
Abstract
Key message
The study carry out comprehensive transcriptome analysis of C. deltoidea and exploration of BIAs biosynthesis and accumulation based on UHPLC-MS/MS and combined sequencing platforms.
Abstract
Coptis deltoidea is an important medicinal plant with a long history of medicinal use, which is rich in benzylisoquinoline alkaloids (BIAs). In this study, Ultra performance liquid chromatography-electrospray ionization tandem mass spectrometry (UHPLC-ESI-MS/MS) and combined sequencing platforms were performed for exploration of BIAs biosynthesis, accumulation and comprehensive transcriptome analysis of C. deltoidea. By metabolism profiling, the accumulation of ten BIAs was analyzed using UHPLC-MS/MS and different contents were observed in different organs. From transcriptome sequencing result, we applied single-molecule real-time (SMRT) sequencing to C. deltoidea and generated a total of 75,438 full-length transcripts. We proposed the candidate biosynthetic pathway of tyrosine, precursor of BIAs, and identified 64 full length-transcripts encoding enzymes putatively involved in BIAs biosynthesis. RNA-Seq data indicated that the majority of genes exhibited relatively high expression level in roots. Transport of BIAs was also important for their accumulation. Here, 9 ABC transporters and 2 MATE transporters highly homologous to known alkaloid transporters related with BIAs transport in roots and rhizomes were identified. These findings based on the combined sequencing platforms provide valuable genetic information for C. deltoidea and the results of transcriptome combined with metabolome analysis can help us better understand BIAs biosynthesis and transport in this medicinal plant. The information will be critical for further characterization of C. deltoidea transcriptome and molecular-assisted breeding for this medicinal plant with scarce resources.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coptis deltoidea C.Y. Cheng et Hsiao., a medicinal plant belonging to the family Ranunculaceae, has been used for preventing and treating human diseases for centuries in China. Termed “Yalian”, its dried rhizome is one of the sources of the traditional Chinese medicine “Huanglian”, which is a widely used medicinal material with definitive pharmacological roles in anti-inflammatory, anti-bacteria, anti-diabetic and neuroprotection (Li et al. 2009; Wang et al. 2014; Xiang et al. 2016). To date, phytochemical analysis has illustrated that the major bioactive constituents of C. deltoidea are benzylisoquinoline alkaloids (BIAs) (He et al. 2014; Qiao et al. 2009; Qi et al. 2018), such as berberine, palmatine, jatrorrhizine, coptisine, columbamine, epiberberine, and magnoflorine. These metabolites play important roles in plant physiology, particularly in defence responses. In addition, BIAs display many medicinal properties. For example, the most abundant alkaloid in C. deltoidea, berberine shows significant antimicrobial, anti-inflammatory, antidiabetic, and cardiovascular activities (Vuddanda et al. 2010). However, C. deltoidea is only distributed in Southwest China (Chen et al. 2017a, b) and it is a slow-growing plant with low production, requiring more than 5 years to obtain a crude drug conforming to the Chinese Pharmacopoeia. Due to the growth characteristics and overexploitation, the wild resources of C. deltoidea are almost endangered. In order to protect this medicinal plant, elucidating the transcriptome of C. deltoidea and identifying the putative genes for the biosynthesis of active constituents will provide the foundation for the reasonable utilization of resources and the application of biotechnology to improve active ingredients accumulation and biosynthesis.
BIAs are a diverse group of specialized plant metabolites derived from tyrosine (Hagel and Facchini 2013). Currently, the biosynthetic pathways for BIAs have been extensively investigated in some species, such as Thalictrum flavum, Coptis japonica, Eschscholzia californica, Papaver somniferum (Desgagné-Penix et al. 2010; Morishige et al. 2010; Samanani et al. 2005). The persistent effort has allowed the complete, or near-complete elucidation of several metabolic pathways of BIAs, such as those synthesizing berberine, sanguinarine and morphine. At the same time, a number of enzymes involved in BIA biosynthesis have been characterized (Inui et al. 2012; Lee and Facchini 2011; Takemura et al. 2013). Previous studies have demonstrated that various classes of BIAs have the same steps in their early biosynthetic pathways. The biosynthesis of BIAs begins with the conversion of tyrosine to both dopamine and 4-hydroxyphenylacetaldehyde, which are then condensed by (S)-norcoclaurine synthase (NCS) to yield (S)-norcoclaurine, the central precursor to all BIAs in plants (Samanani et al. 2004). A 6-O-methyltransferase, a N-methyltransferase, one cytochrome P450 (CYP450) and a 4′-O-methyltransferase are involved in catalyzing the conversion of (S)-norcoclaurine to (S)-reticuline, which is a branch-point intermediate in the biosynthesis of many BIAs (He et al. 2017, 2018; Inui et al. 2012; Ziegler and Facchini 2008). Sequentially, multistep transformations of the basic BIA backbone for the biosynthesis of different end-products in branch pathways (such as sanguinarine, berberine, palmatine and codeine) are catalyzed by oxidative enzymes including members of the O-methyltransferase (OMT) family and P450s (Ikezawa et al. 2009; Morishige et al. 2010; Mizutani and Sato 2011). Although some cDNA sequences of biosynthetic enzyme genes in some plant species have been cloned, the current understanding of molecular mechanisms catalyzing and regulating BIAs biosynthesis in C. deltoidea are still largely unknown because of unfinished work of genome sequencing and limited information about transcripts.
With the fast development of next-generation high-throughput sequencing technologies (NGST), it is low-cost and time effective to survey the putative genes and stimulate the construction of genome and transcriptome resources (Kamps et al. 2017). RNA-Seq has been used to analyze transcriptome and differential gene expression and understand the regulatory mechanisms for medicinal plant species with or without a reference genome sequence, such as Rhodiola rosea, Camptotheca acuminate, Salvia miltiorrhiza and Dendrobium huoshanense (Sadre et al. 2016; Torrens-Spence et al. 2018; Wenping et al. 2011; Yuan et al. 2018). Recently, gene characterization at transcriptome scale was carried out on Coptis plants (Chen et al. 2017a, b; He et al. 2018). Based on the second-generation sequencing platforms, transcriptome studies of C. chinensis and C. teeta have been conducted, building de novo transcriptome assemblies from short-read RNA-sequencing data, which identified 78,499 and 81,823 unigenes respectively and characterized many genes related to biosynthesis of secondary metabolites. However, the short reads from second-generation sequencing bring about incompletely assembled transcripts and loss of some important information, which cannot provide full-length sequence.
Recently, a novel single molecule real-time (SMRT) technology carried out in PacBio RS (Pacific Biosciences of California, Inc, https://www.pacificbiosciences.com/) has provided a third-generation sequencing platform used to obtain full-length transcripts that do not need to be assembled (Dong et al. 2015; Huddleston et al. 2014). Isoform sequencing (Iso-Seq) based on SMRT platform overcomes the limitations of short-read sequences, which confers long reads length, high consensus accuracy and permits efficient analysis of exon–intron structure and alternative splicing (Lou et al. 2019; Roberts et al. 2013). Despite the concern of the higher error rate (up to 15%) observed in SMRT sequencing, it can be addressed by self-correction via circular-consensus (CCS) and/or correction with short reads data (Au et al. 2012; Li et al. 2014). Therefore, third-generation sequencing has been used to analyze full-length transcriptomes in multiple plant species and proven useful for identification of putative genes for bioactive components biosynthesis (Chen et al. 2018; Sun et al. 2018; Xu et al. 2015). For instance, Iso-Seq has been applied to obtain the full-length transcriptomes of two widely used medicinal plants, Salvia miltiorrhiza (Xu et al. 2015) and safflower (Carthamus tinctorius) (Chen et al. 2018), and provide the information on the biosynthetic pathway of tanshinone and flavonoid. Besides, SMRT sequencing was performed to identify the key genes and alternative splicing related to secondary metabolites biosynthesis in Camellia sinensis (Qiao et al. 2019).
So far, several transcriptome analyses of Coptis plants have been carried out. Nevertheless, there are some differences in the genomes of different Coptis plants. Furthermore, few studies on C. deltoidea has been conducted. Here, we combined long read SMRT sequencing and short read RNA-Seq to analyze C. deltoidea transcriptome. In order to study the BIAs biosynthesis of C. deltoidea, we determined the content of alkaloids in different tissues (leaves, rhizomes and roots) based on ultra-high performance liquid chromatography-electrospray ionization tandem mass spectrometry (UHPLC-ESI-MS/MS) firstly. SMRT sequencing was used to generate full-length transcriptomes of C. deltoidea derived from five different tissues. We then carried out functional annotation of obtained full-length transcriptomes and identified the putative genes involved in tyrosine and BIAs biosynthesis, ABC transporters and MATE transporters. Based on RNA-Seq, the expression levels of the identified genes were analyzed and the validity of the transcriptome sequencing data were further verified by real-time quantitative PCR (qRT-PCR). Herein, the transcriptome data provide sufficient full-length sequences and valuable resources for investigating the biosynthesis of important bioactive compounds in C. deltoidea.
Materials and methods
Plant materials and RNA sample preparation
Nine plant materials of four-year-old C. deltoidea (Fig. S1) with consistent genetic background and growth were collected from the cultivation base in Hongya, Sichuan, China and were randomly divided into three groups (three plants per group). Each plant was divided into five different tissues (leaf, petiole, rhizome, root and stolon) and each tissue sample was obtained by mixing three plants equally. Therefore, each tissue (leaf, petiole, rhizome, root and stolon) obtained three biological replicate samples. Subsequently, each sample of these tissues was ground into powder and used for RNA extraction and UPLC-MS/MS analysis, respectively. For RNA extraction, the OmniPlant RNA Kit (CWbio, Beijing, China) was used, according to the manufacturer’s protocol. The quality and quantity of RNA were determined using the Nanodrop micro-spectrophotometer (Thermo Scientific, Waltham, DE, USA) and Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, USA). For metabolite analysis, the samples of leaf, rhizome and root were dried at 60 °C to constant weight for alkaloids extraction.
