
Abstract
Tartary buckwheat is an important edible crop as well as medicinal plant in China. More and more research is being focused on this minor grain crop because of its medicinal functions, but there is a paucity of molecular markers for tartary buckwheat due to the lack of genomics. In this study, a genome survey was carried out in tartary buckwheat, from which SSR markers were developed for analysis of genetic diversity. The survey generated 21.9 Gb raw sequence reads which were assembled into 348.34 Mb genome sequences included 204,340 contigs. The genome size was estimated to be about 497 Mb based on K-mer analysis. In total, 24,505 SSR motifs were identified and characterised from this genomic survey sequence. Most of the SSR motifs were di-nucleotide (67.14 %) and tri-nucleotide (26.05 %) repeats. AT/AT repeat motifs were the most abundant, accounting for 78.60 % of di-nucleotide repeat motifs. SSR fingerprinting of 64 accessions yielded 49.71 effective allele loci from a total of 63 with the 23 polymorphic SSR primer combinations. Analyses of the population genetic structure using SSR data strongly suggested that the 64 accessions of tartary buckwheat clustered into two separate subgroups. One group was mainly distributed in Nepal, Bhutan and the Yunnan-Guizhou Plateau regions of China; the other group was mainly derived from the Loess Plateau regions, Hunan and Hubei of China and USA. The cluster analysis of these accession’s genetic similarity coefficient by UPMGA methods strongly supported the two subgroup interpretation. However accessions from Qinghai of China could be grouped into either of the two subgroups depending on which classification method was used. This region is at the intersection of the two geographical regions associated with the two subgroups. These results and information could be used to identify and utilize germplasm resources for improving tartary buckwheat breeding.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
There are two species of cultivated buckwheat, common buckwheat (Fagopyrum esculentum Moench.) and tartary buckwheat (Fagopyrum tartaricum Gaertn.). They belong to the genus Fagopyrum (family Polygonaceae), which consists of two monophyletic groups—the cymosum and urophyllum—and cluster in the cymosum clade (Sangma and Chrungoo 2010). Tartary buckwheat seeds and leaves are used mainly for human consumption and animal fodder, and contain proteins with high biological value, a balanced amino acid composition, starch, mineral compounds, bioflavonoid rutin, relatively high crude fiber and vitamins (B1, B2, B6) (Fabjan et al. 2003; Lee et al. 2013; Wang et al. 2014). The seeds contain more rutin than common buckwheat, and tartary buckwheat has recently also attracted attention as a pharmaceutical plant. Tartary buckwheat seeds have very high nutritional value, and have been promoted in several brands of tea products and as a health-care supplement food in China. There are abundant buckwheat germplasm resources in China, cultivated mainly in the Loess Plateau and Yunnan-Guizhou Plateau regions. The importance of preservation of these tartary buckwheat genetic resources has attracted attention in China, Russia and India, and germplasm collections have been characterized, evaluated and utilized by various centers (Arora 1995). A total of 879 tartary buckwheat accessions was collected and stored in the national germplasm repository of China. Among the collections, those from Sichuan, Yunnan and Shanxi Province accounted for 19.77 %, 15.14 % and 13.06 %, respectively (Zhao and Shaan 2008). Evaluation and identification of these germplasm resources by a molecular marker approach is very important for elucidating inter- and intra-specific variations and for the use of these stocks for breeding.
Different types of molecular markers have been applied for buckwheat genotyping and to identify genetic diversity. In 2001, based on amplified fragment length polymorphism (AFLP) markers, a study of phylogenetic relationships among 42 accessions from 21 populations revealed that tartary buckwheat probably originated in eastern Tibet or northwestern Yunnan in China. (Tsuji and Ohnishi 2001). In 2002, random amplified polymorphic DNA (RAPD) markers were used to analyze 28 different accessions belonging to 14 species and two subspecies of Fagopyrum; the results showed that F. tartaricum is closer to its wild ancestor F. tartaricum ssp. potanini Batalin (Sharma and Jana 2002). Inter-simple sequence repeats (ISSR) were also used to analyze genetic diversity of 15 germplasms of F. tartaricum in the Western Himalayas, which revealed that the genetic diversity of F. tartaricum correlated significantly with altitude (Kishore et al. 2013). Variation of rutin content among 195 tartary buckwheat accessions has been studied using AFLP fingerprinting, which showed that they clustered into two separate groups, one with high and the other with low rutin content (Gupta et al. 2012). Simple repeat sequence (SSR) markers are used widely for high-throughput genotyping and map construction, due to their great abundance, random distribution within the genome, high polymorphism information content (PIC) and stable co-dominance (Zietkiewicz et al. 1994; Iwata et al. 2005). Despite this, there has not been much research on the application of SSRs to the study of the genetic diversity of tartary buckwheat, due to the high cost if its development and the lack of genome sequence data.
