
Abstract
Moringa oleifera Lam. (drumstick) belongs to the family Moringaceae that is originated from sub-Himalayan tracts of Northern India distributed worldwide in the tropics and sub-tropics. Immature pods and fresh leaves are widely used as vegetable and are rich source of minerals and vitamins. In the present work, we made an attempt to develop and use a set of RAPD-SCAR marker for the identification of superior germplasm of M. oleifera from the accessions collected from South India. Initially, 120 trees were surveyed based on total fruit yield, and single fruit weight from Karnataka, Kerala, and Tamil Nadu states of India; 23 plants had 50% higher fruit yield and single fruit weight than average and were selected as Candidate Plus Trees (CPTs). On the basis of morphological and biochemical analysis, CPT17 was selected as elite germplasm. Random amplified polymorphic DNA (RAPD) analysis of CPTs indicated 89.61% polymorphism among 23 CPTs. These markers could be used in marker-assisted selection and breeding programs in M. oleifera. Further, an attempt to develop a set of RAPD-SCAR marker for the identification of superior germplasm of M. oleifera was made. RAPD primer OPA-19 (CAAACGTCGG) revealed a unique band (1500-bp) in CPT17. The specific RAPD band was recovered from the gel, cloned, and sequenced. BLAST analysis of the CPT17 specific sequences revealed that no considerable similarity with known protein. Based on these unique characterized sequences, specific primers for CPT17 were designed. Specific amplification profile of this primer proved it as a SCAR marker (F2R2) for CPT17 genotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Moringa oleifera Lam. has naturalized in at least 70 countries across the tropical and subtropical regions of the world. The correct origin of this plant is somewhat vague due to its widespread cultivation since early times (Navie and Csurhes 2011). Most authors commonly agree and report that M. oleifera originated in India and Arabia (Qaiser 1973; Somali et al. 1984; Mughal et al. 1999; Palada and Ebert 2012). M. oleifera is indigenous to the sub-Himalayan tracts of Northern India (Qaiser 1973; Olson 2002). Drumstick belongs to the monogeneric family Moringaceae and genus Moringa. The taxonomic position of the family Moringaceae is not yet clear. Though it has some features similar to those of Brassicaceae and Capparidaceae (Edwards et al. 2000), the seed structure does not agree with either of these families. M. oleifera grows best in the tropical regions of the world that have semi-arid or monsoonal climates (Navie and Csurhes 2011). Duke (1983) reported the plant has a wide climatic tolerance and can grow in conditions that range from humid to arid in tropical and subtropical regions.
M. oleifera has been used for such a broad variety of purpose that it has been reported as a “multipurpose tree.” Fruits and leaves of drumstick are rich source of proteins, vitamins, and minerals. Different parts of this plant contain a profile of important minerals and good source of protein, vitamins, β-carotene, amino acids, and various phenolics (Palada and Ebert 2012). All parts of the drumstick can be used in a variety of ways. M. oleifera fruits and leaves are rich in nutrients and vitamins thus consumed and used as fodder. Extracts derived from various parts of M. oleifera especially seeds, leaves and bark havewide, and diverse medicinal properties including antihypertensive, diuretic, cholesterol lowering activities, antispasmodic, antiulcer, hepatoprotective activities, antibacterial, antifungal, antitumor, and anticancer activities (Anwar et al. 2007).
Sequence Characterized Amplified Region (SCAR) markers are one of the PCR-based marker system introduced by Paran and Michelmore (1993). SCAR markers have proven their utility in detection of Fusarium oxysporium sp. Ciceris and cubense (Kiran et al. 2010; Durai et al. 2012; Cunha et al. 2015), selection of hybrid strains of Saccharomyces cerevisiae (Wang et al. 2011), validation of sex of plants in early stage (Korekar et al. 2012), and identification of powdery mildew resistance genes (Srivastava et al. 2012). Kiran et al. (2010) reviewed the utility of SCAR markers in authentication of medical herbs. SCAR markers have been developed for authentication of various plants, viz., two species of Artemisia (A. princeps, A. argyi) (Lee et al. 2006), Phyllanthus emblica (Dnyaneshwar et al. 2006), Curcuma alismatifolia (Anuntalabhochai et al. 2007), and so on. Busconi et al. (2006) reported on the development of SCAR markers for cultivar identification. Kim et al. (2015) developed SCAR markers for selecting rind stripe pattern in Citrullus lanatus L. Primers can be synthesized based on the sequence of the ends of RAPD fragments to generate specific PCR primers which can be used for amplification giving rise to specific PCR markers called SCARs (Paran and Michelmore 1993).