Alkaloids extraction and UHPLC-MS/MS analysis
Dried powder samples (0.02 g) of leaves, rhizomes and roots isolated from C. deltoidea were accurately weighed and subsequently extracted with 10 mL of hydrochloric acid–methanol solution (1:100, v/v) for 45 min in an ultrasonic bath. In order to determine the content of main BIA components among different tissues, analyses were carried out on the UPLC-MS/MS system equipped with a Waters ACQUITY UPLC H-Class connected online to a Waters Xevo triple-quadrupole (TQD) mass spectrometer (Waters, Milford, MA, USA). Samples were chromatographic separated on an ACQUITY UPLC BEH C18 (1.7 μm particle size) 2.1 mm × 50 mm Column with the column temperature kept at 25 °C and a mobile phase flow rate of 0.4 mL/min. The mobile phase consisted of water containing 0.3% (v/v) formic acid (A) and acetonitrile (B) and the initial composition was 85% A and 15% B. The following gradient program was employed: 0–2 min, 85–76% A; 2–4 min, 76–75% A; 4–6 min, 75–73% A; 6–8 min, 73–85% A. The injection volume was 2 μL.
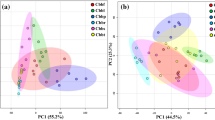
The Xevo TQD mass spectrometer was operated in positive ion mode. High purity nitrogen (N2) was used as nebulizing gas and helium (He) was the collision gas. The source parameters were as follows: capillary voltage 3.50 kV; desolvation temperature 400 °C; source temperature 150 °C; desolvation gas flow 800 L/h; cone gas flow 50 L/h. In order to obtain molecular weight information about BIAs in C. deltoidea, the mass spectrometer was first scanned from m/z 100 to 500 in full scan mode. The extracts of different C. deltoidea tissues were analyzed in multiple reaction monitoring (MRM) mode and the optimized cone voltages and collision energies were listed in Table S1. The contents of ten alkaloids in each sample were calculated from standard curves (Table S2). Reference standards of palmatine, coptisine, epiberberine, columbamine, jatrorrhizine, magnoflorine, groenlandicine, demethyleneberberine and berberrubine were purchased from Chroma-Biotechnology Co., Ltd. (Chengdu, China), and berberine was provided by Chinese National Institute for Food and Drug Control (Beijing, China). The purities for all compounds are higher than 98%. Principal component analysis (PCA) was carried out to visualize the differences regarding different tissues of C. deltoidea.
Localization of BIAs in different tissues
Quaternary protoberberine alkaloids, such as berberine, show a strong yellow fluorescence under UV irradiation and can be stained orange or reddish brown by Dragendorff's reagent. Fresh sections of C. deltoidea leaf, rhizome and root were cross-sectioned at a thickness of < 0.5 mm according to Yeung (Yeung 1998). Then, the fresh hand sections were used to visualized alkaloids under visible light and then sections were treated with Dragendorff's reagent, and the results were viewed and recorded using a Leica M165 FC stereoscope (Leica Microsystems, Wetzlar, Germany).
Sections used to detect the autofluorescence of BIAs were prepared by frozen section. Small sections of leaf, rhizome and root were embedded into a tissue freezing medium. Then, they were placed on a cutting platform in the cryobar of a cryostat and slices of 20 μm in thickness were cut at −20 °C. The Leica TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany; excitation wavelength 340 to 380 nm) was used to detect the autofluorescence of BIAs in different tissue sections.
PacBio SMRT sequencing library preparation and sequencing
The isolated RNAs (from leaves, petioles, rhizomes, roots and stolons) were pooled to provide the total RNA of C. deltoidea. Then, mRNA was isolated from the total RNA using the oligo d(T) magnetic bead binding method and reversely transcribed into cDNA using Clontech SMARTer PCR cDNA Synthesis Kit (Clontech Laboratories, Inc. CA, USA). Size selection of the PCR products was carried out using BluePippin™ Size Selection System (Sage Science, Beverly, MA, USA),and the fragments with 0.5–6 kb was retained. Then, Large-scale PCR was performed to amplify the full-length cDNA. The end of cDNA were repaired and the sequencing adapters were ligated to the cDNA. SMRTbell template libraries were created from the obtained cDNA and sequenced on the PacBio Sequel platform using P6-C4 chemistry with 10 h movie times.
Illumina RNA-Seq library preparation and sequencing
RNA samples from leaves, rhizomes and roots were used for Illumina library construction and sequencing. The cDNA libraries for Illumina HiSeq™ 2500 sequencing were constructed as the following steps. mRNA was enriched from total RNA using the oligo d(T) magnetic beads and fragmented into short fragments using fragmentation buffer. Then, first-strand cDNA was synthesized using random primers by reverse transcription. The products were taken as templates and used to synthesize the second-strand cDNA. The cDNA fragments were purified with QiaQuick PCR extraction kit (Qiagen, Venlo, Netherlands) and ligated with Illumina sequencing adapters. The ligation products were size selected by agarose gel electrophoresis and enriched by PCR to create the cDNA libraries, which were sequenced on the Illumina HiSeq™ 2500 platform. All the sequencing works were carried out at Gene Denovo Biotechnology Co. (Guangzhou, China). After RNA-Seq, raw reads were further filtered to obtain clean reads by removing adaptors, reads containing more than 10% of unknown nucleotides and low-quality reads. The Q30 and GC content of clean reads were calculated.
Analysis of the Iso-Seq data
The SMRT Link v5.0.1 pipeline (Pacific Biosciences, Menlo Park, CA, U.S.A.) was used to process raw sequencing data. Subreads were obtained and subjected to circular consensus sequence (CCS). Then CCS reads were classified into full-length non-chimeric (FLNC) reads, full-length chimeric reads, non-full-length reads, and short reads according to whether the 5′ primer-, 3′primer-adapters and polyA tail signal were simultaneously observed. The CCS reads with all three elements are classified as FLNC. Subsequently, short reads were discarded and the FLNC reads were clustered using the algorithm of iterative clustering for error correction (ICE) to generate the cluster consensus isoforms. To improve accuracy of full-length transcripts, two strategies were employed. First, the non-full-length reads were used to polish the above obtained cluster consensus isoforms using Quiver to obtain the full-length polished high quality consensus sequences (accuracy ≥ 99%). Second, the low quality isoforms were further corrected using filtered Illumina RNA-Seq reads by Long-Read De Bruijn Graph Error Correction (LoRDEC) tool (https://atgc.lirmm.fr/lordec/). Then the final transcriptome isoform sequences were filtered by removing the redundant sequences with software CD-HIT (v4.6.7, https://github.com/weizhongli/cdhit/releases) using a threshold of 0.99 identities.
For comprehensive functional annotation, the full-length transcripts were blasted against public protein databases, including the National Center for Biotechnology Information (NCBI) non-redundant protein (Nr) database (https://www.ncbi.nlm.nih.gov/), the Swiss-Prot database (https://www.expasy.ch/sprot/), the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/) and the Cluster of Orthologous Groups of proteins (COG/KOG) database (https://www.ncbi.nlm.nih.gov/COG) using BLASTX program (https://www.ncbi.nlm.nih.gov/BLAST/) with a cut-off E-value ≤ 10−5. Gene Ontology (GO) annotation was analyzed by Blast2GO software (Conesa et al. 2005) and GO function categories were performed using WEGO software (Ye et al. 2006).
Profiling of differentially expressed genes (DEGs) using RNA-Seq data
Using the full-length transcripts generated from SMRT sequencing as reference sequences, the unigene expression levels among the different tissues (leaves, rhizomes and roots) of C. deltoidea were further analyzed based on the short reads data yielded from RNA-seq. The expression value of each sample was determined by software RSEM (version 1.2.19) (Li and Dewey 2011). Briefly, clean data from RNA-Seq were mapped onto the reference sequences and the resulting alignments were used to estimate gene abundances. Then, the gene expression levels were normalized by using FPKM (Fragment Per Kilobase of transcript per Million fragments mapped). Different expression analysis of three tissues (leaf, rhizome and root) was performed using the edgeR (version 3.12.1, https://www.r-project.org/). Herein, a false discovery rate (FDR) < 0.05 and a fold change ≥ 2 were used as the thresholds for differentially expressed genes (DEGs). The identified DEGs were used for GO and KEGG enrichment analyses.
Reverse transcription quantitative real-time PCR (qRT-PCR) analysis
The expression profiles of 10 randomly selected genes (including nine transcripts related to BIAs biosynthesis pathway and one transcript involved in biosynthesis of secondary metabolites) were analyzed by qRT-PCR to confirm the RNA-Seq results. The cDNAs were synthesized with 0.3 μg total RNA using Master Premix for first-strand cDNA synthesis (FOREGENE, Chengdu, China) according to the manufacturer’s protocol. qRT-PCR was performed in 20 μL solution system composed of 2× Real PCR Easy™ Mix-SYBR (FOREGENE) on a Bio-Rad CFX96 system (Bio-Rad, CA, USA). The 18S ribosomal RNA was used as an internal control gene for normalization. The primers of each genes are listed in Table S5. PCR amplification reaction was conducted under the following conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 61 °C for 30 s. The relative expression levels of target genes were calculated using the 2−ΔΔCt comparative threshold cycle (Ct) method. All analyses were carried out with three biological replicates. Pearson correlation analysis was performed using R package to calculate the consistency of RNA-Seq and qRT-PCR data.
