
Abstract
Key message
Transcriptomic analysis resulted in the upregulation of the genes related to common defense mechanisms for black spot and the downregulation of the genes related to photosynthesis and cell wall modification for powdery mildew.
Abstract
Plant pathogenic fungi successfully colonize their hosts by manipulating the host defense mechanisms, which is accompanied by major transcriptome changes in the host. To characterize compatible plant pathogen interactions at early stages of infection by the obligate biotrophic fungus Podosphaera pannosa, which causes powdery mildew, and the hemibiotrophic fungus Diplocarpon rosae, which causes black spot, we analyzed changes in the leaf transcriptome after the inoculation of detached rose leaves with each pathogen. In addition, we analyzed differences in the transcriptomic changes inflicted by both pathogens as a first step to characterize specific infection strategies. Transcriptomic changes were analyzed using next-generation sequencing based on the massive analysis of cDNA ends approach, which was validated using high-throughput qPCR. We identified a large number of differentially regulated genes. A common set of the differentially regulated genes comprised of pathogenesis-related (PR) genes, such as of PR10 homologs, chitinases and defense-related transcription factors, such as various WRKY genes, indicating a conserved but insufficient PTI [pathogen associated molecular pattern (PAMP) triggered immunity] reaction. Surprisingly, most of the differentially regulated genes were specific to the interactions with either P. pannosa or D. rosae. Specific regulation in response to D. rosae was detected for genes from the phenylpropanoid and flavonoid pathways and for individual PR genes, such as paralogs of PR1 and PR5, and other factors of the salicylic acid signaling pathway. Differently, inoculation with P. pannosa leads in addition to the general pathogen response to a downregulation of genes related to photosynthesis and cell wall modification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungal pathogens have developed specific strategies to obtain nutrients from their hosts depending on their lifestyle. Biotrophic fungi develop nutrient-absorbing structures, such as haustoria, to establish a long-term feeding relationship without killing their hosts. In contrast, necrotrophic fungi kill the host cells, often by secreting toxins, to feed on the dead tissue, while hemibiotrophic pathogens often change their lifestyle during development from an early biotrophic to a later necrotrophic stage (Horbach et al. 2011).
In their natural environment, roses are confronted with many different pathogens. The biotrophic fungus Podosphaera pannosa, which causes powdery mildew, and the hemibiotrophic Diplocarpon rosae, which causes black spot, are the most common and damaging pathogens in roses cultivated in the greenhouse and the field, respectively (Debener and Byrne 2014).
The life cycle of D. rosae begins on the surface of rose leaves with the germination of its conidia within 8–9 h after infection, and 3 h later, appressoria may already have formed. After an additional 3 h, subcuticular hyphae and the first haustoria may be developed (Aronescu 1934). In this biotrophic phase, all the fungal structures in the host cells are surrounded by the plasma membrane of the host. The resulting circular black spots that are typical infection symptoms are mostly surrounded by living host cells, so-called “green islands” (Gachomo et al. 2006). Few details about the infection process of P. pannosa in roses are available. Germ tubes may be observed 2–6 hpi (hours post inoculation), rapidly developing into mycelia that grow on the leaf surface. After approximately 1 dpi, multilobed haustoria develop under the appressoria, which are formed in regular distances (Linde and Shishkoff 2003). Under favorable conditions, asexually produced spores are released after 3–5 days, completing the life cycle on the living host tissue (Coyier 1983).
The recognition of a pathogen can either be mediated by pattern-recognition-receptors in the cell membrane, which recognize highly conserved pathogen- or microbe-associated molecular patterns (PAMP or MAMP) on the surface of a pathogen, e.g., fungal chitin or bacterial flagellin, or by R proteins that recognize the effectors secreted from the pathogen into the host cell. These two mechanisms are called PAMP-triggered immunity (PTI) and effector-triggered immunity (ETI), respectively, and the latter is often accompanied by a so-called “hypersensitive response” (Bent and Mackey 2007; Jones and Dangl 2006).
The recognition of a pathogen by the plant cells leads to drastic changes in the transcriptome and activates a diverse set of immune responses, including the synthesis of secondary metabolites, cell wall modifications and the expression of pathogenesis-related (PR) genes (Slusarenko et al. 2000). In addition to common immune responses in both, compatible and incompatible interactions, the responses to fungal infections may involve specific reactions depending on the life style of the infecting fungus (González et al. 2013; Silvia Sebastiani et al. 2017).
