanged with 80 µM Fe(III) EDTA. Plants were grown in the growth chamber set at 21 ± 1 °C, 50–65% relative humidity, and a photon rate of 300 μmol quanta m−2 s−1 with a 16-h day/8-h night cycle. For sampling, whole roots and whole shoots were harvested separately at 6, 9, 12 and 15 days after Fe-deficiency treatment. RNA extractions were performed by pooling of 12 plants (in order to reduce the genetic variance) from three biological replicates at each time point of respective tissue and treatment. Each of the sample was prepared by pooling equimolar ratios of total RNA from each time point of respective tissue and treatment. A total of four samples were prepared for library construction and sequencing, viz., + Fe root, − Fe root, + Fe shoot and − Fe shoot.
Preparation of Small RNA Library and RNA Sequencing
The total RNA of control and treated roots and shoots was extracted (three biological replicates) using Trizol Reagent (Invitrogen, ThermoFisher, USA) according to the manufacturer’s instructions. The RNA quantity and purity was assessed using NanoDrop™ One (ThermoFisher Scientific) and denatured agarose gel electrophoresis. RNA integrity number (RIN) was checked using Bioanalyzer 2100 (Agilent Technologies, USA). The pooled samples from the different time points were proceeded for small RNA library preparation. The library was constructed by TruSeq Small RNA Sample Prep Kit (Illumina, USA). Small RNAs were ligated with 3′ and 5′ adapters, followed by reverse transcription, PCR enrichment, purification and size selection. The sRNA libraries were sequenced on the NovaSeq6000 Illumina Sequencing platform. Transcriptome sequencing data was deposited at NCBI (Submission: SUB12485490 with BioProject: PRJNA916207).
Bioinformatics Analysis and miRNA Identification
The data obtained from high throughput sequencing was processed into raw sequencing reads by CASAVA base recognition. Low-quality raw reads containing adaptors, 5′ primer contamination, polynucleotide tails and reads with > 50% bases having a Qphred less than or equal to 20 and the ones in which > 10% base information were indeterminable were discarded. The clean reads ranging from 18 to 30 nucleotides were mapped to the reference genome sequence of T. aestivum (ftp://ftp.ensemblgenomes.org/pub/plants/release-50/fasta/triticum_aestivum/dna/Triticum_aestivum.IWGSC.dna.toplevel.fa.gz) using Bowtie v0.12.9 (Langmead et al. 2009) to analyze their expression level and distribution on the genome. Rfam database v14 (Kalvari et al. 2021) and RepeatMasker 4.0.3 (Smit et al. 2013) were used to remove non-coding RNAs — rRNA, tRNA, snRNA, snoRNA and repeat sequences. The unmatched reads were classified as putative miRNAs and subsequently aligned against miRbasev22 (Kozomara et al. 2019) (http://www.mirbase.org/) using ncbi-blastv2.2.28 to obtain detailed information on mapped reads such as secondary structure, their length and number of occurrences etc. Matched sequences were identified as conserved miRNAs, and the characteristic hairpin structure of other remaining miRNAs (marked as novel miRNAs) was predicted by miRDeepv2_0_0_5, miREvo v1.1 software and RNA fold server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (Friedländer et al. 2012; Lorenz et al. 2011; Wen et al. 2012). Structures of miRNAs were predicted using their minimal folding free energy (MFEI) index as suggested previously (Jeffries et al. 2010).
Identification of Differentially Expressed miRNAs
To investigate the differentially expressed miRNAs between (+ Fe) control or (− Fe) Fe-deficiency libraries, the expression of known and unique miRNAs in each sample was statistically analysed by transcripts per million (TPM) (L. Wang et al. 2010). Read counts were normalized to TPM as follows: The normalized expression = (read count * 1,000,000)/Mapped reads. The differential expression level of miRNAs was calculated using DEGseqv1.2.2, and miRNAs with \({\mathrm{log}}_{2}\) fold change > 1 and q-value < 0.01 were considered as differentially expressed.
miRNA Target Prediction and Annotation
Wheat target genes for known and novel microRNAs were predicted using the psRobot_v1.2 and psRNATarget tool (zhaolab.org/psRNATarget/) with default parameters (Dai et al. 2018; Wu et al. 2012). To determine the functional categorization of miRNA target genes, Gene Ontology analysis was carried out using PANTHER classification system (http://pantherdb.org/) (Mi et al. 2019). p-values were corrected using the Bonferroni method, and GO terms with adjusted p-value ≤ 0.05 were considered as significantly enriched. GO terms enriched by more than threefold were plotted using the ggplot2 package from R studio (Wickham 2016). To gain insight into the metabolic pathways of target genes, KEGG pathway enrichment was carried out by KOBASv3.0 software (Mao et al. 2005) in an offline mode. The docker image of latest version of KOBAS was used to annotate and identify the significant pathways using following command:
sudo docker run –rm -v /media/genomics/HDD2/nkobas/seq_pep:/seq_pep -v /media/genomics/HDD2/nkobas/sqlite3:/sqlite3 -v $(pwd):/work-dir agbase/kobas:3.0.3_3 -j -t fasta:nuc -T 40 -e 1e-10 -i miRNA_fasta/nuc_stv3.fasta -s osa -o output/nuc_stv3.
Owing to lack of wheat dataset in KOBAS, rice dataset was used to identify significantly enriched KEGG pathways (Kaur et al. 2023; Zhang et al. 2021). Wheat nucleotide sequences were annotated with RefSeq rice protein database using BLASTX and e value 1e − 10. The p-value of enrichment significance was evaluated using Fisher’s exact test for each pathway and corrected using the Benjamini and Hochberg (BH) method to obtain the q-value. The significant pathways were obtained with cutoff score of 0.05.
Quantification of miRNAs by Real Time-PCR
To get an insight into the spatiotemporal expression patterns of differentially expressed miRNAs and to validate the transcriptome data, stem-loop qRT PCR analysis was conducted. Primers were designed as reported previously (Chen et al. 2005) and are listed in Table S5. Briefly, 1 μg of DNase-treated total RNA was reverse-transcribed using TaqMan microRNA reverse transcription kit (Applied Biosystems™) according to the manufacturer’s instructions. The real-time PCR program was set as follows: 95 °C for 3 min, 45 cycles (95 °C for 10 s, 55 °C for 20 s,72 °C for 20 s). All reactions were performed in triplicate for each time point. The relative expression levels of the miRNAs were calculated by the 2−∆∆CT method (Livak and Schmittgen 2001). For normalization, the wheat gene U6 (GenBank: X63066.1) was used as an internal control (Gasparis et al. 2017). All qRT-PCR was performed using SYBR Green I (Takara SYBR Premix Ex Taq) on the Bio-Rad CFX96 Real-time PCR detection system. Furthermore, to validate our transcriptomic data, qRT-PCR was conducted on the eight randomly selected differentially expressed miRNAs. The pooled total RNA samples used for library construction were also employed for expression analysis via qRT-PCR, and correlation was drawn between the Log2FC values of miRNAs in RNA-Seq and qRT-PCR.