In this study, we obtained approximately 25,000 SSR loci from a genome survey of tartary buckwheat by using next generation sequencing with the Illumina HiSeq 2000 platform, and developed SSR markers and tested their effectiveness for analyzing genetic diversity with 64 accessions of tartary buckwheat.
Results
Genome Size Prediction and Sequence Assembly
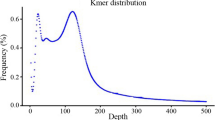
A total of 21.9 Gb nucleotide raw reads were obtained by sequencing a tartary buckwheat genome library with 500 bp inserts, and the average read length and sequencing coverage were 100 bp and 40 fold, respectively. According to17-mer analysis, the K-mer frequency distribution curve conformed to a Poisson distribution, and had a peak at K = 37 (Fig. 1). A total of 18,387,842,676 unique k-mers were identified, and the genome size of tartary buckwheat was estimated to be 497 Mb by the Lander-Waterman formula. We also observed a smear phenomenon after the main peak, suggesting a relatively high abundance of repetitive sequences in this genome. A total of 204,340 contigs and 348 Mb assembled sequences were obtained by removing redundancy from this genome survey information. The contig and scaffold length of N50 were 1492 and 7862 bp in this assembly, and the number of contigs (longer than 2 kb) was 31,787 (Supplementary Table S1).

K-mer (17mer) frequency analysis of the Fagopyrum tartaricum sequence. Data produced from 500 bp insert libraries. The peak k-mer frequency was 37. Genome size was estimated with the formula: estimated genome size (bp) = total number of K-mers /peak k-mer frequency
Identification and Characteristic of SSR Motif in Genome Survey
From the 348,339,842 bp genome survey sequence, 24,505 SSR loci were identified, which included 18,285 SSR-containing sequences (Supplementary additional files S1, S2). However, only 4440 sequences contained more than 1 SSR motif, and 898 SSR loci were present in compound formation. The motif types of SSR repeats included 67.14 % di-nucleotide, 26.05 % tri-nucleotide, 5.29 % tetra-nucleotide, 0.93 % penta-nucleotide and 0.59 % hexa-nucleotide repeats (Fig. 2a). Of the di-nucleotide repeats, AT/AT was the most abundant, accounting for 78.60 % (12,931) of all di-nucleotide repeats, whereas AG/CT and AC/GT repeats accounted for 14.47 %(2381) and 6.67 %(1098), respectively (Fig. 2b). GC/CG repeats were rather rare (0.26 %, 42). Among the tri-nucleotide repeats, AAT/TTA was the most abundant, accounting for 31.83 % (2032) of all tri-nucleotide repeats, followed by AAG/CTT (29.20 %, 1864) and ATC/TAG (11.84 %, 756) (Fig. 2c). AAAT/ATTT was the most abundant tetra-nucleotide repeats, accounting for 56.25 % (729) of the total, followed by AAAG/CTTT (12.96 %, 168). Among penta- and hexa-nucleotide repeats, AAAAT/ATTTT and AAACCG/CGGTTT were the most abundant, accounting for 25.11 % (57) and 0.08 % (12), respectively. Moreover, 98.84, 93.72, 93.60, and 76.65 %, of di-, tri-, tetra- and penta-nucleotide repeats, respectively, were less than 30 bp in length, and only < 0.3 % tri-, tetra-, pentra-, or hexa-nucleotide repeats were longer than 35 bp.