Plant hormones play an important role as signaling molecules in plant defense. Salicylic acid (SA) is involved in the defense response to biotrophic and hemibiotrophic pathogens, while jasmonate (JA) and ethylene (ET) are the primary hormones involved in the response to necrotrophic pathogens. Both pathways have often been described as antagonistic. In addition, crosstalk with other phytohormones can modulate the responses to pathogens/stress (Derksen et al. 2013). In the compatible interaction between Erysiphe necator and Vitis vinifera, significant alterations in the host transcriptome were induced, essentially genes involved in signaling and secondary metabolite biosynthetic pathways (Borges et al. 2013). The analysis of the compatible interaction between Malus × domestica Borkh. and Alternaria alternata showed that genes associated with photosynthesis and oxidation–reduction were downregulated, while transcription factors (i.e., WRKY, MYB, NAC, and Hsf) and genes involved in cell wall modification, defense signaling, the synthesis of defense-related metabolites, including pathogenesis-related (PRs) genes and phenylpropanoid/cyanoamino acid/flavonoid biosynthesis, were activated (Zhu et al. 2017).
PR-proteins are pathogen or stress-related induced plant proteins, which are classified into 17 families based on their structural or functional similarities (van Loon et al. 2006). PR1 is one of the primary marker genes for the SA-mediated defense response, but its overall function is not yet clear. The PR2 family (endo-1,3-β-d-glucanase) and the PR5 family (thaumatin-like) are also associated with this pathway, while the other PR-protein families, such as PR4 (hevein-like protein), PR6 (proteinase inhibitor) or PR9 (peroxidase) are more closely associated with the JA-ET pathway (van Loon et al. 2006; Vidhyasekaran 2015; Derksen et al. 2013).
Among the secondary metabolites involved in plant pathogen interactions, flavonoids are widely distributed with different biological functions, such as protecting against harmful radiation and phytopathogens, binding phytotoxins and controlling auxin transport. They are synthesized through the phenylpropanoid pathway, which transforms phenylalanine into 4-coumaroyl-CoA, which finally enters the flavonoid biosynthesis pathway (Falcone Ferreyra et al. 2012; Winkel-Shirley 2002). A transcriptome analysis of Fragaria vesca infected with either Colletotrichum fragariae or Phytophtora cactorum showed the induction of several genes of the flavonoid biosynthetic pathway (Guidarelli et al. 2011; Toljamo et al. 2016).
In this study, we analyzed differences in the manipulation of the leaf transcriptome of roses after successful colonization of the leaves with either the hemibiotrophic fungus D. rosae or the biotrophic fungus P. pannosa.
The goal of these analyses is to gain initial insights into the changes that the two pathogens inflict on the host transcriptomes as the first step to understand host–pathogen interactions in the two systems. For this, we analyzed the transcriptomic responses against the two pathogens in the early stages of infection (0 hpi, 24 hpi and 72 hpi) using the massive analysis of cDNA ends (MACE) data from three independent inoculation experiments.
Materials and methods
Plant material
For the MACE analysis and the multiplication of the fungal isolates, the susceptible rose cultivar ‘Pariser Charme’ (PC) was propagated in vitro as previously described (Davies 1980; Debener et al. 1998; Linde and Debener 2003; Dohm et al. 2001), rooted, and transplanted into fertilized substrate (“Einheitserde T”, Einheitserdewerke Gebr. Patzer, Sinntal-Altengronau, Germany) in 9-cm pots and cultivated under semicontrolled conditions (12 h light/12 h darkness). The plants used to validate the MACE analysis were cultivated in climate chambers under short-day conditions (8 h light/16 h darkness) at 22 °C.
Disease assays
The D. rosae isolate DortE4 was used for the black spot inoculations. The single conidial isolate was maintained on detached leaves of the susceptible cultivar PC as previously described (von Malek and Debener 1998). Young unfolded leaves were infected with a suspension of 500,000 conidia/mL with a vaporizer and kept on moist tissue paper in translucent plastic boxes in an air-conditioned laboratory at 20 °C.