Expression Analysis of Differentially Expressed miRNAs in Wheat Database
For expression analysis of differentially expressed miRNAs in parent wheat cultivars, we utilized our in-house plant miRNA expression atlas database (PmiRExAt, http://pmirexat.nabi.res.in/) (Gurjar et al. 2016). The expression from the parent lines was collated for the whole plant, including T. aestivum (AABBDD), T. durum c.v. Langdon TTR16 (AABB) and Aegilops tauschii, TQ113 (DD). The table was collated using the TPM values, and comparative analysis was done using pie charts. The contribution of different subgenomes was calculated based on the number of miRNAs detected in any specific genome (penetrance) and the level of miRNA expression in any particular subgenome (expressivity).
Results
Analysis of Wheat sRNA During Fe Deficiency
To get an insight into sRNA (small RNA)-mediated regulation of Fe-deficiency response in wheat, we performed whole-genome sRNA sequencing from the root and shoot tissues of seedlings subjected to Fe deficiency. A total of 14.54 million and 14.46 million reads were obtained for iron-sufficient (+ Fe) and iron-deficient (− Fe) shoots, respectively, whereas 14.97 million and 16.03 million reads were obtained for + Fe and − Fe root libraries. After removing low-quality reads, 14.31 million and 13.72 million clean reads yielded for + Fe and − Fe shoot, whereas 14.77 million and 15.77 million for + Fe and − Fe root, respectively. After further refining the sequence reads, we ended with 6.4 million to 10.01 million total sRNA reads with 1.74 million to 2.28 million unique reads encompassing different types of small RNAs (Table S1). Consequently, these sRNA reads were analysed for their distribution in the wheat genome between the sense (“ + ”) and antisense (“ − ”) strands of both coding (exon) and non-coding regions (intron). This positional mapping of sRNA reads from the root and shoot accounted for 11.15% and 6.24% of reads mapping to exon and 2.54% and 3.90% to intron, respectively (Table S2).
Identification of Fe-deficiency–induced miRNAs
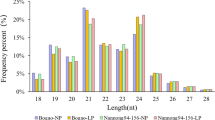
To characterize the sRNA reads into different subfamilies, we annotated all the sRNAs with Rfam v14 database (https://rfam.org/) (Kalvari et al. 2021) into rRNA, snoRNA, snRNA, Trans-acting siRNAs (TAS) and miRNA classes (Fig. 1A–D). This analysis extended our observation of inverse relations in sRNA reads in shoot and root tissues in response to Fe deficiency. This categorized data showed a significant decrease in miRNA representation in response to Fe deficiency in shoot tissues. In contrast, we observed increased miRNA reads in response to Fe deficiency in root tissues (Fig. 1A–D). This suggests that miRNAs might be acting in a tissue manner in regulating Fe-deficiency response in wheat. While analysing the length-based classification of unique sRNA reads, we found that the 20–24 nucleotide sRNAs were the most abundant classes in our datasets, representing around 5–30% of our data irrespective of the treatment conditions (Fig. 1E). The sRNAs with 24 nucleotide length were the predominant category. To our interest, the abundance pattern of sRNAs observed in shoot and root tissues with respect to Fe deficiency was also found to be length biased. We observed that 24 nucleotide-long sRNAs follow decreased abundance in the shoot while showing an increased abundance in root tissue in response to Fe deficiency (Fig. 1E). Also, the length distribution of miRNAs ranged from 20 to 24 nucleotides (Table S3). miRNAs with 21 nucleotide length represented the highest category occupying 59.04%. The per cent distribution of other miRNAs with 20, 22, 23 and 24 lengths was 8.6%, 19.04%, 4.76% and 8.57%.

Expression of Fe-regulated sRNAs in hexaploid wheat tissues. Doughnut pie chart showing a differential abundance of sRNAs families in wheat A Fe-sufficient shoot, B Fe-deficient shoot, C Fe-sufficient root and D Fe-deficient root tissues. E The total length distribution of Fe-deficiency responsive sRNAs identified from Triticum aestivum. The abscissa is the length of sRNA reads; the ordinate is the percentage of one length read accounted for total sRNAs. F Venn diagram showing comparative analysis of the total number of root- and shoot-specific miRNAs and the ones shared among these tissues in + Fe and − Fe sRNA libraries. G Comparative analysis of the differentially expressed miRNAs in the root and shoot tissues under Fe deficiency. UP, upregulated (miRNAs) and Down, downregulated (miRNAs)
Subsequently, our analysis identified 105 miRNAs in our transcriptome analysis, out of which nine were novel (Table S3_Sheet1). The number of miRNAs specific to the tissues and treatments detected in + Fe Shoot was 97, − Fe Shoot 88, + Fe Root 90, and − Fe Root 95 (Fig. 1F). There were 80 miRNAs common among all the treatments and tissues (Table S3_Sheet_readcountTPM). As evident from previous reports, most active miRNAs prefer U at the first nucleotide at 5′ end, which in addition to high A + U content provides them low internal stability, promoting them to be processed into mature miRNA through RISC complex (RNA-induced silencing complex). Additionally, A or U at the 10th position is over-represented in natural plant miRNAs, further contributing to their processibility (Reynolds et al. 2004; Schwab et al. 2005). In agreement with these previous reports, we observed that around 27% of the identified miRNAs showed first nucleotide preference for U, and around 45% of miRNAs have nucleotide preference for A/U at 10th position (Table S3). Hairpin analysis of these identified miRNAs classified them into 36 miRNA encoding families. Interestingly, members for 35 out of 36 hairpin families are represented in the wheat genome, which was the maximum variability observed among all the 66 plant species analysed (Table S4). As MFEI values are characteristic of different classes of RNAs like tRNAs (0.64), mRNAs (0.65) and rRNAs (0.59) while miRNAs are known to have higher MFEI values ⁓ 0.90 and above. To validate our report further, we predicted the secondary structure of the identified miRNAs with an RNAfold web server (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi) with a minimum free energy index (MFEI) algorithm (Jeffries et al. 2010; Lorenz et al. 2011). In accordance to the previous reports, we observed an average MFEI value for all 105 miRNAs as 1.08 with the lowest value of 0.58 and highest value of 2.04 (Table S3). Characteristic stem-loop hairpin formation in all nine novel miRNAs validated their secondary structure and strengthened their putative function as miRNAs (Figure S2).