The simplicity, rapidity, need of low amount of genomic DNA, no requirement of radioisotopes, and prior genetic knowledge are the major advantages of the RAPD technique (Williams et al. 1990). However, there are some technical issues with RAPD that have posed its accuracy as a genetic marker and avoided its widespread use in recent years. The reproducibility of RAPD profiling often influenced DNA consistency, primer and template concentrations, make of thermo cyclers, and source of DNA polymerase (Ellsworth et al. 1993; Muralidharan and Wakeland 1993). As such RAPD technique is highly sensitive to modifications inside the response situations. Consequently, there exists a gap between the capacity to gain connected markers to a gene of interest in a quick time and the use of these markers for a map-based cloning and for ordinary screening technique (Bhagyawant 2016). For instance, Dahlberg et al. (2002) analyzed 42,000 Sorghum bicolor accessions with RAPD markers; the clusters developed have been no longer close to those obtained on the premise of morphological and agronomic statistics. Many examples demonstrate the ambiguous existence of RAPD markers that must be authenticated using interface methods such as SCAR markers. According to Saiki et al. (1988), the polymorphic fragment produced by RAPD-PCR amplification is not always consistent. This restriction can be overcome in RAPD-PCR by transforming the polymorphic bands from RAPDs into sequence-characterized amplified regions. Li et al. (2008a, b) validated that SCAR markers are greater reproducible for genotype authentication and identification than revealing genetic differences between the samples. Amplified fragment length polymorphism (AFLP), simple-sequence repeats (SSRs) and inter simple sequence repeats (ISSRs) are other PCR-based methods that have more repeatable amplification patterns than RAPD markers. However, creating SCAR markers from these markers poses practical difficulty. In contrast, polymorphic regions that differ in size between the samples revealed by RAPD technique can be efficiently used to develop SCAR markers to sample authentication based on SCAR shifts. As a result, the SCAR marker has become the best marker technology for crop species authentication.
Though RAPD-based SCAR widely applied, few reports available on development of SCAR markers on more repeatable tags such as AFLP (Vos et al. 1995), SSR (Litt and Luty 1989; Tautz 1989; Weber and May 1989), and ISSR (Zeitkeinicz et al. 1994). The creation of SCAR markers from these markers, on the other hand, is extremely expensive, complex, and time-consuming, and may necessitate whole-genome sequence details.
In the present study, we used 27 RAPD markers for the diversity analysis in 23 accessions of M. oleifera. One of the major drawbacks of RAPDs is the lack of specificity and reproducibility that can be ruled out by further converting them into far more reproducible and specific markers such as SCAR markers. Li et al. (2008a, b) developed SCAR marker associated with high rooting ability in Lentinula edodes. Tatikonda et al. (2009) surveyed molecular diversity in 48 germplasm collections of Jatropha curcas from six states by using seven AFLP primer combinations showing the occurrence of higher numbers of unique/rare fragments and with greater variation in percentage oil content. These new data have now been employed by them for the development of SCAR marker for the identification of higher oil content J. curcus. Sujatha et al. (2005) utilized RAPD markers to assess the genetic similarity between toxic Indian and non-toxic Mexican accessions. Basha and Sujatha (2007) further extended this study by converting RAPD markers into SCAR markers that differentiates Indian and Mexican accessions. Likewise, in this paper, we have made an attempt to develop and use a set of RAPD-SCAR marker for the identification of superior germplasm of M. oleifera from the accessions collected.
Materials and Methods
Plant Material
An exploration survey was carried out in Karnataka, Kerala, and Tamil Nadu states of India during May–June 2014 primarily to record healthy, morphologically, and biochemically distinct drumstick trees. In the primary survey, two prime yield traits, viz., total fruit yield (kg) and single fruit weight (g) were recorded in 120 trees, ranging 8–15-year-old trees from the study area. Based on fruit yield data and single fruit weight, mean fruit yield and mean fruit weight among surveyed 120 trees were determined. The primary survey revealed that among 120 trees, 23 trees gave more than 50% fruit yield and single fruit weight than average of 120 trees. Each selected tree was designated with CPT number for future identification. Fruit yield-based screening continued two more years on CPTs on various yield attributes (Table 1) and recorded CPT17, an accession from Cherthala representing the agro climatic zone of coastal sandy region, is the elite tree with maximum yield (163.27 kg/plant/season). In biochemical analysis, overall ranking (nutritive value and vitamin C) indicates that CPT17 is the best with remarkably high leaf nutritive value, leaf, and fruit vitamin C content. In addition to these, CPT17 contained moderately high level of vitamin A, B1 and B2 (Ravi et al. 2020).
The limb cuttings of 50–100 cm long and 4–5 cm in diameter are typically planted and were grown in plastic pots (50 cm × 35 cm) containing soil, and FYM (Farm Yard Manure), and maintained in a greenhouse at ambient temperature (̴26–28 °C) (Saini et al. 2012). After 2 months, the rooted plants were transferred to Department Garden for the analysis of diversity.
DNA Extraction
Immature leaves from 3-month-old plants were used for DNA extraction. Genomic DNA of all the 23 genotypes was extracted by cetyl-tri-methyl ammonium bromide (CTAB) method with modification, and 2% polyvinyl pyrrolidone (PVP) was added to extraction buffer for removal of phenolics present in the sample. The DNA concentration and purity were determined by using Biophotometer (Eppendorf, Hamburg, Germany). DNA quality was further checked by gel electrophoresis on 0.7% agarose gel (1X TAE buffer, EtBr). A working DNA concentration of 10 ng/µl was prepared and stored at 4 °C until use.