A multispore isolate of powdery mildew originating from the host genotype PC, which was grown in the greenhouses at Leibniz Universität Hannover (Germany), was used for inoculation. Very young, recently unfolded PC leaves were placed in an infection box covered with a 14 cm tall-100 µm nylon mesh. Infected leaves were rubbed over the mesh to cover the leaves with conidia. The conidia were allowed to settle for 30 min to ensure that they all reached the leaf surfaces. To estimate the density of the conidia, a microscope slide was placed next to the leaves, and the conidia were counted under a microscope at 100-fold magnification (20 conidia/mm2, 25 conidia/mm2, 60 conidia/mm2 for the three biological replicates respectively, and the validation experiment: 16 conidia/mm2). Infected leaves were stored in translucent plastic boxes on moist tissue paper at 20 °C. Control leaves were used and stored in conditions identical to those of the samples inoculated. The treatments and infection time points for both pathogens are listed in Table 1. Three completely independent inoculation experiments for each time point were conducted for the MACE experiment, and independent sets of three additional experiments were conducted for the qPCR experiment. These experiments were treated as biological repeat experiments.
Microscopic analysis
The control leaves and leaves infected with either D. rosae or P. pannosa were sampled at 0, 24, and 72 hpi for the microscopic analysis. Leaf pieces of approximately 1 cm2 were fixed, stained with Alexa Fluor 488-conjugated wheat germ agglutinin (Invitrogen, Carlsbad, USA) and examined as previously described (Menz et al. 2017).
MACE
Transcriptomic data was generated using the MACE technique (Kahl et al. 2012). A particular feature of this technique is that only one sequence (tag) per cDNA molecule is generated, so normalization to the length of the respective transcript/gene model is not necessary. The RNA for this analysis was extracted from the independent biological replicates using an RNeasy® Plant Mini Kit from Qiagen (Hilden, Germany) following the manufacturer’s instructions with an additional DNase digestion step using an Ambion VR DNA-free™ Kit from Life Technologies (Carlsbad, CA, USA) to remove the remaining genomic DNA. The cDNA synthesis and sequencing was performed by GenXPro (Frankfurt am Main, Germany). The sequencing data were already quality- and adapter-trimmed by the provider. The raw reads of this study were placed in the NCBI Sequence Read Archive (SRA) under the accession numbers SRR6879138 to SSR6879164. Additional processing of the sequences was performed using CLC Genomic Workbench 9.0.1 (Qiagen, Hilden, Germany). The remaining pieces of the poly-A tail were removed by an additional adapter trimming step using a poly-A-adapter. Reads shorter than 35 bp were discarded. The trimmed sequences were mapped to the genomic sequence of Rosa chinensis var. ‘Old Blush’ (Hibrand Saint-Oyant et al. 2018) using the following parameters: mismatch cost = 2, insertion cost = 3, deletion cost = 3, length fraction = 0.9, similarity fraction = 0.9, strand specific = both, maximum number of hits = 10 and expression value = unique counts. The expression values were normalized using the tags per million (TPM) normalization and were log2 transformed. Differential gene expression was analyzed using the EdgeR package (Robinson et al. 2010) with a false discovery rate (FDR) adjustment.
Only the genes detected in all three repeat experiments of at least one condition that had at least 1 TPM and a minimum mean fold change ± threefold, with an FDR-adjusted p value ≤ 0.05, were considered to be differentially expressed.
Additional data analysis
In addition to the annotation presented by Hibrand Saint-Oyant et al. (2018), Blast2GO 4.1.9 software (Conesa et al. 2005) was used to classify sequences with GO terms and generate an automatic functional description derived from a BLASTp search (E-Value cut-off 1E-10) against a subset of the NR database that contained only plant sequences (Ashburner et al. 2000). These GO terms were used for an enrichment analysis of the differentially expressed genes (DEGs) using a Fisher’s exact test implemented in Blast2GO. The figure was designed in Microsoft Excel 2013.
To visualize of the regulated genes in heatmaps, R 3.4.0 (R Core Team) and the package “pheatmap” (Version 1.0.10; Kolde 2018) were used. The clustering default settings were used.
Based on the normalized expression values, the Spearman rank correlations between the samples and the biological repeat experiments were calculated in R 3.4.0 (R Core Team).
Validation of the MACE analysis using high-throughput qRT-PCR
To validate the MACE analysis, an independent set of three inoculations representing independent biological replicates was performed. From 20 to 30 mg of infected leaf tissue (0 hpi, 24 hpi and 72 hpi), the total RNA was isolated using a Quick-RNA™ MiniPrep Plus kit (Zymo Research, Irvine, USA) according to the manufacturer’s instructions with some minor modifications. The leaf material was frozen in liquid nitrogen and disrupted using a bead mill. Dithiothreitol (DTT) was added to the lysis buffer to a final concentration of 50 mM.