a–c Identification and characteristics of simple repeat sequence (SSR) motifs. a Frequency of different SSR motif repeat types. b Frequency of different di-nucleotide SSR motifs. c Frequency of different tri-nucleotide SSR motifs
The distribution of genome-SSRs with various motifs was also studied, and the frequencies of genome-SSRs occurring with different repeats numbers are shown in Fig. 3. Our results show di-nucleotide and tri-nucleotide repeats were more abundant than tetra-, penta-, and hexa-nucleotide repeats. The frequency distribution range of SSR motif repeats among genome-SSR markers in tartary buckwheat ranged from 6 to 18 repeats for di-nucleotide, from 5 to 12 repeats for tri-nucleotide, from 5 to 9 repeats for tetra-nucleotide, from 5 to 7 repeats for penta-nucleotide, and from 5 to 6 repeats for hexa-nucleotide. There were 8962 SSRs with 6 tandem repeats, which was the highest repeat number (36.57 %), followed by 5 tandem repeats (19.86 %, 4866), 7 tandem repeats (19.81 %, 4854), 8 tandem repeats (10.76 %, 2636), 9 tandem repeats (5.89 %, 1443) and over 10 tandem.

The distribution and frequency of SSR motif repeat numbers. x-axis SSR repeat numbers, y-axis Frequency of this SSR type. Red bars Di-nucleotide repeat motifs, yellow-green bars tri-nucleotide, green bars tetra-nucleotides, blue bars penta-nucleotides, purple bars hexa-nucleotides
SSR Loci and Sequence Length Variation
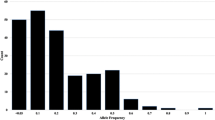
We designed 221 pairs of primers from the above SSR loci in the tartary buckwheat genome, and tested their PCR product polymorphism by polyacrylamide gel electrophoresis (PAGE) analysis in 64 tartary buckwheat accessions (Supplementary additional files S3, S4). The amplification products of 193 pair primers showed 170 monomorphic and 23 polymorphic SSR loci. Finally, 23 polymorphic SSR primer combinations were used for analyzing genetic diversity among these cultivars. These polymorphic SSR loci included 4 di-nucleotide, 13 tri-nucleotide, 1 tetra- and 1 penta-nucleotide and 4 hexa-nucleotide repeats. The PCR products of these SSR primers showed a range of PIC from 0.030713 to 0.747487, and Shannon’s information index from 0.0795 to 1.3828. Only 11 SSR primer combinations had PIC values higher than 0.5. The Shannon’s information indices for (AAG)10, (AAT)10, (AGT)10 and (TTC)11 repeats were higher than 1.0, indicating their abundant genetic variance among these different cultivars. Most importantly, (TTC)11 and (AT)11 motifs had the highest PIC value (0.742356 and 0.747487) and allele loci (4), which could be used as core primers to distinguish these seed accessions (Fig. 4).

PCR-amplified products of markers SXAU97 (a) and SXAU134 (b) for the 64 F. tartarcium accessions resolved on 6 % PAGE. Lanes: M 20 bp DNA marker; 1, 10, 20, 30, 40, 50, 60 number of F. tartarcium accessions
We re-sequenced some of these polymorphic SSR locus amplicons using TA cloning strategy and found allelic length variations among these different accessions. For example, sequence alignment showed that the locus SXAU97 amplified variant allelic from 206 to 234 bp, and revealed a variable number of repeats in the microsatellite motifs (TTC)n accompanied by an insertion or deletion mutation (Fig. 5).