RAPD-PCR Amplification
A total of 35 RAPD primers (BR Biochem Life Sciences, New Delhi, India) were used for the polymorphism survey. RAPD assay was carried out in 25-µl reaction volume, containing 12.5-µl 2 X-PCR smart mixes (Origin Diagnostics, Karunagappally, India), 3-µl DNA (10 ng/µl), 1-µl primer (20 pmol/µl), and 8.5-µl double distilled water. The polymerase chain reaction (PCR) was performed with a thermal cycler (Eppendorf, Mastercycler gradient, Hamburg, Germany). The standardized amplification was performed (Saini et al. 2013) at an initial denaturation at 94 °C for 4 min, followed by 40 cycles of denaturation at 94 °C for 30 s; primer annealing based on Tm for 1 min; primer extension at 72 °C for 2 min; and final extension at 72 °C for 10 min. The annealing temperature for each primer was standardized by performing gradient PCR.
RAPD-PCR products were then analyzed by electrophoresis (V-GEL, Warwickshire, UK) with 100-bp DNA ladder ranged from 100 to 5000-bp (Origin Diagnostics, Karunagappally, India) on a 1.8% agarose gel with 1X TAE buffer stained with 2-µl EtBr. DNA banding patterns were then visualized using Gel documentation system (BIORAD Gel DocTMXR + , California, USA).
SCAR Marker Development
Genomic DNA isolated from all the CPTs were amplified using 27 random primers. A degree of polymorphism was obtained in each RAPD profile. There were numerous similar bands present in all accessions, whereas several unique bands were also obtained in superior genotype CPT17. Based on this a high-intensity unique amplicon was selected in CPT17 samples for the development of SCAR markers. These amplicons were selected by comparing the RAPD profile of the all the samples.
The selected unique bands were excised from the agarose gel (1.8%) with a sterilized scalpel and eluted with gel elution kit (Mfg. Biomatrix Co. Ltd.). The eluted samples were again checked on agarose gel and stored at −20 °C for further use.
The eluted putative specific DNA amplicons were cloned into pGEM-T easy vector (Promega Corporation, Madison, WI, USA). The ligated plasmid was introduced into E. coli strain DH5α by heat shock method. The competent cells were prepared for transformation by the CaCl2 method (Sambrook and Russell 2001). White colonies were picked from LB-X-gal-IPTG plates and grown overnight in LB medium containing ampicillin. The recombinant plasmid was then isolated from the bacterial culture using plasmid isolation kit obtained from Fermentas, USA. Confirmation of successful cloning was carried out by restriction digestion of the recombinant plasmid using the enzyme EcoRI. The cloned amplicons were sequenced by employing primer walking technique by M/s Bangalore Genei, Bangalore, India (ABIPRISM BigDye Terminator v3.1).
The nucleotide sequences of RAPD amplicons were used for similarity searches against NCBI database using Blastn program (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The unique amplicons were also submitted to NCBI, USA. Further, the amplicons were used to design specific primer.
SCAR primers were designed as an extension of the original RAPD primer to which a few arbitrary bases were added after analyzing the sequence (BR Biochem Life Sciences, New Delhi, India). The designed primers of unique DNA sequences (SCAR primers) specific to CPT17 were used for amplification of genomic DNA from all accessions. SCAR marker assay was carried out in 0.2-ml PCR vials containing 12.5-µl 2 X-PCR smart mixes (Origin Diagnostics, Karunagapally, Kerala), 3-µl sample DNA (10 ng/µl), 1-µl forward primer, and 1-µl reverse primer (20 pmol/µl), and 7.5-µl double distilled water. The polymerase chain reaction (PCR) was performed with a gradient thermo cycler (Eppendorf, Mastercycler gradient, Hamburg, Germany). The standardized amplification was performed at an initial denaturation at 94 °C for 4 min, followed by 40 cycles of denaturation at 94 °C for 30 s, primer annealing based on Tm for 1 min, primer extension at 72 °C for 2 min, and final extension at 72 °C for 10 min. The annealing temperature for each primer was standardized by performing gradient PCR. The annealing temperature was first calculated as fourfold the number of GC (guanidine/cytosine) plus twofold the number of AT (adenine/thymine) (Marieschi et al. 2016). Each primer pair was tested with different annealing temperature to obtain the standard temperature.
PCR products were separated on 1.8% agarose gel having 3-μl/100 ml ethidium bromide. 1X TAE buffer was used as both gel and tray buffer. After completion of PCR amplification reaction, 3-μl of 6X loading dye (Bromophenol blue) was added to each PCR tube. About 1.8% agarose gel in 1X TAE buffer (pH 8.0) was prepared, and the contents of the tube were loaded into the gel. A total of 100-bp DNA ladder (100–5000-bp, Origin Diagnostics, Karunagapally, India) was used as molecular marker. Electrophoresis was carried out on 80 V until the loading dye reached the gel front and DNA banding patterns were then visualized using gel documentation system (BIORAD Gel DocTMXR + , California, USA).
Results and Discussion
RAPD primers (35 Nos.) were initially tested with 23 accessions of M. oleifera for the RAPD profile. The most suitable set of primers was however selected for RAPD analysis. The primary screening of 27 primers revealed a total of 519 reproducible bands ranging from 100 to 5000-bp in the accessions of Moringa species generated 89.61% polymorphism (Table 2). Primer RAPD3 showed 100% polymorphism. The number of polymorphic loci ranged from 7 to 29, with a mean of 17.44 loci/primer. The maximum number of polymorphic bands scored by primer RAPD3 followed by primer KFP21. The total number of polymorphic loci obtained was high (29) in the primer RAPD3 and low (7) in primer OPJ13. Out of the 49 bands, the high number of monomorphic bands was observed in KFP7, KFO8, and KFP9 (3).