Results
Accumulation of BIAs in different tissues of C. deltoidea
Benzylisoquinoline alkaloids are nitrogen-containing plant secondary metabolites and occur mainly in the plant families, including Papaveraceae, Ranunculaceae, Berberidaceae and Magoliaceae (Liscombe et al. 2005). According to previous reports, magnoflorine, groenlandicine, columbamine, epiberberine, coptisine, jatrorrhizine, palmatine, berberine constitute the main BIAs of C. deltoidea (He et al. 2014; Qiao et al. 2009). Herein, the content of these major BIAs and other two derivatives of berberine (demethyleneberberine and berberrubine) in leaves, rhizomes and roots was quantified by UPLC-MS/MS (Fig. 1). The MS/MS fragmentation patterns of these quantified BIAs were shown in Fig. S2. As shown in Fig. 2a and Table S4, all tissues accumulate very high levels of berberine (22.67 ± 1.31–40.09 ± 4.87 mg/g, DW). With respect to other BIAs, coptisine (7.34 ± 0.88–17.15 ± 1.53 mg/g, DW), jatrorrhizine (3.66 ± 0.43–12.77 ± 0.82 mg/g) and magnoflorine (3.05 ± 0.42–7.97 ± 0.79 mg/g, DW) are also abundant in C. deltoidea. In terms of different tissues, roots have the highest content of groenlandicine, demethyleneberberine, epiberberine, coptisine, jatrorrhizine and berberine. Besides, the highest accumulation of magnoflorine (7.97 ± 0.79 mg/g, DW) is found in leaves and berberrubine (0.09 ± 0.04 mg/g, DW) is only detected in rhizomes, which are also the best source for columbamine and palmatine. Therefore, based on the quantitative determination and the principal component analysis (PCA) (Figs. 2b, c and S3), the present results further illustrate the similarities of BIA types and differences of contents among different C. deltoidea tissues.

Typical MRM chromatograms of magnoflorine, groenlandicine, demethyleneberberine, columbamine, epiberberine, coptisine, jatrorrhizine, berberrubine, palmatine and berberine

The contents of 10 BIAs in different tissues and PCA analysis of leaves, rhizomes and roots of C. deltoidea based on quantitative determination of BIAs. a The contents of 10 BIAs. b PCA score plot. c Loading plot of PCA
In order to clarify the accumulation sites of alkaloids in different C. deltoidea tissues, optical microscopy and fluorescence microscopy were used to obtain the micrographs of leaf, rhizome and root. Berberine, most abundant alkaloid in C. deltoidea, is a yellow compound and shows yellow fluorescence under UV irradiation. Moreover, Dragendorff's reagent can stain alkaloids orange or reddish brown. The sections and fluorescence characteristics of different tissues are displayed in Figs. 3 and S4. In leaf, alkaloids was abundant throughout the vascular tissues and sclerenchyma. In rhizome, alkaloids were detected in the vascular bundles and almost no yellow fluorescence was detected in the cortex and pith. The cortex cells that accumulate starch was stained dark purple with the iodide in Dragendorff's reagent. In root, yellow fluorescence was mainly distributed in vascular cylinder and it was stained reddish brown by Dragendorff's reagent. Therefore, we speculated that alkaloids mainly accumulate in the vascular cylinder of the root.

Localization of BIAs in different C. deltoidea tissues. Sections of leaf (a–c), rhizome (d–f) and root (g–i) were observed using light microscopy and confocal microscope. Fresh hand sections of leaf (a), rhizome (d) and root (g) (cross-sectioned at a thickness of < 0.5 mm) were observed under visible light. Frozen sections of leaf (b, c), rhizome (e, f) and root (h, i) (cross-sectioned at a thickness of 20 μm) were observed by a confocal microscope. Berberine is a yellow compound and it shows yellow autofluorescence under UV irradiation. c, f, i showed the micrographs of different tissues under UV irradiation and b, e, h only showed the yellow fluorescence of different tissue sections. Number in the figure: 1, epidermis and cork; 2, cortex; 3, pericyclic fiber bundles; 4, phloem; 5, xylem; 6, pith; 7, sclerenchyma; 8, endodermis
Coptis deltoidea transcriptome analysis using RNA-Seq and PacBio Iso-Seq
To comprehensively characterize C. deltoidea transcriptome, short read RNA-Seq and long read PacBio Iso-Seq were combined. Nine RNA samples from different tissues (leaves, rhizomes and roots) were sequenced on Illumina HiSeq™ 2500 platform. After quality filtering, 427 million 150 bp-long reads were generated (Table 1). To obtain a wide coverage of C. deltoidea transcriptome, a pooled sample representing high-quality RNA from five tissues (leaves, petioles, rhizomes, roots and stolons) were sequenced using PacBio RS II platform. A total of 15.5 Gb raw reads were generated and 8,882,132 subreads were obtained after filtering. Then SMRT Link v5.0.1 pipeline was performed to process raw sequencing data. In total, 532,835 CCS reads were obtained, including 436,531 FLNC reads and 94,603 non-FL reads (Table S6 and Fig. 4). To solve the high error rate and improve accuracy of PacBio reads, Interactive Clustering and Error Correction (ICE) algorithm combined with the Quiver program was applied for sequence clustering. In total, 190,214 full-length consensus isoforms, including 104,640 polished high-quality (HQ) and 85,574 low-quality (LQ) transcripts were generated. After error correction using the RNA-Seq data derived from three different tissues (leaves, rhizomes and roots) of C. deltoidea and removing the redundant sequences via CD-Hit program, 75,438 non-redundant transcript isoforms were yielded (Table 2). The length of transcripts was in the range of 106 bp to 11,325 bp with N50 of 2517 bp and GC content of 41.28%.

Summary of PacBio single-molecule real-time (SMRT) sequencing. a The number and length distributions of 532,835 CCS reads. b The number and length distributions of 436,531 FLNC reads. c The number and length distributions of 75,438 non-redundant transcript isoforms. d The classification of CCS reads in C. deltoidea
BLASTx similarity analysis against the Nr database demonstrated that the C. deltoidea full-length transcripts were similar to several plant species (Fig. S5). Among them, 21,888 (29.01%) transcripts showed significant homology with that of Nelumbo nucifera and 5484 (7.27%) and 3158 (4.19%) transcripts had high similarity with sequences of Vitis vinifera and Anthurium amnicola, respectively. With respect to Nelumbo nucifera, all parts of this plant have been used in traditional Chinese medicine and the main bioactive components are BIAs richly accumulating in tissues such as leaf and embryo (Deng et al. 2018a, b; Itoh et al. 2011).
Function annotation of full-length C. deltoidea transcriptome
To obtain a comprehensive annotation of C. deltoidea transcriptome, 75,438 full-length transcripts was annotated by searching against four protein databases (Nr, Swiss-Prot, KEGG and KOG). A total of 70,383 transcripts were annotated and the details of overall functional annotation is described in Table S7 and Fig. 5. In addition, 5055 unannotated unigenes might represent novel C. deltoidea genes.

Annotation of the full-length transcripts to public databases
GO enrichment analysis was used to classify the functions of the full-length transcripts to molecular function, cellular component and biological process terms (Fig. 6a and Table S8). Among them, biological process was the majority of the GO terms. In addition, 34,425 and 44,236 transcripts were assigned to molecular function and cellular component, respectively. A high proportion of genes was assigned to the classes such as metabolic process, cellular process and catalytic activity of these GO categories, which are important activities in plants and involved in metabolites biosynthesis. The COG analysis demonstrated that 50,173 transcripts were assigned to 25 functional clusters. As shown in Fig. 6b, the five largest categories were “General function prediction only” (15,528, 16.31%), “Signal transduction mechanisms” (11,837, 12.43%), “Posttranslational modification, protein turnover, chaperone” (11,211, 11.77%), “RNA processing and modification” (6256, 6.57%) and “Translation, ribosomal structure and biogenesis” (5474, 5.75%). With respect to KEGG analysis, it is helpful for functional genes identification, understanding the functions and interactions of genes in the biosynthetic pathways (He et al. 2018). In the KEGG classification, 34,477 transcripts from C. deltoidea were annotated in the KEGG database and assigned to 133 biological pathways (Table S9). The largest pathway was the metabolic pathways containing 9542 transcripts. Moreover, a number of transcripts were assigned to other significant pathways, such as biosynthesis of secondary metabolites, biosynthesis of antibiotics, microbial metabolism in diverse environments and carbon metabolism.

Function annotation and classification of full-length transcripts in C. deltoidea. a GO enrichment analysis of transcripts. The annotated transcripts were distributed in biological process, cellular component and molecular function. b COG classification analysis of transcripts
Overview of differentially expressed genes among different tissues of C. deltoidea
To investigate and understand the variation of transcript abundance and expression patterns of genes among leaf, rhizome and root of C. deltoidea, the Illumina RNA-Seq reads were mapped to the SMRT transcripts to determine expression level using FPKM-normalized read counts. The average of mapped reads was 87.47% (Table 1) and the FPKM distribution of all samples was shown in Fig. 7. Then, we carried out a comparative analysis of the differential genes of leaf, rhizome and root (CdL vs. CdRh, CdL vs. CdRo and CdRh vs. CdRo) and the results were displayed in Table 3 and Fig. 8. In CdL vs. CdRh and CdL vs. CdRo, a total of 24,937 and 25,391 differentially modulated transcripts were identified, respectively. Between rhizome and root, 16,762 differentially expressed genes were identified (Fig. S6). Moreover, as shown in Fig. 8a, leaf and rhizome had the most specifically expressed differential genes (4154), while rhizome and root had fewer differential genes (1597), suggesting that there was a larger biological differences between leaf and the underground part of this plant, and fewer differences between rhizome and root. 5335 genes were differentially expressed in all comparison groups, suggesting that these genes may play an important role in the metabolism of different tissues of C. deltoidea. GO (Table S10) and KEGG enrichment analysis were performed to further analyze the identified transcripts. As shown in Figs. 8 and S7, the widest metabolism class occurred in the three different tissues of C. deltoidea and involved carbohydrate metabolism, biosynthesis of secondary metabolites, energy metabolism, amino acid and lipid metabolism.