Genetic analysis of SXAU97 PCR amplicon sequence variant length with (TTC)n SSR motif repeat
Population Genetic Structure of Tartary Buckwheat
Bayesian modelling of the number of homogeneous genepools (K) in STRUCTURE gave a minimum mean probability of In P(D) = −1156.3 at K = 1 and a maximum mean probability of In P(D) = −604.6 at K = 7. Evaluation of the optimum number of K following the procedure of Evanno et al. (2005) found one clear maximum for ΔK, at K = 2 (Fig. 6a). The 64 accessions from 15 geographical regions were classified into two subgroups (Fig. 6b). One subgroup contained 31 accessions collected from China (19 accessions), Nepal (3 accessions), Bhutan (8 accessions) and the United States (1 accession). The residual 33 accessions belonged to the other subgroup, which included 29 China accessions and 4 US accessions. Forty-eight accessions from China, however, had no obvious geographical distribution characteristics, but 10 out of 15 accessions from the Yunnan-Guizhou Plateau (Sichuan, Yunnan and Guizhou of China) and 10 out of 11 accessions from the Loess Plateau regions (Shanxi, Shaanxi, Qinghai and Gansu of China) were clearly classified into two separate subgroups. Meanwhile, according to similarity coefficient, we found that these accessions also clustered into two subgroups by the UPMGA method (Fig. 7). One subgroup included 26 accessions collected from Gansu (4 accessions), Hunan (3 accessions), Hubei (7 accessions), Shanxi (2 accessions), Shaanxi (1 accession), Qinghai (1 accession), Anhui (1 accession), Jiangxi (1 accession), Guizhou (1 accession), Guangxi (1 accession) regions of China, and the US (4 accessions). The other subgroup contained 38 accessions, which were derived from Bhutan (8 accessions), Nepal (3 accessions), and, from China, Qinghai (3 accessions), Anhui (2 accessions), Sichuan (4 accessions), Guizhou (7 accessions), Yunnan(3 accessions) and Guangxi (5 accessions).

a, b Population genetic structures and relationships of 64 Tartary buckwheat accessions (K = 2). a x-axis K values from 2 to 7, y-axis delta K. b x-axis 64 accessions, y-axis Q assignments for each sample under K = 2. Colored dots/number key of sources of accessions: Orange/1 Bhutan; red/2 Nepal; dark green/3 Guizhou, China; green/4 Yunnan, China; yellow/5 Sichuan, China; blue/6 Qinghai, China; light blue/7 Gansu, China; dark blue/8 Anhui, China; purple/9 Guangxi, China; dark purple/10 Hubei, China; dark gray/11 Hunan, China; light gray/12 Shaanxi, China; light pink/13 Jiangxi, China; dark pink/14 shanxi, China; black/15 USA

Genetic similarity coefficient analysis of 64 F. tartaricum accessions by the unweighted pair-group method with arithmetic means (UPGMA) method. Background bar color/number indicating source of accession: Brown/1 Bhutan; red/2 Nepal; dark green/3 Guizhou, China; green/4 Yunnan, China; yellow/5 Sichuan, China; blue/6 Qinghai, China; light blue/7 Gansu, China; dark blue/8Anhui, China; violet/9 Guangxi, China; darkmagenta/10 Hubei, China; light cyan/11 Hunan, China; dark gray/12 Shaanxi, China; light pink/ 13 Jiangxi, China; steel blue/14 shanxi, China; dimgray/15 USA
Discussion
The genome sizes of 20 species of the genus Fagopyrum were determined by flow cytometry with rice as a reference, and the C-values of F. tartaricum (2n = 16) was 0.55, which corresponds to a genome size for tartary buckwheat estimated as about 540 Mb (Nagano et al. 2000). A draft genome sequence of tartary buckwheat was reported during the meeting Plant & Animal Genome XXII in 2014, indicating that the total number of bases included in the assembly was 372 Mb, corresponding to 70 % of the genome (Logacheva 2014), and the genome size of tartary buckwheat could be estimated to be 530 Mb. From our genome survey data, the genome size of F. tartaricum was estimated to be 497 Mb, close to the size predicted by flow cytometry and the drafted genome. The different estimates of the genome size for F. tartaricum might be due to different experiment materials or methods used. With the development of next generation sequencing technology, genome sequencing of these minor crops such as tartary buckwheat should not only help to understand genome organization and key genes associated with important traits such as higher rutin content, self-compatibility, indeterminate inflorescence and other agronomic traits, but also make it possible to generate molecular markers for subsequent application in molecular breeding (Mardis 2008; Tan et al. 2014; Du et al. 2015).