These RAPD profiles thereafter analyzed for identification of unique amplicons of superior genotype (CPT17) for developing SCAR markers. There were a number of polymorphic as well as monomorphic bands obtained in all the genotypes. Unique fragments are the specific fragments, present in only one accession for the given primer combination (Tatikonda et al. 2009). Out of the unique amplicons obtained in RAPD profile of CPT17, the most prominent and intense band was selected for developing SCAR markers in this study. The selection criterion for unique band also included its size, and the smaller fragments were selected for ease of sequencing. These genetic differences were successfully detected by RAPD technique, and it was quite evident that this technique can be employed for identification of correct trait.
Identification of CPT17
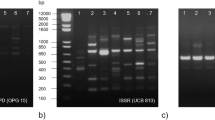
In RAPD analysis, a number of specific RAPD markers were scored specific to superior genotype (CPT17) (Fig. 1). RAPD-PCR reaction was repeated 3 times to make it free from technical errors. In all the trails of PCR reactions, the CPT17 specific marker OPA19 (CAAACGTCGG) consistently amplifies a product of interest 1500-bp (Fig. 1a). Basha and Sujatha (2007) have identified unique bands specific to three accessions of Jatropha, viz., non-toxic Jc, Kerala, and Neemuch-1. After unique band identification, the next step in RAPD-PCR analysis is to validate the marker by ensuring its reproducibility via RAPD to SCAR conversion; i.e., a more efficacious pair of primers of approximately 15–30-bp in length is designed from the RAPD marker sequence, perfectly capable of yielding dependable results with each amplification (Bhagyawant 2016). In order to create a reliable marker for superior germplasm of M. oleifera, the sequence of the 1500-bp DNA fragment from the RAPD analysis was used for further designing a pair of specific primers. Likewise, Cunha et al. (2015) converted high molecular size (1700-bp) RAPD fragment to SCAR primer to discriminate resistant and susceptible genotypes to Foc infection. CPT17 being the superior genotype on the basis of morphological, nutritional, and molecular level.

Identification of CPT17 (superior genotypes) using the primers. a OPA-19, b OPF-02, and c RAPD-2. L-Ladder, 1–23 M. oleifera accessions
Cloning and Sequencing of Unique Band
Unique DNA amplicons of CPT17 was cloned into the pGEMT Easy vector. The presence of a single 3-terminal thymidine at both ends was the reason for selecting the pGEM T Easy vector for cloning. By preventing the circularization of the vector and providing compatible overhangs for PCR products produced by certain thermostable polymerases, T-overhangs at the insertion site significantly improved the efficiency of ligation of PCR products (Mezei and Storts 1994; Robles and Doers 1994). Furthermore, pGEM-T Easy vectors are of high copy number vectors with T7 and SP6 RNA polymerase promoters flanking a multiple cloning region within the enzyme β-galactosidase’s-peptide coding region. The inactivation of the α-peptide during insertion enables recombinants that were identified using blue/white screening on indicator plates. Within various cloning areas, they also contain numerous restriction sites. In the α-peptide coding region of the enzyme β-galactosidase, the desired RAPD fragment was inserted at multiple cloning sites. Each transformed plasmid was isolated, and the existence of the cloned DNA insert was confirmed via restriction digestion with the restriction enzyme EcoRI. The plasmid was sequenced at the Bangalore Genei. The sequence (Fig. 2) was then subjected to homology searches with the sequenced RAPD amplicons obtained from CPT17 using the nonredundant database at GenBank and BLAST at NCBI. The BLAST review revealed no significant similarities recognized in protein-coding sequences. In studies of eight pine species, Nkongolo et al. (2002) discovered RAPD markers unique to one or two species. Several species demonstrated high sequence similarity between amplified sequences of the same molecular weight in this research. We found no correlation between the cloned RAPD band (1500-bp) and any other sequences previously released. In this regard, our findings are consistent with those of Amicucci et al. (1997) and Solieri and Giudici (2010), who discovered that the sequence of RAPD fragments in their analysis was not similar with the previously decided sequences.

Sequence data of amplified product of 1500-bp band generated by primer OPA-19. The highly bold redion indicating the RAPD (OPA19) primer sequence
Primer Designing
BLAST results revealed that the selected RAPD sequences of each plant sample did not have any similarity with the sequences in the NCBI database and also were not contaminated with the vector sequences. Based on these unique characterized sequences, specific primers for CPT17 were designed. The suitable length of primers and annealing temperature, results in sharpest and most intense products, was empirically determined for each primer pair.