The FPKM distribution of different tissue samples

Functional annotation of differentially expressed genes in different comparisons. a Venn diagram of DEGs in different comparisons. b–d Scatterplot of KEGG pathway enrichment of DEGs (top 20) (CdL-vs-CdRh, CdL-vs-CdRo and CdRh-vs-CdRo, respectively)
Identification of full-length transcripts putatively involved in shikimate pathway and tyrosine biosynthesis
In plants, tyrosine is aromatic amino acid required for protein synthesis and serve as precursors of a variety of secondary metabolites, such as BIAs and pigment betalains, which play crucial roles in plant growth, defense and environment responses (Tzin and Galili 2010). Tyrosine is synthesized via the shikimate pathway leading to chorismate and it is converted by chorismate mutase (CM) to prephenate, whose subsequent conversion to tyrosine may be via two possible routes (Maeda and Dudareva 2012). Herein, we discovered the most likely full-length transcripts encoding known enzymes involved in tyrosine biosynthesis according to sequence functional annotations (Fig. 9 and Table S11). A total of 18 full-length transcripts encoding six enzymes catalyzing seven enzymatic reactions of the shikimate pathway were identified, including two, three, six, four, two and three transcripts encoding 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase (DAHPS), 3-dehydroquinate synthase (DHQS), bi-functional 3-dehydroquinate dehydratase/shikimate dehydrogenase (DHD/SDH), shikimate kinase (SK), 5-enolpyruvylshikimate 3-phosphate synthase (EPSPS) and chorismate synthase (CS), respectively. As shown in Fig. 9, most of transcripts participating in the shikimate pathway had high expression levels in root of C. deltoidea. With respect to tyrosine biosynthesis, we discovered 6 transcripts which encode CM, prephenate aminotransferase (PAT) and arogenate dehydrogenase (TyrA) catalyzing the conversion of chorismate to arogenate and subsequent production of tyrosine. However, the second possible route of tyrosine biosynthesis was not discovered in the SMRT sequencing data. We also found a transcript encoding phenylalanine-4-hydroxylase (PAH), which catalyzes the conversion of phenylalanine into tyrosine and is expressed only in the roots of C. deltoidea.

Putative biosynthetic pathways of tyrosine in C. deltoidea and expression patterns of tyrosine biosynthetic genes in different tissues. Full names of enzymes represented by their abbreviated names were showed in Table S11
Identification of full-length transcripts putatively involved in BIAs biosynthesis
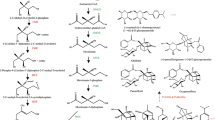
Bioactive constituents of C. deltoidea are BIAs, which were derived from tyrosine and then form (S)-reticuline (the branch-point intermediate) via a series of enzymatic reactions. To date, the biosynthesis pathway of BIAs has been more clearly characterized in other plant species (Hagel et al. 2015; He et al. 2017, 2018). However, the pathway in C. deltoidea has not yet been determined. To identify candidate BIA biosynthetic pathway in C. deltoidea, genes encoding enzymes of previous reported BIA biosynthetic pathway were compared against those of the 75,438 full-length sequences in C. deltoidea. On the basis of known pathway and the present SMRT sequencing data, we proposed the putative biosynthesis pathways for detected BIAs, including berberine, palmatine, jatrorrhizine, columbamine, epiberberine, coptisine and magnoflorine (Fig. 10). We discovered 40 transcripts encoding almost all known enzymes putatively involved in the biosynthesis of the BIA precursor (S)-reticuline from the SMRT sequencing data, including two, three, nine, twelve, five, three, two and four full-length transcripts encoding tyrosine aminotransferase (TyrAT), tyrosine decarboxylase (TYDC), polyphenol oxidase (PPO), (S)-norcoclaurine synthase (NCS), (S)-norcoclaurine 6-O-methyltransferase (6OMT), (S)-coclaurine N-methyltransferase (CNMT), (S)-N-methylcoclaurine-3′-hydroxylase (NMCH), and 3′-hydroxy-N-methyl-(S)-coclaurine 4′-O-methyltransferase (4′OMT), respectively. In addition, a total of 24 transcripts encoding enzymes putatively involved in multistep transformations of the basic BIA backbone for the biosynthesis of different end-products in branch pathways.

Putative biosynthetic pathways of BIAs in C. deltoidea and heatmap depicting the expression patterns of candidate genes encoding enzymes involved in BIAs biosynthesis. a Putative biosynthetic pathways of BIAs. Full names of enzymes represented by their abbreviated names were showed in Table S12. The enzymes found in this study are marked in green. b The expression patterns of candidate genes involved in BIAs biosynthesis in C. deltoidea leaves, rhizomes and roots
Berberine is widely distributed in various plants species and its biosynthesis pathway has been clearly defined (Hagel and Facchini 2013; Sato et al. 2007; Vuddanda et al. 2010). Several candidate transcripts encoding enzymes associated with berberine biosynthesis were identified, which included berberine bridge enzyme (BBE), (S)-scoulerine 9-O-methyltransferase (SOMT), (S)-canadine synthase (CAS), and (S)-tetrahydroprotoberberine oxidase (STOX) (Table S12). However, the pathways of jatrorrhizine and epiberberine, which are closely related to berberine in structure, have not been determined. Previous reports have suggested that the formation of jatrorrhizine results from the methylenedioxy ring-opening of berberine (Rüffer et al. 1983). Nevertheless, the enzyme catalyzing the reaction has not been identified to date. Furthermore, Hagel (2010) proposed that 3-O-demethylation of (S)-scoulerine combined with 2-O- and 9-O-methylation may lead to jatrorrhizine. In support of the hypothesis, codeine-O-demethylase (CODM) isolated from P. somniferum was shown to efficiently catalyze 3-O-demethylation of (S)-scoulerine (Hagel and Facchini 2010). Herein, one full-length transcripts of CODM was identified in the SMRT data (Table S12), which may be involved in the biosynthesis of jatrorrhizine in C. deltoidea. With respect to epiberberine biosynthesis, previous study has considered that members of OMT, CYP719 and oxidoreductase (OX) families may play role in 2-O-methylation of (S)-scoulerine and subsequent oxidation to yield epiberberine (He et al. 2018). For the biosynthesis pathways of coptisine, columbamine and palmatine, (S)-scoulerine is first formed from (S)-reticuline by BBE. The biosynthesis of coptisine begins with the catalysis of two CYP719 subfamily members, (S)-cheilanthifoline synthase (CFS) and (S)-stylopine synthase (SPS), which have been isolated from Eschscholzia californica (Ikezawa et al. 2009). However, the full-length transcripts encoding these two enzymes were not identified in our data. Finally, the oxidation of (S)-stylopine by STOX yields coptisine. In the biosynthesis of the other two components, six candidate transcripts encoding columbamine O-methyltransferase (CoOMT) were identified, which catalyzes the conversion of columbamine to palmatine (Pienkny et al. 2010). In magnoflorine biosynthesis, (S)-reticuline subsequently yields magnoflorine with the catalysis of (S)-corytuberine synthase (CTS) and (S)-corytuberine N-methyltransferase (SCNMT) or reticuline N-methyltransferase (RNMT) (Ikezawa et al. 2008; Morris and Facchini 2016). Three candidate transcripts encoding CTS were identified and no SCNMT was identified in the SMRT sequencing data. In rat, CYP450 enzymes which appeared to transform berberine to demethyleneberberine were identified (Li et al. 2011). However, the enzymes involved in biosynthesis of demethyleneberberine and berberrubine in C. deltoidea need to be further explored.
In order to further define the annotation of candidate BIAs biosynthesis genes and characterize the phylogenetic relationships of BIAs biosynthesis enzymes from C. deltoidea and known enzymes from other BIA-producing plant species, the neighbor-joining (NJ) tree was constructed and their conserved motif structure was analyzed (Fig. 11). Based on ORFs prediction, the putative coding regions of candidate full-length transcripts were identified, and then they were analyzed on the protein family database (Pfam, https://pfam.xfam.org/). As shown in (Fig. 11), most enzymes encoded by putative BIAs biosynthesis genes of C. deltoidea and known enzymes from other BIAs biosynthesis plants have the similar conserved domains. The PPO family contain three domains, which are involved in the major early stages in the biosynthesis of BIAs, such as tyrosine hydroxylation. Most of CdNCS have non-haem dioxygenase in morphine synthesis N-terminal and 2OG-Fe (II) oxygenase superfamily domain. Previous study has reported that NCS isolated from C. japonica shows sequence similarity to 2-oxoglutarate-dependent dioxygenases of plant origin and its catalytic reaction depends on ferrous ion (Minami et al. 2007). S-adenosylmethionine-dependent O- and N-methyltransferases (OMT and NMT) also play an important role in BIAs biosynthesis (Morris and Facchini 2019), which contain O-methyltransferase domain and mycolic acid cyclopropane synthetase domain utilizing S-adenosyl-l-methionine as a substrate for transfer of methyl. The non-haem dioxygenase in morphine synthesis N-terminal and 2OG-Fe (II) oxygenase superfamily domain were also predicted in CdCODM and PsCODM. It was reported that the baine 6-O-demethylase (T6ODM) and CODM were the only known enzymes belonging to the 2OG/Fe (II)-dependent dioxygenase family which can catalyze O-demethylation reaction in plant (Hagel and Facchini 2010). BBE and STOX (from C. deltoidea and other BIA-producing plant species) contain flavin adenine dinucleotide (FAD) binding domain and Berberine and berberine like domain. Both enzymes belong to Flavin-dependent oxidoreductases (FADOXs) family. NMCH, CTS and CAS contain cytochrome P450 domain, which belong to Cytochrome P450 superfamily and play an important part in the creation and modification of several BIA backbones (Dastmalchi et al. 2018; Takemura et al. 2013).