Next, to pinpoint miRNAs responsive to Fe deficiency, all four libraries were profiled for differential expression patterns using DEGseq v1.2.2 (Wang et al. 2010). Our analysis revealed that 26 miRNAs were differentially expressed in the shoot; 17 were shoot specific. Furthermore, seven miRNAs showed downregulation, with 10 miRNAs upregulated in a shoot-specific manner (Fig. 1G). We extended a similar analysis for root tissues where we observed that 11 miRNAs showed upregulation and 12 miRNAs showed downregulation with six and eight miRNAs with root-specific behaviour (Fig. 1G). Overall, this suggests miRNAs are differentially expressed in wheat seedlings in a tissue-specific manner.
Analysis of miRNA for their Temporal Expression Responses
Analysis of the miRNAs was done in a tissue-specific manner. This allows to identify the miRNAs with similar expression response in the respective tissue. We found that tae-miR -1138 commonly upregulated while four miRNAs (tae-miRNA5049-3p, novel_1, 395b, 395a) commonly downregulated in either tissue in response to Fe deficiency. Furthermore, four miRNAs (tae-miRNA-1117, tae-miRNA-9674a-5p, tae-miRNA-9654a-3p and novel_10) showed inverse behaviour in terms of their transcriptional induction during Fe deficiency when shoot and root tissues are compared (Fig. 2; Table S3). Next, eight randomly miRNAs were picked from the transcriptomic data, and stem-loop qRT-PCR analysis with gene-specific primer set was performed (Table S5). We found that the expression pattern of all the selected miRNAs was very similar to the one observed in transcriptomic analysis with Pearson’s correlation coefficient (R2 = 0.9803) (Fig. 3). To further understand miRNA-mediated Fe-deficiency response temporally, we studied expression patterns of multiple miRNAs in either of the tissues after subjecting them to Fe-deficiency stress for 6, 9, 12 and 15 days. We selected miRNAs that were found to be differentially regulated in the wheat shoot (tae-miR6201, -5050, -9774, -1122a, -1137b-5p and -9671-5p) and root (tae-miR1138, -167c-5p, -444a, -9652-5p, -9654a-3p and -397-5p) tissues. Although we found a perfect correlation between our transcriptome and stem-loop qRT-PCR data, varying temporal transcriptional responses of miRNAs were observed during early and prolonged Fe-deficiency stress, irrespective of the tissue considered (Fig. 4). This approach helped us to characterize the tissue-specific expression responses into three categories. The first includes early responsive miRNAs, where most of the miRNAs showed early induction in the shoot, though, in the root, few miRNAs showed a low early response (tae-miR444a, -9654a-3p and -397-5p). The second category comprises late responsive miRNAs mainly accumulated in root tissues, including tae-miR444a, -167c-5p, -9654a-3p and -397-5p. Additionally, in shoot tissues, most of them showed weak late responsive nature except two miRNAs (tae-miR1137b-5p and tae-miR9671-5p) which showed constantly induced and late responsive behaviour, respectively, in response to Fe deficiency (Fig. 4). The third category highlights the miRNAs with mixed temporal expression during Fe deficiency, which included tae-miR9774 in shoot and tae-miR1138 and tae-miR-9652-5p in the root. Altogether, our study observed a time-dependent regulation of the wheat miRNAs and potentially established the molecular responses during Fe deficiency.

Expression of Fe-regulated miRNAs in hexaploid wheat tissues. Heat map showing differentially expressed miRNAs in shoot and root. A Significantly up and downregulated miRNAs in the shoot with their expression values in root in response to Fe deficiency. B Significantly up and downregulated miRNAs in root with their expression values in the shoot. Heat maps were plotted using MeV (https://mybiosoftware.com/mev-4-6-2-multiple-experiment-viewer.html v4.9.0) against the log2FC values of each miRNA in response to Fe deficiency. Log2FC > 1.0 was considered as the criteria for significance

Stem-loop qRT-PCR-based validation of transcriptome data. Expression profile of selected miRNAs in A shoot and B root in response to Fe deficiency in wheat. C Line graph showing correlation of qRT-PCR-based analysis with transcriptome expression of selected miRNAs in shoot and root tissues of wheat in Fe deficiency response. Relative fold change in the expression was calculated (n = 3) after performing the qRT PCR analysis. Two-tailed student’s t test (p ≤ 0.05) was used to determine the significant change

Temporal expression profiling of selected Fe-deficiency responsive miRNAs in wheat. Expression profile of selected miRNAs in A shoot and B root tissues of wheat grown under Fe deficiency for different time points using qRT-PCR. Relative expression levels were calculated w.r.t. wheat reference gene U6 snRNA transcript abundance (CTgene/CTU6) n = 3; two-tailed Student’s t test (p ≤ 0.05) was used to determine the significant change
Sub-genomic Expression of miRNAs in Wheat
To further comment on the involvement of different genomes of wheat in the regulation of miRNA expression, we analysed the expression of the differentially expressed miRNAs through in silico expression analysis with PmiRExAt database (http://pmirexat.nabi.res.in/) (Gurjar et al. 2016). The database provided us with the expression values for 62 out of 105 differentially expressed miRNAs in T. aestivum (AABBDD), T. durum c.v. Langdon TTR16 (AABB) and Aegilops tauschii, TQ113 (DD) genomes (Figure S3A). Our analysis suggested that the DD genome progenitor A. tauschii expressed the least number of miRNAs (27%) while the incorporation of the DD genome into AABB genome (T. durum) only partially increased the number of miRNAs expressed (Figure S3B). Therefore, though we did not find any negative impact of the DD genome, the AABB genome contributed most to the expression of the selected miRNAs. Although, when we analysed the expression levels of selected miRNAs in different genomes, the AABBDD genome contributed the most to the expression levels of miRNAs (48%), while the AABB genome was the least contributor (21%) while the DD genome contributed around 31% for the expression levels of the miRNAs (Figure S3C). Therefore, we concluded that the DD genome has the lowest penetrance in miRNA expression levels and the highest expressivity.