To synthesize the cDNA, 500 ng of total RNA was processed using a High Capacity cDNA Reverse Transcription Kit from Applied Biosystems VR (Carlsbad, USA) according to the manufacturer’s instructions. A set of 28 genes was chosen among the differentially regulated genes identified by the MACE analysis and subjected to BioMark high-throughput qPCR (Fluidigm Corperation, San Francisco, USA). Primers for the genes were constructed using Primer3plus (Rozen and Skaletsky 2000) and are listed in Supplementary Table 1. The primer efficiency was tested with a dilution series (1:4, 1:16, 1:64, 1:256) using a StepOnePlus™ system from Applied Biosystems (Austin, USA) as described by Menz et al. (2017). The expression of the differentially regulated genes and the three reference genes TIP, SAND and UBC (Klie and Debener 2011) were analyzed using a Fluidigm Dynamic Array IFC (96.96) (Fluidigm Corperation, San Francisco, USA) following the manufacturer’s instructions. The specific target amplifications (STA) were diluted fivefold, and the qPCR conditions were as follows: 60 s at 95 °C, 30 cycles of 95 °C for 5 s and 20 s at 64 °C, and a final melting curve analysis. The data were processed using Fluidigm Real-Time PCR Analysis Software (4.3.1, Fluidigm Corporation, San Francisco, USA). A quality threshold of 0.65, a linear baseline correction and an auto global cycle threshold (Ct) were used. Ct values were used to calculate the expression ratios with using the REST 2009 software (V2.0.13, Qiagen, Hilden, Germany). The expression ratios were log2 transformed, and the coefficient of correlation (Pearson) of the significantly up- or downregulated genes in both the MACE and BioMark analysis were calculated in R 3.4.0 (R Core Team).
Results
Fungal development in the early stages of compatible interactions
To analyze the fungal development in the early stages of the rose interaction with the hemibiotrophic fungus D. rosae and the obligate biotrophic fungus P. pannosa, a microscopic analysis was performed. The development of both pathogens was similar at the beginning. At 24 hpi, the spores germinated, and the first haustoria formed (Fig. 1B, F). At 72 hpi, D. rosae developed long-range hyphae, and numerous haustoria were formed in the epidermis and the underlying mesophyll layer (Fig. 1C, D). However, after 72 hpi, the development progressed more in P. pannosa than in D. rosae. More fungal hyphae spread across wide parts of the leaf, and a high number of haustoria formed in the epidermis of the plant (Fig. 1G, H).

Microscopic analysis of the two interaction systems. The interaction between the susceptible genotype PC and D. rosae (black spot, A–D) and P. pannosa (powdery mildew, E–H) at different time points [0 hpi (A, E), 24 hpi (B, F) and 72 hpi (C, D, G, H)]. Co conidia, Gt germ tube, Ha haustoria, Hy hyphae. Samples were stained with Alexa Fluor 488-conjugated wheat germ agglutinin (WGA)
MACE sequencing results and validation
MACE sequencing was performed for the inoculated and control samples for all the time points (0, 24, 72 hpi) in the three biological repeats, each represented by an independent inoculation experiment. The sequencing resulted in 6–30 million high-quality reads per library with an average output of 12.7 million reads per library (Supplementary Table 2). Between 79.2 and 94.2% of these reads could be mapped to the genome sequence of the ‘Old Blush’ rose variety (Hibrand Saint-Oyant et al. 2018). The majority of the sequenced reads (76.3–92.9%) uniquely mapped to the genome (Supplementary Table 1).
To validate the results of the MACE analysis, three additional inoculation experiments were analyzed using a high-throughput RT-qPCR system. For this purpose, a set of 28 significantly up- and downregulated genes was chosen based on the MACE results. The selection primarily focused on genes that showed similar differences in expression between the infected and noninfected samples in all three biological replicates. A close correlation (r = 0.82) between the significant log2-fold changes in the MACE and the RT-qPCR results was detected, and except for the three data points that were downregulated in the qPCR results and upregulated in the MACE results (left upper quadrant), the same expression trends were observed (Fig. 2). The selected genes, primer sequences, PCR amplification efficiencies and expression data of the MACE and RT-qPCR analysis are listed in Supplementary Table 2.