To design CPT17 specific primer, the RAPD marker was used plus first 11 or 15-bp bases in the sequence. Thus, internal primers referred as SCAR primers, designed (Table 3). The three primer pairs (forward and reverse), viz., F1R1 (TCAAACGTCGGGTTTATGGAT/TCAAACGTCGGTGGAAGAGGG), F2R2 (TCAAACGTCGGGTTTATGGATGG/TCAAACGTCGGTGGAAGAGGGAG), and F3R3 (TCAAACGTCGGGTTTATGGATGGAA/TCAAACGTCGGTGGAAGAGGGAGA) designed were used for PCR amplification using the 23 accessions of M. oleifera under varied annealing temperatures ranging from 40 to 65 °C. Nine combinations of primer pairs (viz., F1R1, F1R2, F1R3, F2R1, F2R2, F2R3, F3R1, F3R2, and F3R3) were tested to find out better primer combination.
Amplification of DNA Samples Using SCAR Primers
Out of the nine SCAR primer pair combinations, the SCAR primer F2R2 showed an amplification of single, distinct, and brightly resolved band of the same size as the original RAPD fragment of the primer OPA at an annealing temperature 63 °C (Fig. 3) for CPT17 (superior germplasm). This band was absent in all other accessions. But the decamer RAPD primer OPA produced polymorphic bands in all the 23 accessions. According to Parasnis et al. (2000), SCAR primers deduced from internal sequences are less polymorphic than those including initial RAPD primer sequences, suggesting that the polymorphism is only present in the decamer sequences derived from the RAPD primer sequence. The RAPD markers identified a large number of non-specific fragments, whereas the SCAR markers only identified the specific RAPD fragments (Abraham et al. 2013). It indicates that the SCAR primer F2R2 is specific to identity CPT17.

Amplification of 1500-bp band generated by SCAR primer F2R2 in CPT17. L-Ladder, 1–23 M. oleifera accessions
Primers F2 and R2, both contain 23-bp and the annealing temperature of these primers, were 63 °C. At 63 °C, the primer F2R2 produced the most prominent and intense band for CPT17. SCAR primers of 22–25 nucleotide long were designed for stringent conditions of annealing. According to Innis et al. (1990) using longer primers and higher annealing temperatures during PCR reaction, makes the amplification more stringent and more reproducible. It overcomes the main limitation of RAPD (Smith and Register 1998). The longer annealing site and higher annealing temperature avoid mismatching (Innis et al. 1990; Ferreira and Grattapaglia 1996). GC content of F2 and R2 were 48% and 57%, respectively. According to Ferreira and Grattapaglia (1996), GC content was kept between 55 and 60% to maintain the strength and fidelity of the primers. Also, specificity, temperature and time of annealing is partly dependent on primer length and its GC content (Kiran et al. 2010). SCAR markers were generally a dominant marker. Each marker amplified one fragment; however, there were other cases that one marker produces more than one fragment. In our study, only one fragment was amplified, but in Paraseriathes falcataria, five SCAR markers produced both types of polymorphisms (dominant and codominant) (Yuskianti and Shiraishi 2010). In Asparagus, SCAR markers were scored as adominant marker (Kiran et al. 2010).
Combining RAPD and SCAR markers provides a simple and reliable tool for the genetic characterization of plant species (Bhagyawant 2016). Dyneshwar et al. (2006) developed a SCAR marker (343-bp) for correct genotype identification of Phyllanthus emblica from a species-specific amplicons developed by comparative analysis of RAPD profiles of different cultivars of Phyllanthus. Wang et al. (2011) developed SCAR marker from RAPD fragment to select hybrid strains of Saccharomyces cerevisiae. SCAR marker linked to powdery mildew resistance gene er1 in Pisum sativum was developed by Srivastava et al. (2012). Liriope platyphylla and Ophiopogon japonicas are closely related medicinal plants. Li and Park (2012) developed specific SCAR sequence primers for the identification of closely related these species. Korekar et al. (2012) converted a 470-bp RAPD fragment of Hippophae rhamnoides to a SCAR and used it successfully to determine the sex of plants in early stage. Yang et al. (2013) developed a SCAR marker for variety authentication. The Oryza sativa plants were screened using RAPD and SCAR techniques for discrimination of Thai jasmine rice mutants (Semsanget al. 2013). Litchi chinensis Sonn, L. var., chinensis, belongs to the family Sapindaceae. Litchi is an edible fruit having many medicinal properties. Numerous litchi cultivars grown across the world creates confusion regarding their naming and identification. In this regard, Cheng et al. (2015) has developed a SCAR marker for the authentication of Litchi species by improved RAPD-SCAR amplification. Cunha et al. (2015) have developed a RAPD SCAR marker as a tool to discriminate resistant and susceptible genotypes of Foc infected banana plants.
Due to the use of longer PCR primers, SCARs do not have low reproducibility as generally encountered in RAPDs. The length of the primer determines the specificity and reproducibility of SCAR marker (Vanichanon et al. 2000). Oligonucleotides between 18 and 24 bases are extremely sequence specific and more reproducible than RAPD primers (Kiran et al. 2010). The high specificity of SCAR markers become an important interface tool of DNA fingerprinting. More numbers of SCAR markers are needed to develop various biological systems for discrimination of traits of importance.