KEGG and GO Enrichment Analysis
Next, we performed the KEGG enrichment and Gene Ontology (GO) analysis to predict the biological functions of miRNA target genes. This was done to identify the molecular pathways or processes that could be affected by differentially expressed miRNAs under Fe deficiency in a tissue-specific manner. Firstly, the target genes of differentially expressed miRNAs were subjected to KEGG enrichment to identify significantly enriched metabolic pathways. Most of the target genes in both root and shoot were significantly enriched in the biosynthesis of secondary metabolites (Fig. 5). However, some highly significant enrichment was found to be specific in shoots and roots. For instance, metabolic pathways such as ubiquitin-mediated proteolysis, carbon metabolism, steroid biosynthesis and RNA degradation and were enriched in shoots (Fig. 5). In contrast, taurine metabolism, fatty acid metabolism, glutamate metabolism, nicotinate and nicotinamide metabolism, inositol phosphate metabolism, ABC transporters, RNA transport and homologous recombination were significantly enriched (Fig. 5) in roots.

KEGG enrichment for target genes of Fe-responsive miRNAs in the root and shoot. KEGG pathway is displayed along the Y-axis, and X-axis represents all genes enriched in a particular pathway (the ratio of genes annotated to all genes in a pathway). The colour of the circle indicates the q-value, and the size of the dot correlates with the number of DEGs mapped to the specific pathway. *the most distinct pathway genes as indicated
GO enrichment analysis suggested that in both tissues, miRNA-targeted genes are significantly involved biological, cellular and molecular functions that remained largely conserved (Figure S4). Although these functions were largely conserved, they differ in fold enrichment and the representative gene counts in the pathway in these tissues. We observed a distinct pattern for the molecular function in shoot and root under Fe deficiency (Fig. 6 and Figure S4). Specifically, GO terms of molecular functions such as ion transmembrane transporter activity, ABC type transporter activity, metal-ion binding and heme binding were predominant in shoots. In contrast, roots show predominance of GO function including pyrophosphatase activity, transferring phosphorus-containing group and peptidase activity. Multiple similar GO terms were also observed in molecular functions. We concluded that functional pathway genes in the GO process can overlap in different tissue and be targeted by different miRNAs expressed in a tissue-specific manner.

Gene Ontology (GO) categorization of targets of differential expressed Fe-deficiency responsive miRNAs for molecular functions. Targets predicted with psRobot and psRNATarget tools were analysed using PANTHER classification system, and GO terms with adjusted p-value ≤ 0.05 with more than threefold enrichment were considered for plots. Data was plotted using ggplot2 (R-studio). The y-axis represents the category of miRNA targets, and the x-axis shows fold enrichment (Figure S4). The bubble size indicates the number of genes, with the colour representing the significance as denoted by the p-value
Target Prediction of Wheat miRNAs Revealed an Adaptive Response Against Fe Deficiency
To identify the regulatory network involved in Fe-homeostasis by these distinct miRNAs, we predicted their targets and proposed them for their involvement in Fe-deficiency response (Table S6). Targets of these miRNAs revealed a complex regulatory network involving multicopper oxidases (MCOs), transcription factors like GRAS and MADS-box, major facilitator superfamily (MFS) transporters, E3 ubiquitin-protein ligases, oxidoreductases, protein kinases, etc. (Table S6). In prediction analysis, we observed that miRNA397-5p could target transcripts encoding for MCOs. Tae-miRNA171b was predicted to target transcripts encoding for GRAS TF. On similar lines, sulphate transporters are predicted to be targeted by Fe-deficiency–induced miRNA395a and 395b. As we proposed to understand the miRNA-mediated regulatory network against Fe deficiency, we predicted the differential response of shoot- and root-expressed miRNAs in terms of their targets. Interestingly, we observed the targets for root-induced miRNAs, GRAS family TFs, NAC and MADS TFs, SCR-like genes, serine/threonine phosphatases and sugar transporters. While the targets of root downregulated miRNAs included potassium transporter, CNX (molecular chaperon), bHLH TFs, MFS transporters, Zn finger TFs, F-box related, sulphate transporters, kinases and cell wall-related genes. Our analysis supported our previous report suggesting over-accumulation of bHLH TFs during Fe deficiency (Kaur et al. 2019). miRNA-regulated expression of CNX type of molecular chaperones suggested an adaptive response for root against the detrimental effect of ROS accumulation during Fe deficiency. Apart from it, we observed that a significantly downregulated miRNA in the root (tae-miR5049-3p) was found to target S-adenosyl-L-methionine-dependent methyltransferase while the S-adenosyl-methionine is the precursor for mugineic acid (MAs) family of phytosiderophores (PS) (Kobayashi and Nishizawa 2012). This indicates that PS biosynthesis in wheat might be regulated through tae-miR 5049-3p. In the shoot, however, we observed that targets associated with redox enzymes, kinases and phosphatases were over-represented. Among the TF families, NAC and Myb were among the targets of Fe-responsive miRNAs, and phytohormone (like auxin and JA)-associated genes were also targeted through these miRNAs.
To further validate miRNA-mediated regulation of Fe homeostasis genes, we checked the expression of the predicted target genes during Fe deficiency from our previously published datasets (Kaur et al. 2019, 2023). Correlation was drawn for the expression of the target gene and the miRNA (Figure S5 and Table S7). Our validation again strengthened the proposed model as we observed that root-induced miR397-5p targets MCOs (TraesCS1D02G283000). Whilst downregulated root-specific miRNAs like miR9783, miR5384-3p, miR1118 targets expansins, Gamma-glutamyltranspeptidases, endo-1–4-beta-xylanases, cytochrome P450, glutathione-S-transferases, Aspartyl-protease family proteins (TraesCS6D02G037500, TraesCS3B02G090400, TraesCS4A02G045800, TraesCS5A02G397900, TraesCS3D02G133100, TraesCS6D02G263900) (Figure S5). In Fe deficiency, responsive shoot-induced miRNAs like tae-miR9774 and tae-miR1122b-3p are expected to target ATP-dependent RNA helicase (TraesCS1D02G089100) and Jasmonate biosynthesis-related proteins like allene oxide cyclase (Traes4D02G290600). On the other hand, downregulated tae-miR1137b-5p is expected to target GDSL esterase/lipase, cytochrome P450 (TraesCS4A02G426700, TaesCS3A02G074500). Our analysis supports the involvement of miRNAs in Fe-deficiency response in wheat in a tissue-specific manner.