Scatter plot for 28 genes analyzed with both MACE and RT-qPCR at three time points. Only the expression ratios of the significantly up- or downregulated genes from the MACE and RT-qPCR data are shown, resulting in 58 data points. The Pearson’s correlation coefficient is also shown
Common responses of roses to both pathogens
To provide an overview of the functions and processes linked to the differentially expressed genes in the transcriptome, we performed a GO enrichment analysis (Fig. 3 and Supplementary Table 3) using Blast2GO with a particular interest in visualizing the pathogen-related reactions of the rose. In addition, the expression of single and typical marker genes and gene families were analyzed (Fig. 4).

Gene ontology enrichment analysis in significant down- and upregulated gene sets. This figure shows the number of genes associated with GO terms, which exhibited statistically significant enrichments in at least one of the time points or infection treatments. Interesting Go terms in the category of “biological process”, “cellular component” and “molecular function” are shown

Heatmaps showing the expression of genes associated with different keywords or gene families. Fold changes, compared to the control leaves, of down- (blue) and upregulation (red) are shown on a log2 scale for all the time points and infection treatments. At least in one data point, the expression is significantly different according to our criteria. “DR” stands for inoculations with D. rosae and “PP” for P. pannosa at “0”, “24” and “72” hpi. The clustering default settings were used
The response of the susceptible rose PC to infection with P. pannosa and D. rosae leads to major changes in the leaf transcriptome (Supplementary Tables 4 and 5). The most strongly differentially regulated genes (Supplementary Table 6) comprised some defense-related genes, such as the major allergen Pru ar1, belonging to the PR10 family. In addition, some genes involved in signaling pathways, such as kinases, transcription factors and a calmodulin, as well as genes encoding cytochrome P450 proteins, were among the most highly regulated genes.
The GO terms enriched in the set of upregulated genes at 24 and 72 hpi include particular terms such as “chitinase activity”, “chitin catabolic process”, “defense response” and “response to biotic stimulus”(Fig. 3). Consistent with the drastic changes in the transcriptome, the GO term “transcription factor activity, sequence-specific DNA binding” was also overrepresented in the upregulated sequences.
In addition to these upregulated terms, other GO terms were enriched in the sets of significantly downregulated genes, such as terms related to auxin signaling, such as “auxin efflux”, “auxin-activated signaling pathway”, “cellular response to auxin stimulus” and “response to auxin”, which were enriched at 24 hpi for D. rosae and at 72 hpi for P. pannosa.
In addition, GO terms representing photosynthesis-related (“chlorophyll binding”, “photosynthesis, light harvesting”) and cell wall organization-related mechanisms (“cellular polysaccharide catabolic process”, “pectin metabolic process”) were overrepresented in the gene sets for both pathogens. In addition, walls are thin 1 (WAT1) gene with a general low expression level, which is also found in the control samples, was downregulated under both types of infections.
Pathogen-related (PR) genes
Different PR genes were strongly upregulated in rose leaves during the interactions with both pathogens (Fig. 4). In particular, many PR10 genes and major allergens (Pru av1 and Pru ar1) showed very high levels of upregulation. Of the 39 major allergens found in the genome of ‘Old Blush’, 29 were highly upregulated in response to D. rosae, and the majority of these were already differentially expressed at 24 hpi. Of these, 15 were also significantly upregulated in response to P. pannosa, and 8 were already upregulated at 24 hpi. In addition to PR10, the upregulation of the chitinases, which belong to the PR3 and PR4 classes, was observed in both pathosystems. Eleven chitinases were upregulated in response to D. rosae, and some were more highly upregulated at 24 hpi than at 72 hpi. Six were also upregulated in response to P. pannosa, and four of these were already expressed at 24 hpi. In addition, six genes only annotated as “pathogenesis-related family protein” were induced in response to D. rosae and five in response to P. pannosa.
Non-race specific disease resistance protein 1 (NDR1), which acts as a downstream regulator of resistance (R) protein-derived signalling, was interestingly significantly downregulated at 24 hpi in the interaction with D. rosae and at 72 hpi for P. pannosa.