The phylogenetic relationships and conserved motif structure of transcripts encoding enzymes related to BIAs biosynthesis in C. deltoidea and known enzymes from other plants. The phylogenetic trees were constructed based on the deduced amino acid sequences. The protein sequences were analyzed on Pfam to obtain the conserved motif structure. a Norcoclaurine synthase (NCS), tyrosine aminotransferase (TyrAT), polyphenol oxidase (PPO), tyrosine decarboxylase (TYDC). bO-Methyltransferase (OMT), N-methyltransferase (NMT), codeine-O-demethylase (CODM). c Cytochrome P450 (CYP), Flavin-dependent oxidoreductase (FADOX). The accession numbers for the sequences from other plant species are as follows: CjNCS (Coptis japonica var. dissecta, BAF45337); MnNCS (Morus notabilis, XP_024026516); NnTYDC (Nelumbo nucifera, XP_010245171); TfTYDC (Thalictrum flavum subsp. glaucum, AAG60665); PsTYDC (Papaver somniferum, AAA62347); NnTyrAT (N. nucifera, XP_010250299); PsTyrAT (P. somniferum, ADC33123); AmPPO (Argemone mexicana, ACJ76786); ShPPO (Sinopodophyllum hexandrum, ALG05139); PsPPO (P. somniferum, XP_026389945); Cc6OMT (Coptis chinensis, AXC09386); Cj6OMT (Coptis japonica, BAB08004); Tf6OMT (T. flavum subsp. glaucum, AAU20765); Ps6OMT (P. somniferum, AAQ01669); Ec4′OMT (Eschscholzia californica, BAM37633); Tf4′OMT (T. flavum subsp. glaucum, AAU20768); Cc4′OMT (C. chinensis, ABY75613); Md7OMT (Malus domestica, XP_008365438); Pp7OMT (Prunus persica, XP_020415935); CcSOMT (C. chinensis, ACL31653); CjSOMT (C. japonica, BAA06192); TfSOMT (T. flavum subsp. glaucum, AAU20770); CjCoOMT (C. japonica, BAC22084); CjCNMT (C. japonica, BAB71802); NnCNMT (N. nucifera, AXJ91467); PsCNMT (P. somniferum, XP_026398838); AtCNMT (Arabidopsis thaliana, AAM65762); PsCODM (P. somniferum, XP_026416229); NnCODM (N. nucifera, XP_010250269); CcNMCH (C. chinensis, ABS19627); TfNMCH (T. flavum subsp. glaucum, AAU20767); CjNMCH (C. japonica, BAB12433); CjCTS (C. japonica var. dissecta, BAF80448); ShCTS (Sinopodophyllum hexandrum, AJD20229); CjCAS (C. japonica, BAB68769); CjBBE (C. japonica, BAM44344); EcBBE (E. californica, AAC39358); BsBBE (Berberis stolonifera, AAD17487); BwSTOX (Berberis wilsoniae, ADY15026); AmSTOX (A. mexicana, ADY15027); CjTHBO (C. japonica, BAJ40864)
Moreover, the expression patterns of the full-length transcripts encoding enzymes putatively involved in BIAs biosynthesis among different tissues were analyzed based on FPKM values from Illumina reads datasets (Fig. 10). The results revealed that most of the candidate transcripts displayed differential expression levels in leaves, rhizomes and roots of C. deltoidea. Basically consistent with the accumulation of BIAs, the majority of genes showed the highest expression in roots and the lowest in leaves. For example, NCS, 6OMT and 4′OMT encoding enzymes involved in the upstream of BIA biosynthetic pathway displayed higher expression levels in roots than that in the other two tissues. CdTYDC1, CdTYDC2, CdTYDC3 showed similar expression patterns in rhizomes and roots, which were higher than that in leaves. However, CdCTS1, CdCTS2 and CdCTS3 involved in biosynthesis of magnoflorine had high expression in leaves and roots, which was consistent with the result of the relatively high accumulation of magnoflorine in leaves and roots. To validate the reliability of the transcriptome analysis data, nine transcripts related to BIAs biosynthesis and one transcript involved in biosynthesis of secondary metabolites were randomly selected to carry out qRT-PCR analysis. As shown in Fig. S8, the qRT-PCR results of the selected genes revealed similar expression patterns with the RNA-Seq results (r2 > 0.8), indicating the validity of the transcriptome sequencing data.
Transcription factors prediction
Transcription factors (TFs) are sequence-specific DNA-binding proteins, which play a significant role in plant growth, development and controlling secondary metabolism (Endt et al. 2002). For TFs prediction, the putative protein sequences were aligned to plant transcription factor database (Plant TFDB, https://planttfdb.cbi.pku.edu.cn/). A total of 2147 expressed TFs belonging to 55 TF families were identified from the transcriptome dataset (Table S13). Among them, the most abundant TF family was basic helix-loop-helix (bHLH), which is one of the largest class of plant TFs and participate in the regulation of many essential biological processes including flavonoid biosynthesis, transcriptional activation and stress responses (Feller et al. 2011). With respect to TFs regulating BIAs biosynthesis, CjbHLH1 and CjWRKY1 were identified in C. japonica, which are specific to berberine biosynthesis and do not regulate the expression of genes involved in primary metabolism and stress response (Kato et al. 2007; Yamada et al. 2011). In our transcriptome data, 4 transcripts encoding bHLH which were similar to bHLH (CjbHLH1 and CjbHLH2) isolated from C. japonica. Sequence alignment analysis clarified that 3 of them were similar to CjbHLH1, named CdbHLH1a, CdbHLH1b and CdbHLH1c (Fig. S9). The expression pattern analysis of the three transcripts indicated that TFs regulating BIAs biosynthesis in C. deltoidea displayed high expression in rhizomes and roots.
ABC transporters and MATE transporters in C. deltoidea
In order to reveal the accumulation and membrane transport of BIAs in C. deltoidea, we discovered the putative transcripts encoding ATP-binding cassette (ABC) transporters and multidrug and toxic compound extrusion (MATE) transporters in the SMRT sequencing data. ABC transporters constitute a large protein family which are organized phylogenetically into eight clusters (ABCA-ABCI subfamilies). Previous studies have reported that ABC transporters participate directly in the transport of a wide range of secondary metabolites, such as alkaloids, polyphenols and terpenoids and play important roles in physiological processes of plant growth (Kretzschmar and Burla 2011; Verrier et al. 2008). Here, a total of 149 full-length transcripts were identified and the three largest subfamilies were ABCB, ABCC and ABCG (Table S14). Furthermore, the expression pattern analysis indicated that most of transcripts of ABC transporters were expressed lowly. As shown in Fig. S10, ABC proteins can be roughly divided into three categories based on their expression patterns and the transcripts with relatively high expression levels were mostly distributed in leaves and roots. CjABCB1, CjABCB2 and CjABCB3 which belong to the ABC protein of the B-type are currently known to be responsible for alkaloid transport in C. japonica (Shitan et al. 2013). In our dataset, we obtained 9 transcripts encoding ABCB transporters that were highly similar to CjABCB1, CjABCB2 and CjABCB3 (Fig. 12).

The phylogenetic relationship of ABCB-type ABC transporters and MATE transporters. The phylogenetic tree was constructed using the neighbor-joining (NJ) clustering method with MEGA 7.0 (https://www.megasoftware.net/) using the bootstrap values from 1000 replicates. The accession numbers for the sequences of ABCB-type ABC transporters and MATE transporters from other plant species are shown in Table S15
MATE transporters have been found to mediate the secondary metabolites transport and are involved in a wide range of biological events during plant development. In contrast to ABC proteins, MATE transporters use H+ electrochemical gradient across the localized membrane as the driving force (Takanashi et al. 2017). We identified 28 putative transcripts encoding MATE transporters in C. deltoidea. Phylogenetic analysis showed that 10 unigenes were closely clustered with CjMATE1, NtMATE1, NtMATE2, AtDTX1 and Nt-JAT1, respectively, which were involved in the accumulation of alkaloids (Fig. 12). Moreover, we analyzed the expression levels of identified MATE transporters. Among the candidate alkaloid transporters of MATE family, unigene 0012717 and 0060046 having high sequence homology with CjMATE1 were found to be highly expressed in rhizomes. However, other uingenes had high expression levels in roots (Fig. S10).
Discussion
The first and most comprehensive transcriptome analysis of C. deltoidea
As an important medicinal plant, C. deltoidea has been mainly focused on the researches of pharmacological effects and bioactive components. However, the genome of C. deltoidea is still unknown. RNA-Seq can be used to explore and analyze differentially expression genes, but it is often unable to obtain full-length transcripts. With the fast development of sequencing technologies, SMRT has been successfully applied to analyze full-length transcriptomes in multiple plant species with or without an available reference genome, which increases gene discovery, the accuracy of alternative splice detection, gene structure characterization and lncRNA prediction (Lou et al. 2019; Sun et al. 2018; Xu et al. 2015; Zhang et al. 2019). Herein, to generate a much more complete transcriptome of C. deltoidea, we combined long read SMRT sequencing of five different tissues (leaves, petioles, rhizomes, roots and stolons) and short read RNA-Seq of leaves, rhizomes and roots. With the problem of higher error rate of SMRT sequencing, RNA-Seq reads was used to correct the SMRT reads. Finally, a total of 532,835 CCS reads were obtained (Fig. 4) on PacBio SMRT, yielding 75,438 non-redundant transcripts (N50 = 2517 bp). Besides, alternative splicing events were identified from the Iso-Seq reads (Fig. S11) using Coding GENome reconstruction tool (Cogent v3.1, https://github.com/Magdoll/Cogent) and SUPPA (https://github.com/comprna/SUPPA). Without an available reference genome of C. deltoidea, the combination of RNA-Seq and SMRT analyses could provide a more effective and complete characterization of the C. deltoidea transcriptome and comprehensively understand the biosynthetic pathway of secondary metabolites in C. deltoidea.