Discussion
Earlier plant miRNA-mediated regulation has been reported under different abiotic and biotic stresses (Navarro et al. 2006; Rong et al. 2014; Zeller et al. 2009; Zhou et al. 2010, 2013). Reports have suggested that the expression of miRNAs under different nutrient deficiencies influences the adaptation to different conditions. For example, Arabidopsis miRNAs show overlapping responses during multiple nutrient deficiency conditions. This suggests the multifunctional role of miRNA that may be commonly upregulated or suppressed by certain nutrients (Liang et al. 2015). Reports on miRNAs regulating plant adaptation to Fe deficiency and their functional analysis are limited mainly to the model plant Arabidopsis (Kong and Yang 2010). Exploring the miRNAs has provided information on the network associations that help crop plants to adapt to different abiotic stresses (Zhou et al. 2010, 2013). However, an attempt has yet to be made to unravel the miRNA-mediated control of Fe homeostasis in hexaploid wheat. Although genes involved in Fe homeostasis were reported earlier, the miRNA-mediated targets were not addressed in wheat (Kaur et al. 2019, 2023). Noteworthy, such molecular responses largely depend on the genotype and specific stress condition. This study attempts to identify wheat miRNAs that could be linked to the target pathway functions to identify the critical regulatory miRNA-mRNA interaction involved in Fe-deficiency conditions. A total of 105 Fe-deficiency–responsive miRNAs were identified in shoots and roots. Our work identified nine novel miRNAs with a typical stem-loop structure having distinct expression responses during Fe deficiency (Figure S2).
In this study, sRNA libraries were generated from the root and shoot of wheat seedlings subjected to Fe deficiency for different time points. This was done to generate the inventory of miRNA that could assist in collating miRNAs that may differentially express during Fe deficiency. Our analysis identified multiple root and shoot preferential miRNAs in response to Fe deficiency (Fig. 2; Table S3). qRT-PCR of these tissue-specific miRNAs drew a strong correlation to RNA-Seq data. Furthermore, the validation of differentially expressed miRNAs from root (tae-miR1138, -167c-5p, -444a, -9652-5p, -9654a-3p and -397-5p) and shoot (tae-miR6201, -5050, -9774, -1122a, -1137b-5p and -9671-5p) revealed the spatiotemporal expression of these Fe-responsive miRNAs.
Fe deficiency is a major nutritional disorder that limits crop productivity. In plants, multiple miRNA gene families are involved in the Fe-deficiency responses (Carrió-Seguí et al. 2019; Kong and Yang 2010). Previously, correlation studies were done where the expression of specific miRNA was observed in high and low Fe genotypes of rice (Agarwal et al. 2015; Paul et al. 2016). These studies support that miRNA-mediated control could occur concerning the Fe flux in a tissue-specific manner. These studies highlighted osa-miR156, 164, 167, 171, 395, 398, 399 and 408 for having a regulatory role in Fe homeostasis, while all these miRNAs were represented in our study. Therefore, while validating these previous reports, our study proposed a miRNA-mediated Fe-homeostasis in wheat. Previous reports proposed Fe-deficiency responsive miRNAs targeting TFs and oxidoreductase family of genes in rice which is further a similar kind of response observed in wheat.
Wheat DE miRNA also predicted to target genes from the family of TFs, such as MADS, GRAS, WRKY and F-box-containing proteins. At the transcript level, miRNA targets include ferroxidases, E3 ubiquitin ligases and enzymatic reaction–associated genes. Wheat recruit strategy-II mode of Fe3+ in its chelated form. The generation of the Fe3+ in the periplasmic space is primarily controlled by the ferroxidase activity of the laccase subfamily (Hoopes and Dean 2004; Huston et al. 2002). Consistent to these studies, in our analysis, tae-miR397-5p shows potential targets for multiple wheat MCOs (Table S6). This suggested a mechanistic insight that regulated the turnover of ferroxidase activity in wheat through miR397 at the post-transcriptional level. Another candidate, tae-miR444, was highly upregulated in Fe deficiency. At the mechanistic level, it was shown that miR444 could repress the MADS-box encoding transcript. Our target prediction suggests that tae-miR444 could target multiple wheat MADS-box and F-box encoding transcripts. MADS-box encoding transcripts are differentially regulated during Fe-deficiency conditions. Tae-miR1122 also shows shoot-preferential induction during Fe deficiency. Our prediction supported that tae-miR1122 majorly targets oxidoreductase, JA biosynthesis gene (allene oxide cyclase), ARFs (auxin responsive factors), Zn-finger TFs, Enoyl Co-A hydratase and F-box proteins (Figure S5, Table S6).
Strategy II plants, like rice and wheat, primarily rely on releasing PS in the rhizosphere to mobilize soil Fe. The slow rate of PS biosynthesis and the decreased nicotianamine (NA) level in the cells were linked to the sulfur (S) deficiency. Therefore, sulphate metabolism and plant distribution have been linked with Fe uptake and translocation. Plant sulphate transporters (SULTR) were differentially regulated under the changing regimes of Fe (Kaur et al. 2019; Khan et al. 2022). Our study observed good target scores for multiple SULTR targeted by wheat miR395 (Table S4). miR395 is an integral part of sulphate assimilation that regulates the expression of SULTR to maintain sulphate uptake and utilization in plant tissue (Zhou et al. 2010). We conclude that Fe-deficiency–induced recruitment of post-transcriptional regulation could affect the secondary process regulating nutrient uptake.
Comparison of miRNA repertoires between wheat and its diploid progenitors provides useful information about the changes in miRNA gene content over time and the role of miRNAs in wheat’s adaptation to its environment (Alptekin and Budak 2017). Our study observed a high proportion of sRNA contribution from A and B genomes compared to D. This was consistent for both + Fe and − Fe datasets. Our analysis for the − Fe regulated miRNAs points to a high expression contribution in the ancestral genotypes of wheat, with the highest for the T. aestivum (AABBDD) and T. durum c.v. Langdon TTR16 (AABB) compared to Aegilops tauschii (DD genome) (Figure S3). Our observation agrees with previous reports, suggesting the least involvement of DD genome in miRNA diversity (Alptekin and Budak 2017). It could be possible that incorporating the DD genome into the AABB genome increased the expression of miRNAs, suggesting a synergistic effect or some trans-genomic regulation.