Phytohormones
Supporting the GO enrichment related to auxin signaling within the downregulated gene sets, a transcriptional downregulation of single auxin-induced/responsive genes (AUX/IAA/ARG) could be observed. In detail, 14 genes were expressed at a lower level at 24 hpi and four at 72 hpi in the leaves inoculated with D. rosae and nine at 72 hpi for P. pannosa compared to the control leaves.
In both pathosystems, ethylene-related genes also showed significant regulation; however, they exhibit both up- and down-regulations (Fig. 4). One gene encoding the ethylene-responsive transcription factor (TF) ERF109 was downregulated, while three ABR1-likes were upregulated for interaction with D. rosae at 0 hpi. At 24 hpi six genes encoding for 1-aminocyclopropane-1-carboxylate oxidase 1 homologs (ACO1), and each one ACO4 and ACO5 gene were highly upregulated. In addition, one ERF2, three ABR1s and two ERF113 genes were induced. In contrast, eight ethylene-responsive TF genes (ERFs, ANT and SHINE2) and one ACO gene were expressed at a lower level than in the control plants. A similar scenario emerged after 72 h.
Rose leaves inoculated with P. pannosa also showed transcriptional changes in ethylene-related genes, too. Five ethylene responsive transcription factors were already downregulated at 0 hpi. At 72 hpi, five ethylene-responsive TFs were downregulated, which were different from the others downregulated at 0 hpi. In addition, similarly to an infection with D. rosae, six ethylene responsive TFs were upregulated, including ERF1B, ERF113 and ABR1. At 24 hpi, no ethylene-related genes were significantly differentially expressed.
Transcription factors
Inoculation with both P. pannosa and D. rosae led to a regulation of several WRKY transcription factors (Fig. 4); WRKY31, WRKY47, WRKY48, WRKY61, WRKY 71 and WRKY75 were upregulated. During infection with P. pannosa, upregulation could only be observed after 72 h, but in infections with D. rosae, the majority of the genes were upregulated at both 24 and 72 hpi.
The basic helix-loop-helix (bHLH) transcription factor MYC2 is downregulated in both interactions at 72 hpi and with D. rosae at 24 hpi. The downregulation is tenfold during D. rosae and almost fourfold during P. pannosa infection after 72 h. In addition, one MYC3 gene is also downregulated at 72 hpi. In contrast MYB108 is upregulated at the same time points.
The heat shock factors (Hsf) 4 (also known as HsfB1) and HsfB3 were found to be upregulated in response to D. rosae at 24 hpi and 72 hpi and in response to P. pannosa at 72 hpi.
Two C2H2-type zinc finger family proteins were downregulated in response to both pathogens. In addition, only two are downregulated in response to P. pannosa, and only one was downregulated in response to D. rosae (Fig. 4). In addition, two different zinc finger (CCCH type) helicase family proteins were downregulated in response to either P. pannosa or D. rosae.
Sugar transporters
A number of genes related to the transport of sugars were differentially expressed in roses during pathogen attack. Leaves inoculated with both pathogens upregulated a number of sugar transporters, such as ERD6. During the interaction with D. rosae two hexose carrier 6 (HEX6) genes, a homolog of sugar transporter 13 and one sugar phosphate/phosphate translocator were also upregulated, while other homologues of the same genes are downregulated in both leading to a contradictory scenario.
Response to P. pannosa compared to the response to D. rosae
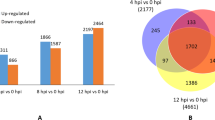
Based on the comparison of the responses of roses to both pathogens (Fig. 5), the majority of the significantly regulated genes were specific to the reaction to one of the pathogens. Only a smaller number of the genes was regulated in response to both pathogens. One of the major differences between the two responses is the time point when the genes were regulated. During the interaction with D. rosae, many of the responses occurred at 24 hpi. In contrast, almost no change in the gene expression was visible in the interaction with P. pannosa at 24 hpi (Fig. 5). In the comparison of the later responses at 72 hpi, it is noticeable that only approximately 25% of the significantly upregulated genes and 21.6% of the significantly downregulated genes were identical between the different pathogen interaction systems. The remaining genes were exclusively regulated in response to only one pathogen.