Conclusions
In conclusion, the present study reveals high degree of genetic diversity in M. oleifera germplasm in terms of yield levels and molecular profiles. RAPD marker used in the study was informative to differentiate the high yielding germplasm from low yielding ones (OPA19, 1500-bp). The identified RAPD marker was converted to SCARs for increasing the reliability and use in marker-assisted programs aimed at development of accessions with high yield and vitamin C content. The SCAR primer pair (F2R2) was designed to find out the superior trait M. oleifera (CPT17) on the basis of morphological, nutritional, and molecular levels. The newly developed SCAR primer pair F2R2 amplified a 15,000-bp fragment superior germplasm (CPT17), while that fragment was absent in all the other accessions. The results of this study also indicated that the improved RAPD analysis has the potential for the genetic analysis of this crop. The RAPD fragments can be cloned to generate to diagnostic SCAR markers that are stable and specific. Thus, the combination of the two techniques provides a simple and reliable tool for the genetic characterization of any crop plant. SCAR markers developed in this study can be used to support tree improvement program of M. oleifera. Further work should also be done in the verification of more accessions of M. oleifera using developed SCAR markers in the future.
References
Abraham G, Pandey N, Mishra V, Chaudhary AA, Ahmad A, Singh R, Singh PK (2013) Development of SCAR based molecular markers for identification of different species of Azolla. Indian J Biotechnol 12:489–492
Amicucci A, Rossi I, Potenza L, Agostini D, Stocchi V (1997) Use of sequence characterised amplified region and RAPD markers in the identification of the white truffle Tuber magnatum Pico. Biotechnol Tech 11(3):149–154. https://doi.org/10.1023/A:1018493111804
Anuntalabhochai S, Sitthiphrom S, Thongtaksin W, Sanguansermsri M, Cutler RW (2007) Hybrid detection and characterization of Curcuma species using sequence characterized DNA markers. Sci Hortic 111:389–393. https://doi.org/10.1016/j.scienta.2006.11.008
Anwar F, Latif S, Ashraf M, Gilani AH (2007) Moringa oleifera: A food plant with multiple medicinal uses. Phytother Res 21(1):17–25. https://doi.org/10.1002/ptr.2023
Basha SD, Sujatha M (2007) Inter and intra-population variability of Jatropha curcas (L.) characterized by RAPD and ISSR markers and development of population-specific SCAR markers. Euphytica 156:271–431. https://doi.org/10.1007/s10681-007-9387-5
Bhagyawant SS (2016) RAPD-SCAR markers: an interface tool for authentication of traits. J Biosci Med 4:1–9. https://doi.org/10.4236/jbm.2016.41001
Busconi M, Sebastiani L, Fogher C (2006) Development of SCAR markers for germplasm characterisation in olive tree (Olea europea L.). Molecular Breeding 17: 59–68. https://doi.org/10.1007/s11032-005-1395-3
Cheng J, Long Y, Khan MA, Wei C, Fu S, Fu J (2015) Development and significance of RAPD-SCAR markers for the identification of Litchi chinensis Sonn. by improved RAPD amplification and molecular cloning. Electron J Biotechnol 18:35–39. https://doi.org/10.1016/j.ejbt.2014.11.004
Cunha CMS, Hinz RH, Pereira A, Tcacenco FA (2015) A SCAR marker for identifying susceptibility to Fusarium oxysporum f. sp. cunense in banana. Scientia Horticulturae 191: 108–112. https://doi.org/10.1016/j.scienta.2015.04.038
Dahlberg JA, Zhang X, Hart GE, Mullet JE (2002) Comparative assessment of variation among sorghum germplasm accessions using seed morphology and RAPD measurements. Crop Science 42: 291–296. https://doi.org/10.2135/cropsci2002.0291
Dnyaneshwar W, Preeti C, Kalpana J, Bhushan P (2006) Development and application of RAPD-SCAR marker for identification of Phyllanthus emblica L. Biol Pharm Bull 29:2313–2316. https://doi.org/10.1248/bpb.29.2313
Duke JA (1983) Moringa oleifera Lam. Handbook of energy crops. Center for New Crops & Plants Products. Purdue University.[Consultado 28 de mayo de2013] http://www.hort.purdue.edu/newcrop/duke_energy/Moringa_oleifera.html
Durai M, Dubey SC, Triphathi A (2012) Genetic diversity analysis and development of scar marker for detection of Indian populations of Fusarium oxysporum f. sp. ciceris causing chickpea wilt. Folia Microbiology 57:229–235. https://doi.org/10.1007/s12223-012-0118-5
Edwards S, Tadesse M, Demissew S, Hedberg I (2000) Flora of Ethiopia and Eritrea, Vol. II part 1: Magnoliaceae to Flacourtiaceae. Addis Ababa, Ethiopia and Uppsala, Sweden: The National Herbarium, Addis Ababa University p532
Ellsworth DL, Rittenhouse KD, Honeycutt RL (1993) Artifactual variation in Random amplified polymorphic DNA banding patterns. Biotechniques 14:214–217
Ferreira ME, Grattapaglia D (1996) Introducaoaouso de marcadoresmolecularesemanalisegenetica. Brasilia:Embrapa-Cenargen p 220