A total of 24,505 SSR loci were obtained from the genome survey sequence data described here. Therefore, the SSR distribution frequency in this genome is estimated to be about 70.34 SSR per Mb, which is lower than in Arabidopsis, where it is about 135.5 SSR per Mb (Zhao et al. 2015). In Arabidopsis, the higher frequency of AT/AT SSRs motifs accounted for 52.6 % di-nucleotide SSRs motifs, in the whole genome, followed by AG/CT motif (36.8 %) (Sonah et al. 2011). Meanwhile, we also found a higher frequency of AT/AT and AG/CT SSRs motifs, accounting for 78.6 % and 14.47 % di-nucleotide SSR motifs in the tartary buckwheat genome, respectively. In this study, the distribution frequency characteristic of di-nucleotide SSR motifs in tartary buckwheat was the similar to that of Arabidopsis, possibly because they are both dicot species. In the tri-nucleotide repeats category, however, the distribution frequency of AAT/ATT repeats (31.83 %) in the tartary buckwheat genome was higher than in Arabidopsis (7.9 %). Moreover, the distribution frequency of all tetra-, penta- and hexa-nucleotide repeats in tartary buckwheat genome was higher than in Arabidopsis. These differences in distribution frequency of SSR motif types suggest that the duplicated regions in each species may be subject to different selection pressures.
Our analysis suggests that two separate genetic populations of F. tartaricum are distributed in two geographical regions of China with different ecological environments and latitude: the Loess plateau (‘Shanxi & Shaanxi’, ‘Gansu & Qinghai’) and the Yunnan-Guizhou plateau (‘Sichuan’, Yunnan & Guizhou, Guangxi). A few reports indicate that tartary buckwheat may have originated from the Himalayas (Ohnishi 1998; Yasui et al. 2004). Interestingly, 3 accessions from Nepal and 8 accessions from Bhutan, which are located in the southern Himalayas, were grouped with 10 accessions from the Yunnan-Guizhou plateau. This result revealed that these accessions from the Yunnan-Guizhou plateau were close relatives of those from Nepal and Bhutan. Additionally, previous research had revealed that the original birthplace of cultivated Tartary buckwheat is eastern Tibet and North-western Yunnan (Tsuji and Ohnishi 2001). Our results provided further evidence to prove that the origin of cultivated tartary buckwheat is the Yunnan-Guizhou plateau in China. The results of cluster analysis of these accessions using UPMGA methods were also consistent with this conclusion, although the accessions from Qinghai appeared to belong to a different subgroup because it was at the intersection of the two geographical regions associated with the two subgroups. In addition, the accessions from Hunan and Hubei had a close genetic relationship and clustered into one group by two different clustering analyses. This finding suggests these accessions from Hunan and Hubei might represent an additional independent ecological population.
The present study provided important new SSR loci and polymorphism markers, and revealed the population genetic structure mapping of 64 tartary buckwheat accessions. These results will be helpful for evaluation of germplasm resources for breeding.
Materials and Methods
Plant Materials, Genome Sequencing and Genome Survey
The plant material used for whole-genome de novo sequencing was “HeiFeng No.1”, an elite tartary buckwheat cultivar planted mainly in Shanxi province and nearby areas of China as a homozygous inbred line. In total, paired-end sequencing libraries with insert sizes of approximately 500 bp were constructed and sequenced using the Illumina Hiseq 2000 platform. The raw genome survey sequence data was first filtered to obtain high quality reads, then assembled using SOAP (http://soap.genomics.org.cn/soapdenovo.html) de novo software to contig and scaffold . K-mer analysis was used to estimate genome size.
Sixty-four accessions of tartary buckwheat collected worldwide (Table 1) were used to evaluate and identify the SSR suitability for genetic distance analysis. Young leaves were collected and frozen in liquid nitrogen prior to genomic DNA extraction using CTAB methods (Porebski et al. 1997). DNA concentrations were measured by spectrophotometry at 260 nm, and the extracts were electrophoresed on 1 % agarose to confirm quality. The purified DNAs were standardized at 100 ng/μL and stored at −20 °C.