Current knowledge of C. deltoidea transcriptome is lack. Based on RNA-Seq, the researches of few Coptis plants transcriptome has been carried out, such as C. chinensis and C. teeta (He et al. 2017, 2018). Trinity program was used for de novo assembly of short-reads into unigenes. In C. chinensis, a total of 78,499 unigenes with an average length of 784 bp were generated and 81,823 unigenes obtained from C. teeta were 810 bp in length on average. In our study, the average length of transcripts obtained by PacBio SMRT from C. deltoidea was 2149 bp. The transcripts from SMRT sequencing were longer than that from RNA-Seq obviously. Moreover, SMRT sequencing is with the advantages in discovering novel species-specific or uncharacterized transcripts or genes. 14.43% of the transcripts (10,484 of 72,648) were identified as novel in Litopenaeus vannamei (Zhang et al. 2019) and 13,935 transcripts in alfalfa (Chao et al. 2019). Here, a total of 70,383 transcripts of C. deltoidea were successfully function annotated in public databases and remaining 5055 transcripts might represent species-specific genes, which will be helpful for accurate characterization of C. deltoidea transcriptome and further exploration of gene functions. In this study, we focused on transcriptome and alkaloids metabolism profiling of C. deltoidea. We found that there were differences in gene expression patterns, BIAs biosynthesis and accumulation in leaves, rhizomes and roots. Based on the analysis of expression levels of genes in different tissues, we obtained 5335 common differentially expressed transcripts in three comparison groups, most of which were assigned to “Metabolism” category (Fig. S7). Moreover, we noticed the “Transport and catabolism” pathway, which suggested that different accumulations of metabolites in different C. deltoidea tissues might be related to the transport and catabolism of metabolites.
BIAs accumulation and gene expression of biosynthesis enzymes
BIAs, one of the most important groups of plant secondary metabolites, are the bioactive compounds of the endangered medicinal plant C. deltoidea. Based on UPLC-MS/MS analysis, the content of BIAs in leaves, rhizomes and roots were compared and the results showed that accumulation of BIAs was various in different tissues. We found that all tissues accumulated very high levels of berberine, and basically, the content of most BIAs in roots is the highest. Besides, the highest accumulation of magnoflorine was found in leaves and rhizomes had the highest content of berberrubine, columbamine and palmatine. Furthermore, fresh hand sections of different tissues were observed by light microscope and stained by Dragendorff's reagent under visible light. It was found that alkaloids accumulation was detected mainly in vascular tissues of C. deltoidea. However, previous studies have reported that distinct and different cell types are involved in the biosynthesis and accumulation of BIAs in different Ranunculaceae plants, such as T. flavum and P. somniferum. Endodermis, pericycle, protoderm, cortex or pith are involved in BIAs accumulation in T. flavum, while BIAs metabolism in P. somniferum is mainly in the vascular cell types (Samanani et al. 2005), which implicates that the metabolism and transport mechanisms of BIAs in different plants may be different.
To clarify the BIAs biosynthesis and accumulation in different plant tissues of C. deltoidea, putative genes responsible for the components biosynthesis must be identified and characterized in the biosynthetic pathway. This requires a detailed understanding of the molecular regulation mechanism of the individual steps in the pathway, especially for the poorly investigated species. We attach importance to the relationship between BIAs biosynthesis and primary metabolic pathways. As the precursor of BIAs, tyrosine is synthesized via the shikimate pathway and amino acid pathway. From the SMRT sequencing data, we proposed the candidate tyrosine biosynthesis pathway in C. deltoidea and discovered 25 full-length transcripts that putatively involved in tyrosine biosynthesis, most of which were highly expressed in the roots. CdDAHPS encoding the first enzyme that converts primary carbon metabolism into the shikimate pathway and CdCS encoding enzyme of the final step in shikimate pathway had high expression level in roots. Besides, CdPAH involved in converting phenylalanine into tyrosine was only expressed in roots of C. deltoidea. The results suggested that the biosynthesis of aromatic amino acids might be relatively active in this tissue. With respect to tyrosine pathway, we assumed that genes highly expressed in roots were be more likely to be involved in tyrosine biosynthesis in the roots of C. deltoidea. Therefore, CdCS1, CdPAT1, CdPAT2 and CdTyrA2 may play a role in tyrosine biosynthesis in the roots.
Based on researches of BIA-producing plant species, such as Coptis Japonica, Papaver somniferum, Eschscholtzia californica, the biosynthesis of BIAs including the pathways and enzymes involved in the synthesis are relatively clear, although not complete (Morishige et al. 2010; Beaudoin and Facchini 2014; Ikezawa et al. 2009). Combined the Iso-Seq transcripts and Illumina short-read data, 64 putative transcripts encoding enzymes involved in BIAs biosynthesis were identified in C. deltoidea, which belong to a relatively limited number of protein families, such as 2-oxoglutarate/Fe (II)-dependent dioxygenases (ODDs), cytochrome P450 (CYPs), Flavin-dependent oxidoreductases (FADOXs), S-adenosylmethionine-dependent O- and N-methyltransferases. There was more than one transcript that assigns to the same enzyme, which indicated that such transcripts may represent different parts of a single gene, different members of a gene family, or both (Deng et al. 2018a, b). In C. deltoidea, transcripts encoding almost all known enzymes putatively involved in the biosynthesis of the BIA precursor (S)-reticuline were identified, and most enzymes encoded by putative BIAs biosynthesis genes of C. deltoidea and known enzymes from other BIAs biosynthesis plants have the similar conserved domains (Fig. 11), indicating that BIA biosynthesis among different BIA-producing plants may share most of common steps, especially those in the upstream pathways (Liao et al. 2016). Although the transcripts encoding 3OHase involved in tyrosine hydroxylation were not identified in the transcriptome of C. deltoidea, we found 9 transcripts encoding PPO containing common central domain of tyrosinase, which can catalyze the tyrosine hydroxylation with the formation of dopa (Lovkova et al. 2006). This enzyme may be involved in the early stages of BIAs biosynthesis in C. deltoidea. Previous report has reported that O- and N-methylations catalyzed by methyltransferase (MT) enzymes are ubiquitous features in the biosynthesis of many specialized metabolites and the MT enzymes may be responsible for the chemical diversity of BIA-producing plants (Morris and Facchini 2019). Several OMTs were involved in BIAs biosynthesis. Therefore, OMT protein sequences of C. deltoidea and other BIAs biosynthesis plants were aligned using Clustal Omega under default parameters (Chojnacki et al. 2017) and the percent identity matrix was built (Fig. S12). We found that 6OMT, 4′OMT, SOMT, CoOMT and 7OMT share relatively low amino acid identity. In addition, OMTs from C. deltoidea shared relatively higher amino acid identity with that from Coptis species than that from other genus and families, which may be responsible for the similar metabolites of BIAs in Coptis plants.
Based on RNA-Sqe data, the expression level of genes in different tissues were determined. Most putative genes involved in BIAs biosynthesis were highly expressed in the roots of C. deltoidea, which suggested that the roots might be the main tissues for the biosynthesis of BIAs. NMCH, CTS and CAS contain cytochrome P450 domain (Fig. 11), which belong to CYPs superfamily and play an important part in the creation and modification of several BIA backbones (Dastmalchi et al. 2018). CdCTS1, CdCTS2 and CdCTS3 involved in biosynthesis of magnoflorine had high expression in leaves and roots. The high contents of magnoflorine were also detected in leaves and roots. STOX catalyze the last steps of several BIAs biosynthesis, such as coptisine, berberine and columbamine. In our study, four genes of STOX (CdSTOX1, CdSTOX2, CdSTOX3 and CdSTOX4) were identified, which were relatively high expressed in rhizomes and roots. The expression levels of STOX were also consistent with the accumulation of coptisine, berberine and columbamine. Multiple sequence alignment analysis of STOX indicated that all STOX identified from C. deltoidea were similar to CjTHBO (Fig. S13). Previous researches have reported that STOX from Berberis wilsoniae exhibited broad substrate specificities for protoberberines and simple BIAs (Amann et al. 1988; Gesell et al. 2011). However, CjTHBO was more substrate specific, which preferentially accepts (S)-canadine (Facchini 2001). This may lead to the higher accumulation of berberine in C. deltoidea plants than coptisine, columbamine and epiberberine (Fig. 2). Three bHLH1 transcription factors which may be involved in regulating BIAs biosynthesis had similar expression patterns to most transcripts encoding enzymes that participate in BIAs biosynthetic pathway. Therefore, we speculate that the biosynthesis of most BIAs, such as berberine, coptisine and columbamine, may mainly found occur in roots. Furthermore, leaves and roots may be the main organ for magnoflorine biosynthesis. However, the subcellular accumulation and intra-organ transport of alkaloids in C. deltoidea need further research.
Alkaloids transport in C. deltoidea
It has been reported that BIAs are biosynthesized in root tissues in C. japonica and transported from root after biosynthesis to the rhizome for accumulation (Chao et al. 2019). In C. deltoidea, most candidate genes involved in BIAs biosynthesis were highly expressed in the roots and had relatively low expression levels in leaves and rhizomes. However, with berberine as an example, the content in roots and rhizomes is similar, which are higher than that in leaves. Hence, the result indicated that transport of BIAs may be present in roots and rhizomes. ABC transporters and MATE transporters play important roles in the transport of secondary metabolites (Chao et al. 2019; Kretzschmar and Burla 2011; Shitan et al. 2013). CjABCB1, CjABCB2 and CjABCB3 are ABCB-type ABC transporters, which are proved to transport alkaloids in C. japonica. Herein, 9 transcripts that clustered closely with CjABCB were identified and the expression levels in roots were relatively higher than that in rhizomes (Figs. 12 and S10). CjMATE1 are found to localize at tonoplasts in C. japonica cells and to be expressed preferentially in rhizomes. The analysis of its orthologous genes in C. deltoidea showed that 2 transcripts which had relatively higher expression level in rhizomes than that in roots were highly similar to CjMATE1. This suggested that they may be responsible for the transport of BIAs and accumulation in vacuoles.