Venn diagram of the quantitative comparison of the significantly regulated genes in response to inoculations with D. rosae and P. pannosa. Downregulated genes are shown in “A” and upregulated genes in “B” each for 24 and 72 hpi
Specific response of rose to P. pannosa infection
In total, 1450 genes (650 up- and 800 downregulated) were differentially expressed (Fig. 6). The majority of these genes, representing approximately 80% of the up- and downregulated genes, were differentially expressed at 72 hpi. The smallest number of differentially expressed genes was detected at 24 hpi. The majority of the up- or downregulated genes at 24 hpi were also regulated at 72 hpi. Directly after the inoculation (0 hpi), 131 genes were upregulated and 142 genes were downregulated in the inoculated leaves compared to the control leaves. However, only a few of those changes were also observed at the other time points. In addition, several GO terms were enriched in the gene sets at the beginning of the experiment (0 hpi), in contrast to the D. rosae infections.

Venn diagram of significantly regulated genes after P. pannosa inoculation. Genes that were significantly up- (A) and downregulated (B) compared to the control samples at different time points (0, 24, 72 hpi) are shown
In addition to the mentioned common GO terms related to photosynthesis, the terms “photosynthetic membrane”, “photosynthesis”, “photosystem II” and “response to light stimulus” were enriched in the set of downregulated genes.
The majority of the significantly downregulated genes associated with photosynthesis encode chlorophyll a/b binding proteins (cab), structural components of photosystem I (subunit l, K, N) and a PsbP gene of photosystem II. In addition, four genes of different steps in chlorophyll biosynthesis were downregulated, including glutamyl-tRNA reductase (HEMA1), a subunit of magnesium-chelatase (CHIH), geranylgeranyl diphosphate reductase (CHLP) and a chlorophyllase (CLH).
Also, several genes related to cell-wall organization were specifically downregulated in response to P. pannosa, including the genes encoding six expansins, three trichome birefringence-like proteins (TBL), two fasciclin-like arabinogalactan (FLA) proteins and two COBRA-like genes and one additional walls are thin 1-like (WAT1) gene (Fig. 4). One cellulose synthase 4 gene, three genes encoding pectin lyases, two xyloglucan endotransglucosylase genes and some genes of the lignin biosynthesis pathway were downregulated.
Furthermore, the heat shock protein (Hsp) 90.1 is specifically upregulated in response to P. pannosa. In contrast to the infection with D. rosae, no other PR genes or other genes related to a defense response specific to the interaction of roses to P. pannosa could be identified.
Specific response of rose to D. rosae infection
Almost 2000 genes were changed in their expression specifically in response to D. rosae, and the majority (1158) of these were upregulated with only 828 that were downregulated (Fig. 7). Less than 100 genes were considered differentially expressed directly after inoculation at 0 hpi. Approximately 80% of both the up- or downregulated genes were observed at 24 hpi. The majority of the upregulated genes and many of the downregulated genes show a similar regulation pattern at 72 hpi.

Venn diagram of significantly regulated genes after D. rosae inoculation. Genes, that were significantly up- (A) and downregulated (B) compared to the control samples at different time points (0, 24, 72 hpi) are shown
The majority of the overrepresented GO terms were the same for both 24 and 72 hpi (Fig. 3, Supp. 3). In the reaction to D. rosae, GO terms related to the defense response (e.g., “chitinase activity”, “defense response”, “response to biotic stimulus”, “response to fungus”) were highly enriched in the set of upregulated genes. In addition, GO terms such as “calcium ion binding”, “kinase activity” or “reactive oxygen species metabolic process”, which are involved in signaling processes, were also significantly overrepresented, as well as the GO terms representing secondary metabolism, especially those related to the phenylpropanoid and flavonoid pathways (“caffeate O-methyltransferase activity”, “flavonoid metabolic process”, “phenylpropanoid metabolic process”, and “secondary metabolite biosynthetic process”).
Remarkably, genes responding to the salicylic acid (SA)-mediated signaling pathway were only upregulated in the samples inoculated with D. rosae, including typical marker genes, such as phytoalexin deficient 4 (PAD4) and PR1 and PR5s (Derksen et al. 2013). In particular, three PR5 genes (thaumatin) were highly upregulated at 24 and 72 hpi, and one PR1 gene was among the most highly upregulated gene (Fig. 4). The upregulation of the PR1 gene was also observed in the qPCR validation. In addition, six senescence-associated carboxylesterase 101 (SAGs 101) genes were upregulated after 72 hpi.