SSR Identification and Primer Analysis
The Perl script MicroSatellite (MISA) was used to identify microsatellites in tartary buckwheat genomes (Parida et al. 2010; Sonah et al. 2011). To identify the presence of SSRs, only two to six nucleotide motifs were considered, and the minimum repeat unit was defined as five reiterations for all of the repeat units. The SSR primers were designed by Primer3 software online (Untergrasser et al. 2012). We designed 221 pairs of primers that met the following parameters (Supplementary additional file S5): 100–300 bp final product length, primer size from 18 to 25 bp and GC content 35–70 %; annealing temperature was 50–65 °C. Primers were synthesized by Sangon (Shanghai, China). We tested 64 cultivars for 221 SSR loci by PAGE to confirm their suitability.
Each 25 μL reaction mixture contained 10× PCR buffer (plus Mg2+), 0.2 mM of each dNTP, 5 pmol of each reverse primer and forward primer, 1.5 units of rTaq polymerase (TaKaRa, Japan) and 100 ng genomic DNA template. DNA amplification was accomplished in a PCR system (PTC-200, MJ Research, Waltham, MA) programmed at 94 °C for 5 min for initial denaturation, then 35 cycles at 94 °C (30 s)/55 °C(30 s)/72 °C (30 s), followed by an extension step for 10 min at 72 °C. Each PCR product was run on 2 % agarose gel at 120 V for a quality check. Subsequently, PCR products were run on a 6 % denaturing polyacrylamide gel at 60 W constant gain, and polymorphism was detected by silver staining.
SSR Data Analysis
Twenty-three high polymorphic SSR primer combinations were selected as core SSR markers. Sixty-four tartary buckwheat varieties were analyzed using these core SSR markers. The observed number of alleles (Na), effective number of alleles (Ne), and PIC were calculated by using PopGen 32 (http://popgen-popgen.software.informer.com). Population genetic structure was calculated using Structure 2.3.4 (Hubisz et al. 2009). Genetic similarity coefficient and UPMGA cluster analysis of these accessions were calculated by NTSYS software (Powell et al. 1996).
References
Arora RK, Baniya BK, Joshi BD(1995) Buckwheat genetic resources in the Himalayas: their diversity, conservation and use. Curr Adv Buckwheat Res 39–45
Du F, Wu Y, Zhang L, Li XW, Zhao XY, Wang WH, Gao ZS, Xia YP (2015) De novo assembled transcriptome analysis and SSR marker development of a mixture of six tissues from Lilium oriental hybrid ‘Sorbonne’. Plant Mol Biol Report 2:281–293
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 8:2611–2620
Fabjan N, Rode J, Kosir IJ, Wang Z, Zhang Z, Kreft I (2003) Tartary buckwheat (Fagopyrum tataricum Gaertn.) as a source of dietary rutin and quercitrin. J Agric Food Chem 51:6452–6455
Gupta N, Sharma SK, Rana JC, Chauhan RS (2012) AFLP fingerprinting of tatary buckwheat accessions (Fagopyrum tataricum) displaying rutin content variation. Fitoterapia 83:1131–1137
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Iwata H, Imon K, Tsumura Y, Ohsawa R (2005) Genetic diversity among Japanese indigenous common buckwheat (Fagopyrum esculentum) cultivars as determined from amplified fragment length polymorphism and simple sequence repeat markers and quantitative agronomic traits. Genome 48:367–377
Kishore G, Pandey A, Dobhal R, Gupta S (2013) Population genetic study of Fagopyrum tataricum from Western Himalaya using ISSR markers. Biochem Genet 51:750–765
Lee CC, Shen SR, Lai YJ, Wu SC (2013) Rutin and quercetin, bioactive compounds from tatary buckwheat, prevent liver inflammatory injury. Food Funct 4:794–802
Logacheva MD, Penin AA, Sutormin RA, Demidenko NV, Naumenko SA, Vinogradov DV, Mazin PV, Kurmangaliev YZ, Gelfand MS, Kondrashov AS (2014) A draft genome sequence of Tatary Buckwheat, Fagopyrum tataricum. Plant & animal genome XXII, San Diego, CA. p 667
Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24:133–141