Altogether, miRNA profiling suggests their involvement in regulating Fe-deficiency responses. Our work proves that miRNA perturbation due to Fe-deficiency targets a subset of previously reported Fe-deficiency responses (Fig. 7). This reflects that miRNA-mediated control of Fe-responsive genes contributes to such regulatory mechanisms in hexaploid wheat. Specifically, the miRNA can target the genes primarily involved in changing the Fe redox status (tae-miR1118, 397-5p, 9678-3p, 1122a, 1137b-5p), Fe uptake, metal transporters (tae-miR1122b-3p, 5049-3p, 395a, 395b), TFs, multi-nutrient homeostasis E3 ubiquitin ligases (tae-miR1133, 1122b-3p, 9674a-5p) and phytohormone (JAs and auxins) regulation (tae-miR171b, 444a, 1123, 5049-3p, 1122a, 1133, 1137b-5p, 1122b-3p) (Fig. 7). Overall, the generated datasets will serve as an important resource to further investigate the transcriptional rearrangements that influence different tiers of molecular responses during Fe deficiency. A further study focusing on the candidate miRNAs function could add a new paradigm into Fe deficiency and other stress to improve plant growth and yield.

A speculated model for miRNA-mediated Fe homeostasis in hexaploid wheat. The model represents the multiple miRNAs that were DE expressed under Fe deficiency. These miRNAs target distinct genes involved in different biological functions, as mentioned. Some major functions include redox-related metal transporters, transcription factors and E3 ubiquitin ligases
Data Availability
The small RNA-Seq raw read data has been submitted at NCBI (Submission: SUB12485490 with BioProject: PRJNA916207).
References
Abadía J, Vázquez S, Rellán-Álvarez R, El-Jendoubi H, Abadía A, Álvarez-Fernández A, Flor López-Millán A (2011). Towards a Knowledge-Based Correction of Iron Chlorosis. https://doi.org/10.1016/j.plaphy.2011.01.026
Agarwal S, Mangrauthia SK, Sarla N (2015) Expression profiling of iron deficiency responsive microRNAs and gene targets in rice seedlings of Madhukar x Swarna recombinant inbred lines with contrasting levels of iron in seeds. Plant Soil 396(1–2):137–150. https://doi.org/10.1007/S11104-015-2561-Y
Alptekin B, Budak H (2017) Wheat miRNA ancestors: evident by transcriptome analysis of A, B, and D genome donors. Funct Integr Genomics 17(2–3):171–187. https://doi.org/10.1007/S10142-016-0487-Y
Bhatnagar T, Sachdev A, Johari RP (2002) Molecular characterization of glutenins in wheat varieties differing in Chapati quality characteristics. J Plant Biochem Biotechnol 11:33–36. https://doi.org/10.1007/BF03263131
Briat JF, Dubos C, Gaymard F (2015) Iron nutrition, biomass production, and plant product quality. Trends Plant Sci 20(1):33–40. https://doi.org/10.1016/j.tplants.2014.07.005
Buckhout TJ, Schmidt W (2013) Iron in plants. John Wiley & Sons, Ltd., In eLS. https://doi.org/10.1002/9780470015902.a0023713
Carrió-Seguí À, Ruiz-Rivero O, Villamayor-Belinchón L, Puig S, Perea-García A, Peñarrubia L (2019) The altered expression of Microrna408 influences the arabidopsis response to iron deficiency. Front Plant Sci 10:324. https://doi.org/10.3389/FPLS.2019.00324/BIBTEX
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33(20). https://doi.org/10.1093/NAR/GNI178
Chen X (2009) Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 25:21–44. https://doi.org/10.1146/ANNUREV.CELLBIO.042308.113417
Colangelo EP, Guerinot ML (2004) The essential basic helix-loop-helix protein FIT1 is required for the iron deficiency response. Plant Cell 16(12):3400–3412. https://doi.org/10.1105/TPC.104.024315
Dai X, Zhuang Z, Zhao PX (2018) psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res 46(W1):W49–W54. https://doi.org/10.1093/NAR/GKY316 FAOSTAT. Food and Agriculture Organization of the the United Nations. http://www.fao.org/faostat/en/#data/FBS
Friedländer MR, MacKowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40(1):37–52. https://doi.org/10.1093/NAR/GKR688
Gao S, Yang L, Zeng HQ, Zhou ZS, Yang ZM, Li H, Sun D, Xie F, Zhang B (2016) A cotton miRNA is involved in regulation of plant response to salt stress. Sci Rep 6. https://doi.org/10.1038/SREP19736
Gasparis S, Yanushevska Y, Nadolska-Orczyk A (2017) Bioinformatic identification and expression analysis of new microRNAs from wheat (Triticum aestivum L.). Acta Physiol Plant 39(10):1–13. https://doi.org/10.1007/S11738-017-2530-6/FIGURES/4
Guo S, Xu Y, Liu H, Mao Z, Zhang C, Ma Y, Zhang Q, Meng Z, Chong K (2013) The interaction between OsMADS57 and OsTB1 modulates rice tillering via DWARF14. Nat Commun 4(1):1–12. https://doi.org/10.1038/ncomms2542
Gurjar AKS, Panwar AS, Gupta R, Mantri SS (2016) PmiRExAt: plant miRNA expression atlas database and web applications. Database: J Biol Database Curation. https://doi.org/10.1093/DATABASE/BAW060
Heim MA, Jakoby M, Werber M, Martin C, Weisshaar B, Bailey PC (2003) The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol Biol Evol 20(5):735–747. https://doi.org/10.1093/MOLBEV/MSG088
Hell R, Stephan UW (2003) Iron uptake, trafficking and homeostasis in plants. Planta 216(4):541–551. https://doi.org/10.1007/s00425-002-0920-4
Hoopes JT, Dean JFD (2004) Ferroxidase activity in a laccase-like multicopper oxidase from Liriodendron tulipifera. Plant Physiol Biochem 42(1):27–33. https://doi.org/10.1016/J.PLAPHY.2003.10.011
Hou G, Du C, Gao H, Liu S, Sun W, Lu H, Kang J, Xie Y, Ma D, Wang C (2020) Identification of microRNAs in developing wheat grain that are potentially involved in regulating grain characteristics and the response to nitrogen levels. BMC Plant Biol 20(1):1–21. https://doi.org/10.1186/S12870-020-2296-7/FIGURES/7
Huston WM, Jennings MP, McEwan AG (2002) The multicopper oxidase of Pseudomonas aeruginosa is a ferroxidase with a central role in iron acquisition. Mol Microbiol 45(6):1741–1750. https://doi.org/10.1046/J.1365-2958.2002.03132.X