Conclusion
In conclusion, we analyzed the contents of ten BIAs in leaves, rhizomes and roots of C. deltoidea using UPLC-MS/MS and first carried out the analysis of C. deltoidea transcriptome with the combination of PacBio SMRT long-read and Illumina short-read sequencing approaches. A total of 75,438 full length transcripts, 2147 transcription factors were obtained. The candidate biosynthesis pathway in C. deltoidea of the precursor of BIAs (tyrosine) was proposed. Furthermore, we screened the genes involved in the BIAs biosynthetic pathway and 64 putative full length-transcripts were identified, which encode a relatively limited number of protein families such as ODDs, CYPs, FADOXs, OMTs and NMTS. We analyzed the expression levels of the candidate genes based on RNA-Seq data and the results indicated that the majority of genes exhibited relatively high expression level in roots. In addition, 3 bHLH1 transcription factors were identified and expression patterns were similar to most transcripts encoding enzymes that participate in BIAs biosynthetic pathway. In order to reveal the accumulation and membrane transport of BIAs in C. deltoidea, a total of 149 and 28 transcripts of ABC transporters and MATE transporters were discovered, respectively. Among them, 9 and 2 transcripts highly homologous to known alkaloid transporters may be related with BIAs transport in roots and rhizomes. Therefore, the work provided important information for characterization of C. deltoidea transcriptome and valuable genetic resources for this medicinal plants with scarce resources.
Abbreviations
- BIA:
-
Benzylisoquinoline alkaloid
- UHPLC-ESI-MS/MS:
-
Ultra-high performance liquid chromatography-electrospray ionization tandem mass spectrometry
- SMRT:
-
Single molecule real-time technology
- CCS:
-
Circular consensus sequence
- FLNC:
-
Full-length non-chimeric read
- DAHPS:
-
3-Deoxy-d-arabino-heptulosonate-7-phosphate synthase
- DHQS:
-
3-Dehydroquinate synthase
- DHD/SDH:
-
3-Dehydroquinate dehydratase/shikimate dehydrogenase
- SK:
-
Shikimate kinase
- EPSPS:
-
5-Enolpyruvylshikimate 3-phosphate synthase
- CS:
-
Chorismate synthase
- CM:
-
Chorismate mutase
- PAT:
-
Prephenate aminotransferase
- TyrA:
-
Arogenate dehydrogenase
- PAH:
-
Phenylalanine-4-hydroxylase
- PDH:
-
Prephenate dehydrogenase
- TyrAT:
-
l-Tyrosine aminotransferase
- TYDC:
-
Tyrosine decarboxylase
- PPO:
-
Polyphenol oxidase
- NCS:
-
(S)-Norcoclaurine synthase
- 6OMT:
-
(S)-Norcoclaurine 6-O-methyltransferase
- CNMT:
-
(S)-Coclaurine N-methyltransferase
- NMCH:
-
(S)-N-Methylcoclaurine-3′-hydroxylase
- 4′OMT:
-
3′-Hydroxy-N-methyl-(S)-coclaurine 4′-O-methyltransferase
- BBE:
-
Berberine bridge enzyme
- SOMT:
-
(S)-Scoulerine 9-O-methyltransferase
- CAS:
-
(S)-Canadine synthase
- STOX:
-
(S)-Tetrahydroprotoberberine oxidase
- CTS:
-
(S)-Corytuberine synthase
- CoOMT:
-
Columbamine O-methyltransferase
- CODM:
-
Codeine-O-demethylase
- CFS:
-
(S)-Cheilanthifoline synthase
- SPS:
-
(S)-Stylopine synthase
- SCNMT:
-
(S)-Corytuberine N-methyltransferase
- bHLH:
-
Basic helix-loop-helix
- ABC:
-
ATP-binding cassette transporter
- MATE:
-
Multidrug and toxic compound extrusion transporter
References
Amann M, Nagakura N, Zenk MH (1988) Purification and properties of (S)-tetrahydroprotoberberine oxidase from suspension-cultured cells of Berberis wilsoniae. Eur J Biochem 175:17–25
Au KF, Underwood JG, Lee L, Wong WH (2012) Improving PacBio long read accuracy by short read alignment. PLoS ONE 7:e46679
Beaudoin G, Facchini PJ (2014) Benzylisoquinoline alkaloid biosynthesis in opium poppy. Planta 240:19–32
Chao Y, Yuan J, Guo T, Xu L, Mu Z, Han L (2019) Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing. Plant Mol Biol 99:219–235
Chen H, Deng C, Nie H, Fan G, He Y (2017a) Transcriptome analyses provide insights into the difference of alkaloids biosynthesis in the Chinese goldthread (Coptis chinensis Franch.) from different biotopes. PeerJ 5:e3303
Chen H, Fan G, He Y (2017b) Species evolution and quality evaluation of four Coptis herbal medicinal materials in Southwest China. 3 Biotech 7:62.
Chen J, Tang X, Ren C, Wei B, Wu Y, Wu Q, Pei J (2018) Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genomics 1:548
Chojnacki S, Cowley A, Lee J, Foix A, Lopez R (2017) Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res 45:W550–W553
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Dastmalchi M, Park MR, Morris JS, Facchini P (2018) Family portraits: the enzymes behind benzylisoquinoline alkaloid diversity. Phytochem Rev 17:249–277
Deng X, Zhao L, Fang T, Xiong Y, Ogutu C, Yang D et al (2018a) Investigation of benzylisoquinoline alkaloid biosynthetic pathway and its transcriptional regulation in lotus. Hortic Res 5:29
Deng Y, Zheng H, Yan Z, Liao D, Li C, Zhou J, Liao H (2018b) Full-length transcriptome survey and expression analysis of cassia obtusifolia to discover putative genes related to aurantio-obtusin biosynthesis, seed formation and development, and stress Response. Int J Mol Sci 19:2476
Desgagné-Penix I, Khan MF, Schriemer DC, Cram D, Nowak J, Facchini PJ (2010) Integration of deep transcriptome and proteome analyses reveals the components of alkaloid metabolism in opium poppy cell cultures. BMC Plant Biol 10:252
Dong L, Liu H, Zhang J, Yang S, Kong G, Chu JS et al (2015) Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics 16:1039
Endt V, Kijne D, Memelink JW (2002) Transcription factors controlling plant secondary metabolism: what regulates the regulators? Phytochemistry 61:107–114
Facchini PJ (2001) Alkaloid biosynthesis in plants: biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu Rev Plant Biol 52:29–66
Feller A, Machemer K, Braun EL, Grotewold E (2011) Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J 66:94–116
Gesell A, Chávez MLD, Kramell R, Piotrowski M, Macheroux P, Kutchan TM (2011) Heterologous expression of two FAD-dependent oxidases with (S)-tetrahydroprotoberberine oxidase activity from Argemone mexicana and Berberis wilsoniae in insect cells. Planta 233:1185–1197
Hagel JM (2010) Biochemistry and occurrence of O-demethylation in plant metabolism. Front Physiol 1:14
Hagel JM, Facchini PJ (2010) Dioxygenases catalyze the O-demethylation steps of morphine biosynthesis in opium poppy. Nat Chem Biol 6:273–275
Hagel JM, Facchini PJ (2013) Benzylisoquinoline alkaloid metabolism: a century of discovery and a brave new world. Plant Cell Physiol 54:647–672
Hagel JM, Mandal R, Han B, Han J, Dinsmore DR, Borchers CH et al (2015) Metabolome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Biol 15:220
He Y, Hou P, Fan G, Arain S, Peng C (2014) Comprehensive analyses of molecular phylogeny and main alkaloids for Coptis (Ranunculaceae) species identification. Biochem Syst Ecol 56:88–94
He SM, Song WL, Cong K, Wang X, Dong Y, Cai J et al (2017) Identification of candidate genes involved in isoquinoline alkaloids biosynthesis in Dactylicapnos scandens by transcriptome analysis. Sci Rep 7:9119
He S, Liang Y, Cong K, Chen G, Zhao X, Zhao Q et al (2018) Identification and characterization of genes involved in benzylisoquinoline alkaloid biosynthesis in Coptis Species. Front Plant Sci 9:731
Huddleston J, Ranade S, Malig M, Antonacci F, Chaisson M, Hon L et al (2014) Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res 24:688–696
Ikezawa N, Iwasa K, Sato F (2008) Molecular cloning and characterization of CYP80G2, a cytochrome P450 that catalyzes an intramolecular C-C phenol coupling of (S)-reticuline in magnoflorine biosynthesis, from cultured Coptis japonica cells. J Biol Chem 283:8810–8821
Ikezawa N, Iwasa K, Sato F (2009) CYP719A subfamily of cytochrome P450 oxygenases and isoquinoline alkaloid biosynthesis in Eschscholzia californica. Plant Cell Rep 28:123–133
Inui T, Kawano N, Shitan N, Yazaki K, Kiuchi F, Kawahara N et al (2012) Improvement of benzylisoquinoline alkaloid productivity by overexpression of 3′-hydroxy-N-methylcoclaurine 4′-O-methyltransferase in transgenic Coptis japonica plants. Biol Pharm Bull 35:650–659
Itoh A, Saitoh T, Tani K, Uchigaki M, Sugimoto Y, Yamada J et al (2011) Bisbenzylisoquinoline alkaloids from Nelumbo nucifera. Chem Pharm Bull 59:947–951
Kamps R, Brandão R, Bosch B, Paulussen A, Xanthoulea S, Blok M, Romano A (2017) Next-generation sequencing in oncology: genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci 18:308
Kato N, Dubouzet E, Kokabu Y, Yoshida S, Taniguchi Y, Dubouzet JG et al (2007) Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol 48:8
Kretzschmar T, Burla B (2011) Functions of ABC transporters in plants. Essays Biochem 50:145–160
Lee EJ, Facchini PJ (2011) Tyrosine aminotransferase contributes to benzylisoquinoline alkaloid biosynthesis in opium poppy. Plant Physiol 157:1067–1078