Nagano M, Aii J, Campbell C, Kawasaki S, Adachi T (2000) Genome size analysis of the genus Fagopyrum. Fagopyrum 17:35–39
Ohnishi O (1998) Search for the wild ancestor of buckwheat III: the wild ancestor of cultivated common buckwheat, and of Tatary buckwheat. Econ Bot 52:123–133
Parida SK, Yadava DK, Mohapatra T (2010) Microsatellites in Brassica unigenes: relative abundance, marker design, and use in comparative physical mapping and genome analysis. Genome 53:55–67
Porebski S, Bailey GL, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Report 15:8–15
Powell W, Morgante M, Andre C, Hanafey M, Vogel J (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) marker for germplasm analysis. Mol Breed 2:225–238
Sangma SC, Chrungoo NK (2010) Buckwheat gene pool: potentialities and drawbacks for use in crop improvement programmes. Eur J Plant Sci Biotechnol 4:45–50
Sharma R, Jana S (2002) Species relationships in Fagopyrum revealed by PCR-based DNA fingerprinting. Theor Appl Genet 105:306–312
Sonah H, Deshmukh RK, Sharma A, Singh VP, Gupta DK, Gacche RN, Rana JC, Singh NK, Sharma TR (2011) Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium. PLoS One 6:e21298
Tan ML, Wu K, Wang L, Yan MF, Zhao ZD, Xu J, Zeng Y, Zhang XK, Fu CL, Xue JF, Wang LJ, Yan XC (2014) Developing and characterising Ricinus communis SSR markers by data mining of whole-genome sequences. Mol Breed 34:893–904
Tsuji K, Ohnishi O (2001) Phylogenetic relationships among wild and cultivated Tartary buckwheat (Fagopyrum tataricum Gaertn.) populations revealed by AFLP analyses. Genes Genet Syst 76:47–52
Untergrasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115
Wang X, Feng B, Xu Z, Sestili F, Zhao G, Xiang C, Lafiandra D, Wang T (2014) Identification and characterization of granule bound starch synthase I (GBSSI) gene of tatary buckwheat (Fagopyrum tataricum Gaertn.). Gene 534:229–235
Yasui Y, Wang Y, Ohnishi O, Campbell CG (2004) Amplified fragment length polymorphism linkage analysis of common buckwheat (Fagopyrum esculentum) and its wild self-pollinated relative Fagopyrum homotropicum. Genome 47:345–351
Zhao G, Shaan F (2008) Chinese Tartary buckwheat. Science, Beijing, pp 85–88
Zhao HS, Yang L, Peng ZH, Sun HY, Yue XH, Lou YF, Dong LL, Wang LL, Gao ZM (2015) Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci Rep 5:8018
Zietkiewicz E, Rafalski A, Labuda DF (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 20:176–183
Acknowledgments
We would like to thank Professor Donald Grierson, University of Nottingham, UK, for discussion and helping with the manuscript. We would like to thank Germplasm Resources Information Network in the US, Chinese Crop Germplasm Resources Information System and Institute of crop variety resources, Shanxi, China. This work was supported by NSFC (National Natural Science Foundation of China) (No. 31301385, 31471556), Science and Technology Project of Shanxi Province (20150311007-1), the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (STIP, 2013115), the Basic Facility Platform Project of Shanxi Provincial Department of Science and Technology (2012091004–0103), Shanxi Agricultural University breeding innovation funds (2010028) and the Key Discipline Foundation of Shanxi Province.
Conflicts of Interest
The authors declare no conflict of interest.
Authors’ Contributions
Siyu Hou, Hongying Li and Yuanhuai Han designed the experiments and drafted the manuscript. Zhaoxia Sun , Lijun Zhang, Zhijun Qiao and Bin Wu collected plant materials. Bin Linghu performed the SSR experiments and analyzed the data. Dongmei Xu modified the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hou, S., Sun, Z., Linghu, B. et al. Genetic Diversity of Buckwheat Cultivars (Fagopyrum tartaricum Gaertn.) Assessed with SSR Markers Developed from Genome Survey Sequences. Plant Mol Biol Rep 34, 233–241 (2016). https://doi.org/10.1007/s11105-015-0907-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-015-0907-5