Jeffries CD, Perkins DO, Guan X (2010) Gene processing control loops suggested by sequencing, splicing, and RNA folding. BMC Bioinformatics 11(1):1–10. https://doi.org/10.1186/1471-2105-11-602/FIGURES/6
Jian H, Wang J, Wang T, Wei L, Li J, Liu L (2016) Identification of rapeseed MicroRNAs involved in early stage seed germination under salt and drought stresses. Front Plant Sci 7(MAY2016):658. https://doi.org/10.3389/FPLS.2016.00658/BIBTEX
Jin LF, Yarra R, Yin XX, Liu YZ, Cao HX (2021) Identification and function prediction of iron-deficiency-responsive microRNAs in citrus leaves. 3 Biotech 11(3):121. https://doi.org/10.1007/S13205-021-02669-Z
Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, Griffiths-Jones S, Toffano-Nioche C, Gautheret D, Weinberg Z, Rivas E, Eddy SR, Finn RD, Bateman A, Petrov AI (2021) Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49(D1):D192. https://doi.org/10.1093/NAR/GKAA1047
Kaur G, Meena V, Kumar A, Suman G, Tyagi D, Joon R, Balk J, Pandey AKA, expression of homoeologous genes in wheat roots modulates the early phase of iron-deficiency signalling, (2023) Environ. Exp Bot 208:105254. https://doi.org/10.1016/j.envexpbot.2023.105254
Kaur G, Shukla V, Kumar A, Kaur M, Goel P, Singh P, Shukla A, Meena V, Kaur J, Singh J, Mantri J, Rouached SH, Pandey AK (2019) Integrative analysis of hexaploid wheat roots identifies signature components during iron starvation. J Exp Bot 70(21):6141–6161. https://doi.org/10.1093/jxb/erz358
Khan MS, Lu Q, Cui M, Rajab H, Wu H, Chai T, Ling HQ (2022) Crosstalk between iron and sulfur homeostasis networks in Arabidopsis. Front Plant Sci 13:1745. https://doi.org/10.3389/FPLS.2022.878418/BIBTEX
Kobayashi T, Nagasaka S, Senoura T, Itai RN, Nakanishi H, Nishizawa NK (2013) Iron-binding haemerythrin RING ubiquitin ligases regulate plant iron responses and accumulation. Nat Commun 4. https://doi.org/10.1038/NCOMMS3792
Kobayashi T, Nishizawa NK (2012) Iron uptake, translocation, and regulation in higher plants. https://doi.org/10.1146/annurev-arplant-042811-105522
Kobayashi T, Nozoye T, Nishizawa NK (2019) Iron transport and its regulation in plants. Free Radical Biol Med 133:11–20. https://doi.org/10.1016/J.FREERADBIOMED.2018.10.439
Kong WW, Yang ZM (2010) Identification of iron-deficiency responsive microRNA genes and cis-elements in Arabidopsis. Plant Physiol Biochem 48(2–3):153–159. https://doi.org/10.1016/J.PLAPHY.2009.12.008
Kozomara A, Birgaoanu M, Griffiths-Jones S (2019) miRBase: from microRNA sequences to function. Nucleic Acids Res 47(D1):D155–D162. https://doi.org/10.1093/NAR/GKY1141
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3):1–10. https://doi.org/10.1186/GB-2009-10-3-R25/TABLES/5
Li C, Zhang B (2016) MicroRNAs in control of plant development. In J Cell Physiol 231(2):303–313. Wiley-Liss Inc. https://doi.org/10.1002/jcp.25125
Liang G, Ai Q, Yu D (2015) Uncovering miRNAs involved in crosstalk between nutrient deficiencies in Arabidopsis. Sci Rep 5(1):1–13. https://doi.org/10.1038/srep11813
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods (San Diego, Calif.) 25(4):402–408. https://doi.org/10.1006/METH.2001.1262
Long TA, Tsukagoshi H, Busch W, Lahner B, Salt DE, Benfey PN (2010) The bHLH transcription factor POPEYE regulates response to iron deficiency in Arabidopsis roots. Plant Cell 22(7):2219–2236. https://doi.org/10.1105/TPC.110.074096
Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA Package 2.0. Algorithms Mol Biol 6(1):1–14. https://doi.org/10.1186/1748-7188-6-26/TABLES/2
Lu W, Li J, Liu F, Gu J, Guo C, Xu L, Zhang H, Xiao K (2011) Expression pattern of wheat miRNAs under salinity stress and prediction of salt-inducible miRNAs targets. Front Mech Eng China 5(4):413–422. https://doi.org/10.1007/S11703-011-1133-Z/METRICS
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics (oxford, England) 21(19):3787–3793. https://doi.org/10.1093/BIOINFORMATICS/BTI430
Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD (2019) Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14(3):703–721. https://doi.org/10.1038/s41596-019-0128-8
Morrissey J, Guerinot ML (2009) Iron uptake and transport in plants: the good, the bad, and the ionome. Chem Rev 109(10):4553. https://doi.org/10.1021/CR900112R
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JDG (2006) A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science (New York, N.Y.) 312(5772):436–439. https://doi.org/10.1126/SCIENCE.1126088
Ogo Y, Nakanishi Itai R, Nakanishi H, Kobayashi T, Takahashi M, Mori S, Nishizawa NK (2007) The rice bHLH protein OsIRO2 is an essential regulator of the genes involved in Fe uptake under Fe-deficient conditions. Plant J 51(3):366–377. https://doi.org/10.1111/J.1365-313X.2007.03149.X
Paul S, Gayen D, Datta SK, Datta K (2016) Analysis of high iron rice lines reveals new miRNAs that target iron transporters in roots. J Exp Bot 67(19):5811. https://doi.org/10.1093/JXB/ERW346
Ravichandran S, Ragupathy R, Edwards T, Domaratzki M, Cloutier S (2019) Microrna-guided regulation of heat stress response in wheat. BMC Genomics 20(1):1–16. https://doi.org/10.1186/S12864-019-5799-6/FIGURES/5
Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A (2004) Rational siRNA design for RNA interference. Nat Biotechnol 22(3):326–330. https://doi.org/10.1038/nbt936
Rong W, Qi L, Wang A, Ye X, Du L, Liang H, Xin Z, Zhang Z (2014) The ERF transcription factor TaERF3 promotes tolerance to salt and drought stresses in wheat. Plant Biotechnol J 12(4):468–479. https://doi.org/10.1111/PBI.12153
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8(4):517–527. https://doi.org/10.1016/J.DEVCEL.2005.01.018