Innis MA, Gelfand DH, Sninsky JJ, White TJ (1990) PCR protocols: a guide to methods and application. San Diego:Academic Press p 482
Kim H, Han D, Kang J, Choi1 Y, Levi A, Pyo GL, Park Y (2015) Sequence-characterized amplified polymorphism markers for selecting rind stripe pattern in watermelon (Citrullus lanatus L.). Horticulture, Environment and Biotechnology 56(3): 341–349. https://doi.org/10.1007/s13580-015-0017-1
Kiran U, Khan S, Mirza KJ, Ram M, Abdin MZ (2010) SCAR markers: a potential tool for authentication of herbal drugs. Fitoterapia 81:969–976. https://doi.org/10.1016/j.fitote.2010.08.002
Korekar G, Sharma RK, Kumar R, Bisht NC, Srivastava RB, Ahuja PS, Stobdan T (2012) Identification and validation of sex-linked SCAR markers in dioecious Hippophae rhamnoides L. (Elaeagnaceae). Biotech Letter 34:973–978. https://doi.org/10.1007/s10529-012-0852-4
Lee MY, Doh EJ, Chae HP, Young HK, Eung SK, Ko BS (2006) Developmentof SCAR marker for discrimination of A. princeps and A. argyi from other Artemisia Herbs. Biology and Pharmaceutical Bulletin 29:629–630. https://doi.org/10.1248/bpb.29.629
Li G, Park YJ (2012) SCAR Markers for discriminating species of two genera of medicinal plants, Liriope and Ophiopogon. Genet Mol Res 11:2987–2995. https://doi.org/10.4238/2012.May.18.14
Li H, Zhang SG, Gao JM, Wang CG, Zhang Y, Qi LW, Chen L, Song WQ (2008) Development of a sequence characterized amplified region (SCAR) marker associated with high rooting ability in Larix. Biol Plant 52(3):525–528. https://doi.org/10.1007/s10535-008-0102-8
Li HB, Wu XQ, Peng HZ, Fu LZ, Wei HL, Wu QQ, Jin QY, Li N (2008) New available SCAR markers: potentially useful in distinguishing a commercial strain of the superior type from other’s trains of Lentinula edodes in China. App Microb Biotech 81: 303–309. https://doi.org/10.1007/s00253-008-1671-3
Litt M, Luty JA (1989) A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am J Hum Genet 44:397–401
Marieschi M, Torelli A, Beghe D, Bruni R (2016) Authentication of Punica granatum L.: Development of SCAR markers for the detection of 10 fruits potentially used in economically motivated adulteration. Food Chem 202:438–444. https://doi.org/10.1016/j.foodchem.2016.02.011
Mezei LM, Storts DR (1994) Purification of PCR products. In: Griffin HG, Griffin AM (eds) PCR Technology: Current Innovations. CRC Press, Boca Raton, FL, p 21
Mughal MH, Ali G, Srivastava PS, Iqbal M (1999) Improvement of drumstick (Moringa pterygosperma Gaertn.)—a unique source of food and medicine through tissue culture. Hamdard Medicine 42:37–42
Muralidharan K, Wakeland EK (1993) Concentration of primer and template qualitatively affects products in RAPD-PCR. Biotechniques 14:362–364
Navie S, Csurhes S (2011) Weed risk assessment horseradish tree Moringa oleifera. Biosecurity Queensland Department of Employment, Economic Development and Innovation, Brisbane 4001:1–22
Nkongolo KK, Michael P, Gratton WS (2002) Identification and characterization of RAPD markers inferring genetic relationships among Pine species. Genome 45(1):51–58. https://doi.org/10.1139/g01-121
Olson ME (2002) Combining data from DNA sequences and morphology for a phylogeny of Moringaceae (Brassicales). Syst Bot 27:55–73. https://doi.org/10.1043/0363-6445-27.1.55
Paran I, Michelmore RW (1993) Development of reliable PCR-based markers linked to downy mildew resistance genes in lettuce. Theor Appl Genet 85:985–993. https://doi.org/10.1007/BF00215038
Palada MC, Ebert AW (2012) Moringa. In: PeterKV HP (ed) Handbook of Vegetables. Stadium Press, New Delhi, India, pp 193–242
Parasnis AS, Gupta VS, Tamhankar SA, Ranjekar PK (2000) A highly reliable sex diagnostic PCR assay for mass-screening of Papaya seedlings. Mol Breeding 6:337–344. https://doi.org/10.1023/A:1009678807507
Qaiser M (1973) Moringaceae. In flora of West Pakistan. Nasir E and Ali SI pp 1–4
Ravi RSD, Siril EA, Nair BR (2020) Proximate and nutritive value screening of Moringa oleifera, a multipurpose tree, and identification of elite germplasm. Abrahamia 6:16–25
Robles J, Doers M (1994) pGEM®-T Vector Systems troubleshooting guide. Promega Notes 45:19–20
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA (1988) Primer directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239: 487–491. https://doi.org/10.1126/science.2448875
Saini RK, Saad KR, Ravishankar GA, Giridhar P, Shetty NP (2013) Genetic diversity of commercially grown Moringa oleifera Lam. cultivars from India by RAPD, ISSR and cytochrome P450-based markers. Plant Syst Evol 299:1205–1213. https://doi.org/10.1007/s00606-013-0789-7
Saini RK, Shetty NP, Giridhar P, Ravishankar GA (2012) Rapid in vitro regeneration method for Moringa oleifera and performance evaluation of field grown utritionally enriched tissue cultured plants. 3 Biotech 2: 187–192. https://doi.org/10.1007/s13205-012-0045-9