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Li CY, Tsai SI, Damu AG, Wu TS (2009) A rapid and simple determination of protoberberine alkaloids in Rhizoma Coptidis by 1H NMR and its application for quality control of commercial prescriptions. J Pharm Biomed Anal 49:1272–1276
Li Y, Ren G, Wang YX, Kong WJ, Jiang JD (2011) Bioactivities of berberine metabolites after transformation through CYP450 isoenzymes. J Transl Med 9:62
Li Q, Li Y, Song J, Xu H, Xu J, Zhu Y et al (2014) High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol 204:1041–1049
Liao D, Wang P, Jia C, Sun P, Qi J, Zhou L et al (2016) Identification and developmental expression profiling of putative alkaloid biosynthetic genes in Corydalis yanhusuo bulbs. Sci Rep 6:19460
Liscombe DK, MacLeod BP, Loukanina N, Nandi OI, Facchini PJ (2005) Evidence for the monophyletic evolution of benzylisoquinoline alkaloid biosynthesis in angiosperms. Phytochemistry 66:1374–1393
Lou H, Ding M, Wu J, Zhang F, Chen W, Yang Y et al (2019) Full-length transcriptome analysis of the genes involved in tocopherol biosynthesis in Torreya grandis. J Agric Food Chem 67:1877–1888
Lovkova MY, Buzuk G, Sokolova S (2006) Regulatory role of elements in the formation and accumulation of alkaloids in Papaver somniferum L. seedlings. Appl Biochem Microbiol 42:420–423
Maeda H, Dudareva N (2012) The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu Rev Plant Biol 63:73–105
Minami H, Dubouzet E, Iwasa K, Sato F (2007) Functional analysis of norcoclaurine synthase in Coptis japonica. J Biol Chem 282:6274–6282
Mizutani M, Sato F (2011) Unusual P450 reactions in plant secondary metabolism. Arch Biochem Biophys 507:194–203
Morishige T, Tamakoshi M, Takemura T, Sato F (2010) Molecular characterization of O-methyltransferases involved in isoquinoline alkaloid biosynthesis in Coptis japonica. Proc Jpn Acad Ser B 86:757–768
Morris JS, Facchini PJ (2016) Isolation and characterization of reticuline N-methyltransferase involved in biosynthesis of the aporphine alkaloid magnoflorine in opium poppy. J Biol Chem 291:23416
Morris JS, Facchini PJ (2019) Molecular origins of functional diversity in benzylisoquinoline alkaloid methyltransferases. Front Plant Sci 10:1058
Pienkny S, Brandt W, Schmidt J, Kramell R, Ziegler J (2010) Functional characterization of a novel benzylisoquinoline O-methyltransferase suggests its involvement in papaverine biosynthesis in opium poppy (Papaver somniferum L.). Plant J 60:56–67
Qi L, Ma Y, Zhong F, Shen C (2018) Comprehensive quality assessment for Rhizoma Coptidis based on quantitative and qualitative metabolic profiles using high performance liquid chromatography, Fourier transform near-infrared and Fourier transform mid-infrared combined with multivariate statistical analysis. J Pharm Biomed 161:436–443
Qiao YL, Sheng YX, Wang LQ, Zhang JL (2009) Development of a rapid resolution liquid chromatographic method for simultaneous analysis of four alkaloids in Rhizoma Coptidis under different cultivation conditions. J AOAC Int 92:663–671
Qiao D, Yang C, Chen J, Guo Y, Li Y, Niu S et al (2019) Comprehensive identification of the full-length transcripts and alternative splicing related to the secondary metabolism pathways in the tea plant (Camellia sinensis). Sci Rep 9:2709
Roberts RJ, Carneiro MO, Schatz MC (2013) The advantages of SMRT sequencing. Genome Biol 14:405
Rüffer M, Ekundayo O, Nagakura N, Zenk MH (1983) Biosynthesis of the protoberberine alkaloid jatrorrhizine. Tetrahedron Lett 24(6):2643–2644
Sadre R, Magallanes-Lundback M, Pradhan S, Salim V, Mesberg A, Jones AD, DellaPenna D (2016) Metabolite diversity in alkaloid biosynthesis: a multilane (diastereomer) highway for camptothecin synthesis in Camptotheca acuminata. Plant Cell 28:1926–1944
Samanani N, Liscombe DK, Facchini PJ (2004) Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J 40:302–313
Samanani N, Park SU, Facchini PJ (2005) Cell type–specific localization of transcripts encoding nine consecutive enzymes involved in protoberberine alkaloid biosynthesis. Plant Cell 17:915–926
Sato F, Inui T, Takemura T (2007) Metabolic engineering in isoquinoline alkaloid biosynthesis. Curr Pharm Biotechnol 8:211–218
Shitan N, Dalmas F, Dan K, Kato N, Ueda K, Sato F et al (2013) Characterization of Coptis japonica CjABCB2, an ATP-binding cassette protein involved in alkaloid transport. Phytochemistry 91:109–116
Sun MY, Li JY, Li D, Huang FJ, Wang D, Li H et al (2018) Full-length transcriptome sequencing and modular organization analysis of the naringin/neoeriocitrin-related gene expression pattern in Drynaria roosii. Plant Cell Physiol 59:1398–1414
Takanashi K, Yamada Y, Sasaki T, Yamamoto Y, Sato F, Yazaki K (2017) A multidrug and toxic compound extrusion transporter mediates berberine accumulation into vacuoles in Coptis japonica. Phytochemistry 138:76–82
Takemura T, Ikezawa N, Iwasa K, Sato FJP (2013) Molecular cloning and characterization of a cytochrome P450 in sanguinarine biosynthesis from Eschscholzia californica cells. Phytochemistry 91:100–108
Torrens-Spence MP, Pluskal T, Li FS, Carballo V, Weng JK (2018) Complete pathway elucidation and heterologous reconstitution of Rhodiola salidroside biosynthesis. Mol Plant 11:205–217
Tzin V, Galili G (2010) New insights into the shikimate and aromatic amino acids biosynthesis pathways in plants. Mol Plant 3:956–972
Verrier PJ, Bird D, Burla B, Dassa E, Forestier C, Geisler M et al (2008) Plant ABC proteins—a unified nomenclature and updated inventory. Trends Plant Sci 13:151–159
Vuddanda PR, Chakraborty S, Singh S (2010) Berberine: a potential phytochemical with multispectrum therapeutic activities. Expert Opin Investig Drugs 19:1297–1307
Wang H, Mu W, Shang H, Lin J, Lei X (2014) The antihyperglycemic effects of Rhizoma coptidis and mechanism of actions: a review of systematic reviews and pharmacological research. Biomed Res Int 3:798093
Wenping H, Yuan Z, Jie S, Lijun Z, Zhezhi W (2011) De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98:272–279
Xiang KL, Wu SD, Yu SX, Liu Y, Jabbour F, Erst AS et al (2016) The first comprehensive phylogeny of Coptis (Ranunculaceae) and its implications for character evolution and classification. PLoS ONE 11:e0153127
Xu Z, Peters RJ, Weirather J, Luo H, Liao B, Zhang X et al (2015) Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J 82:951–961
Yamada Y, Kokabu Y, Chaki K, Yoshimoto T, Ohgaki M, Yoshida S et al (2011) Isoquinoline alkaloid biosynthesis is regulated by a unique bHLH-type transcription factor in Coptis japonica. Plant Cell Physiol 52:1131–1141
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z et al (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297
Yeung E C (1998) A beginner’s guide to the study of plant structure. In: Karcher SJ (ed) Tested studies for laboratory teaching, vol 19. Proceedings of the 19th workshop/conference of the Association for Biology Laboratory Education (ABLE). Purdue University, Lafayette, pp 125–141
Yuan Y, Yu M, Jia Z, Liang Y, Zhang J (2018) Analysis of Dendrobium huoshanense transcriptome unveils putative genes associated with active ingredients synthesis. BMC Genomics 19:978
Zhang X, Li G, Jiang H, Li L, Ma J, Li H, Chen J (2019) Full-length transcriptome analysis of Litopenaeus vannamei reveals transcript variants involved in the innate immune system. Fish Shellfish Immunol 87:346–359
Ziegler J, Facchini PJ (2008) Alkaloid biosynthesis: metabolism and trafficking. Annu Rev Plant Biol 59:735–776
Acknowledgements
This work was supported by Sichuan Science and Technology Support Program (2014SZ0156). The authors also thank the National Wild Plant Germplasm Resources Infrastructure for its support.
Author information
Authors and Affiliations
Contributions
Furong Zhong, Yuntong Ma and Zhuyun Yan conceived and designed the experiments; Furong Zhong performed the experiments and wrote the manuscript; Furong Zhong, Ling Huang and Luming Qi analyzed the data and prepared the figures and tables. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The sequencing data are available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA accession PRJNA556312, https://dataview.ncbi.nlm.nih.gov/object/PRJNA556312?reviewer=t0t8vejmp8s9u1bt38r4qq5rn4).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhong, F., Huang, L., Qi, L. et al. Full-length transcriptome analysis of Coptis deltoidea and identification of putative genes involved in benzylisoquinoline alkaloids biosynthesis based on combined sequencing platforms. Plant Mol Biol 102, 477–499 (2020). https://doi.org/10.1007/s11103-019-00959-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-019-00959-y