Selote D, Samira R, Matthiadis A, Gillikin JW, Long TA (2015) Iron-binding E3 ligase mediates iron response in plants by targeting basic helix-loop-helix transcription factors. Plant Physiol 167(1):273–286. https://doi.org/10.1104/PP.114.250837
Singh A, Mantri S, Sharma M, Chaudhury A, Tuli R, Roy J (2014) Genome-wide transcriptome study in wheat identified candidate genes related to processing quality, majority of them showing interaction (quality x development) and having temporal and spatial distributions. BMC Genomics 15(1). https://doi.org/10.1186/1471-2164-15-29
Smit AFA, Hubley R, Green P (2013) RepeatMasker Open-4.0. http://www.repeatmasker.org
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17(4):196–203. https://doi.org/10.1016/J.TPLANTS.2012.01.010
Valdés-López O, Yang SS, Aparicio-Fabre R, Graham PH, Reyes JL, Vance CP, Hernández G (2010) MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytol 187(3):805–818. https://doi.org/10.1111/J.1469-8137.2010.03320.X
Wang B, Sun Y, fei, Song, N., Wei, J. ping, Wang, X. jie, Feng, H., Yin, Z. yuan, & Kang, Z. sheng. (2014) MicroRNAs involving in cold, wounding and salt stresses in Triticum aestivum L. Plant Physiol Biochem PPB 80:90–96. https://doi.org/10.1016/J.PLAPHY.2014.03.020
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics (oxford, England) 26(1):136–138. https://doi.org/10.1093/BIOINFORMATICS/BTP612
Wang M, Gong J, Bhullar NK (2020) Iron deficiency triggered transcriptome changes in bread wheat. Comput Struct Biotechnol J 18:2709. https://doi.org/10.1016/J.CSBJ.2020.09.009
Wang M, Kawakami Y, Bhullar NK (2019) Molecular analysis of iron deficiency response in hexaploid wheat. Front Sustain Food Syst 3:67. https://doi.org/10.3389/FSUFS.2019.00067/BIBTEX
Waters BM, McInturf SA, Stein RJ (2012) Rosette iron deficiency transcript and microRNA profiling reveals links between copper and iron homeostasis in Arabidopsis thaliana. J Exp Bot 63(16):5903–5918. https://doi.org/10.1093/JXB/ERS239
Wen M, Shen Y, Shi S, Tang T (2012) MiREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13(1):1–10. https://doi.org/10.1186/1471-2105-13-140/FIGURES/5
Wickham H (2016) ggplot2. https://doi.org/10.1007/978-3-319-24277-4
Willmann MR, Poethig RS (2005) Time to grow up: the temporal role of smallRNAs in plants. In Curr Opin Plant Biol 8(5):548–552. NIH Public Access. https://doi.org/10.1016/j.pbi.2005.07.008
Wu HJ, Ma YK, Chen T, Wang M, Wang XJ (2012) PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res 40(Web Server issue). https://doi.org/10.1093/NAR/GKS554
Yan JY, Li CX, Sun L, Ren JY, Li GX, Ding ZJ, Zheng SJ (2016) A WRKY transcription factor regulates Fe translocation under Fe deficiency. Plant Physiol 171(3):2017. https://doi.org/10.1104/PP.16.00252
Yu Y, Zhang Y, Chen X, Chen Y (2019) Plant noncoding RNAs: hidden players in development and stress responses. 35:407–431. https://doi.org/10.1146/ANNUREV-CELLBIO-100818-125218
Zeller G, Henz SR, Widmer CK, Sachsenberg T, Rätsch G, Weigel D, Laubinger S (2009) Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J Cell Mol Biol 58(6):1068–1082. https://doi.org/10.1111/J.1365-313X.2009.03835.X
Zhang H, Li Y, Yao X, Liang G, Yu D (2017) Positive regulator of iron homeostasis1, OsPRI1, facilitates iron homeostasis. Plant Physiol 175(1):543. https://doi.org/10.1104/PP.17.00794
Zhang L, Dong C, Chen Z, Gui L, Chen C, Li D, Xie Z, Zhang Q, Zhang X, Xia C, Liu X, Kong X, Wang J (2021) WheatGmap: a comprehensive platform for wheat gene mapping and genomic studies. Mol Plant 14(2):187–190. https://doi.org/10.1016/J.MOLP.2020.11.018
Zhang Q, Zhao C, Li M, Sun W, Liu Y, Xia H, Sun M, Li A, Li C, Zhao S, Hou L, Picimbon JF, Wang X, Zhao Y (2013) Genome-wide identification of thellungiella salsuginea micrornas with putative roles in the salt stress response. BMC Plant Biol 13(1):1–13. https://doi.org/10.1186/1471-2229-13-180/FIGURES/6
Zheng L, Ying Y, Wang L, Wang F, Whelan J, Shou H (2010) Identification of a novel iron regulated basic helix-loop-helix protein involved in Fe homeostasis in Oryza sativa. BMC Plant Biol 10(1):1–9. https://doi.org/10.1186/1471-2229-10-166/FIGURES/6
Zhou H, Liu Q, Li J, Jiang D, Zhou L, Wu P, Lu S, Li F, Zhu L, Liu Z, Chen L, Liu YG, Zhuang C (2012) Photoperiod- and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res 22(4):649–660. https://doi.org/10.1038/cr.2012.28
Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot 61(15):4157–4168. https://doi.org/10.1093/JXB/ERQ237
Zhou M, Li D, Li Z, Hu Q, Yang C, Zhu L, Luo H (2013) Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol 161(3):1375–1391. https://doi.org/10.1104/PP.112.208702
Acknowledgements
The authors thank the Executive Director of NABI for the facilities and support. DBT-eLibrary Consortium (DeLCON) is acknowledged for providing timely support and access to e-resources for this work.
Funding
This work was funded by the Department of Science and Technology- Science & Engineering Research Board (SERB) Grant number CRG/2020/000940 and the NABI-CORE grant to AKP.
Author information
Authors and Affiliations
Contributions
AKP and SS conceived and designed the research. SS and APS carried out wet lab experiments; SS, APS, RJ, PS, DS, SM and VS carried out wet lab experiments and performed data analyses. AKP, SS, APS and DS wrote and finalized the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
All authors approved the manuscript for publication.
Competing Interests
The authors declare no potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sharma, S., Singh, D., Joon, R. et al. System Analysis of Differentially Expressed miRNAs in Hexaploid Wheat Display Tissue-Specific Regulatory Role During Fe-Deficiency Response. Plant Mol Biol Rep 42, 265–280 (2024). https://doi.org/10.1007/s11105-023-01421-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-023-01421-6