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USApp 6–11
Semsang N, Chundet R, Phanchisri B (2013) Development of a SCAR marker for discrimination of a Thaijasmine rice (Oryza sativa L. cv. KDML105) mutant, BKOS6, and associated with purple color trait in Thai jasmine rice-related varieties. American Journal of Plant Sciences 4: 1774–1783. https://doi.org/10.4236/ajps.2013.49218
Smith JSC, Register JC (1998) Genetic purity and testing technologies for seed quality: a company perspective. Seed Sci Res 8(5):285–293. https://doi.org/10.1017/S0960258500004189
Solieri L, Giudici P (2010) Development of a sequence-characterized amplified region marker-targeted quantitative PCR assay for strain-specific detection of Oenococcus oeni during wine malolactic fermentation. Appl Environ Microbiol 76(23):7765–7774. https://doi.org/10.1128/AEM.00929-10
Somali MA, Bajnedi MA, Al-Faimani SS (1984) Chemical composition and characteristics of Moringa peregrina seeds and seed oil. J Am Oil Chem Soc 61:85–86
Srivastava RK, Mishra SK, Singh AK, Mohapatra T (2012) Development of a coupling-phase SCAR marker linked to the powdery mildew resistance gene ‘er1’in pea (Pisum sativum L.). Euphytica 186:855–866. https://doi.org/10.1007/s10681-012-0650-z
Sujatha M, Makkar HPS, Becker K (2005) Shoot bud proliferation from axillary nodes and leaf sections of non-toxic Jatropha curcas L. Plant Growth Regul 47:83–90. https://doi.org/10.1007/s10725-005-0859-0
Tatikonda L, Wani SP, Kannan S, Beerelli N, Sreedevi TK, Hoisington DA, Devi P, Varshney RK (2009) AFLP-based molecular characterization of an elite germplasm collection of Jatropha curcas L., a biofuel plant. Plant Science 176(4): 505–513. https://doi.org/10.1016/j.plantsci.2009.01.006
Tautz D (1989) Hyper variability of simple sequences as a general source of polymorphic DNA markers. Nucleic Acid Res 17:6463–6471. https://doi.org/10.1093/nar/17.16.6463
Vanichanon A, Blake NK, Martin JM, Talbert LE (2000) Properties of sequence tagged-site primer sets influencing repeatability. Genome 43:47–52. https://doi.org/10.1139/g99-087
Vos P, Hogers R, Bleeker M, Reijans M, Lee TV, Hornes M, Friters A, Pot J, Paleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21):4407–4414. https://doi.org/10.1093/nar/23.21.4407
Wang PM, Wu XC, Chi XQ, Li YD, Zheng DQ, Ding R, Min H (2011) Development and application of RAPD-SCAR markers to identify intra-species hybrids of industrial Saccharomyces cerevisiae. World J Microbiol Biotechnol 27: 185–188. https://doi.org/10.1007/s11274-010-0430-7
Weber JL, May PE (1989) Abundant class of human DNA polymorphisms, which can be typed using the polymerase chain reaction. Am J Hum Genet 44:388–396
Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphism amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Research 18: 6531–6535. https://doi.org/10.1093/nar/18.22.6531
Yang L, Fu S, Khan MA, Zeng W, Fu J (2013) Molecular cloning and development of RAPD-SCAR markers for Dimocarpus longan variety authentication. Springerplus 2:1–8. https://doi.org/10.1186/2193-1801-2-501
Yuskianti V, Shiraishi S (2010) Sequence characterized amplified region (SCAR) markers in Sengon (Paraseriathes falcataria (L.) Nielsen. Hayati Journal of Biosciences 17(4): 167–172. https://doi.org/10.4308/hjb.17.4.167
Zietkiewicz E, Rafalski A, Labuda D (1994) Geome fingerprinting by simple sequence repeats (SSR) anchored polymerase chain reaction amplification. Genomics 20:176–183. https://doi.org/10.1006/geno.1994.1151
Acknowledgements
The authors are grateful to Dr. Suhara Beevy S, Professor and Head, Department of Botany, University of Kerala, for the facilities provided.
Funding
The authors received research fellowship from the University of Kerala (No. Ac.EI/A2/10625/2016-I) to undertake the present work.
Author information
Authors and Affiliations
Contributions
DRS conducted the experiments. DRS and EAS analyzed the data. DRS drafted the manuscript. EAS and BRN designed the experiments. EAS, BRN, and DRS revised the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Highlights
• M. oleifera used for a wide variety of purpose thus regarded as a “multipurpose tree.”
• Specific marker for the identification of superior germplasm of M. oleifera.
• This is the first report regarding SCAR marker development for M. oleifera.
Rights and permissions
About this article

Cite this article
Ravi, D., Siril, E.A. & Nair, B.R. SCAR Marker Development for the Identification of Elite Germplasm of Moringa Oleifera Lam.-A Never Die Plant. Plant Mol Biol Rep 39, 850–861 (2021). https://doi.org/10.1007/s11105-021-01300-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-021-01300-y