The phenylpropanoid and the flavonoid pathways seemed to also be specifically affected. Figure 8 displays the primary steps of the flavonoid synthesis pathway and the significantly regulated genes. It can be seen that genes in almost all the steps were upregulated. In particular, the genes encoding for chalcone isomerases (CHI), dihydroflavonol 4-reductase (DFR) and flavonol synthase (FLS) were strongly upregulated at both time points, indicating that the synthesis of flavonols was induced. In addition, the pathway leading to the synthesis of proanthocyanidins (DFR, LDOX, ANR) was upregulated. The only downregulated genes in this interaction were two genes encoding UDPG-flavonoid glucosyl transferase (UFGT). This is a key enzyme in the synthesis of anthocyanins. In contrast to the reaction of PC to D. rosae, the reaction of PC to P. pannosa displayed a different gene regulation pattern. There were significantly upregulated genes, such as one of the flavanone 3-hydroxylase (F3H) genes, one FLS gene and one leucoanthocyanidin dioxygenase (LDOX) gene. In particular, enzymes acting in the first steps of the flavonoid synthesis pathway (PAL, 4CL, CHS and CHI) seemed to remain unaffected or were even downregulated. In addition, the anthocyanidin reductase (ANR) gene leading to the synthesis of proanthocyanidins was significantly downregulated.

Overview of the regulation of the flavonoid biosynthesis pathway. The primary steps in the flavonoid biosynthesis pathway and the regulation of genes in the pathway in response to the two pathogens are displayed. Each quadrat represents a gene. Significant changes in gene expression are indicated by thicker lines. Enzyme names are abbreviated as follows: PAL phenylalanine ammonia-lyase, C4H cinnamate-4-hydroxylase, 4CL 4-coumaroyl-CoA-ligase, CHS chalcone synthase, CHI chalcone isomerase, F3H flavanone 3-hydroxylase, DFR dihydroflavonol 4-reductase, LDOX leucoanthocyanidin dioxygenase, UFGT UDPG-flavonoid glucosyl transferase, FLS flavonol synthase, LAR leucoanthocyanidin reductase, and ANR anthocyanidin reductase
Many genes encoding key enzymes in the phenylpropanoid pathway were upregulated in most of the cases at both time points, 24 hpi and 72 hpi, in response to D. rosae, with p-coumarate 3-hydroxylase (C3H), caffeic acid O-methyltransferase (COMT), caffeoyl-CoA O-methyltransferase (CCoAOMT) and cinnamoyl-CoA reductase (CCR) showing the strongest upregulation (Fig. 9). Of particular interest are CCoAOMT and COMT, because they are involved in the synthesis of G- and S-lignin, which are the primary components of lignin in dicots (Vanholme et al. 2010). In addition to the biosynthic genes, several peroxidases and laccases, which are involved in the cross-linking of lignin monomers, were upregulated. This pathway seemed to be less affected in response to P. pannosa than in response to D. rosae. One CCR gene and a COMT gene showed higher levels of upregulation, but some key factors were also significantly downregulated. Notably, two genes encoding cinnamyl alcohol dehydrogenase (CAD) were significantly downregulated. This enzyme catalyzes one of the later steps in the synthesis of all three types of lignin monomers.

Overview of the regulation of the phenylpropanoid biosynthesis pathway. The primary steps in the phenylpropanoid biosynthesis pathway and the regulation of the genes in the pathway in response to the two pathogens are displayed. Significant changes in gene expression are marked with thicker lines. Enzyme names are abbreviated as follows: PAL phenylalanine ammonia-lyase, C4H cinnamate-4-hydroxylase, 4CL 4-coumaroyl-CoA-ligase, HCT hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase, C3H p-coumarate 3-hydroxylase, CCoAOMT caffeoyl-CoA O-methyltransferase, CCR cinnamoyl-CoA reductase, F5H ferulate 5-hydroxylase, COMT caffeic acid O-methyltransferase, and CAD cinnamyl alcohol dehydrogenase
Among the exclusively regulated transcription factors WRKY 27, 40, 48 50 and 51 that are all known to increase resistance in other pathosystems were upregulated at 72 hpi, while WRKY 53 was strongly downregulated at both 24 hpi (14.5-fold) and at 72 hpi (10.5-fold). WRKY 33 was also downregulated at both time points (9.3 and 5.6-fold), while it was only regulated 2.2-fold in the interaction with P. pannosa at 72 hpi.
In addition, six heat shock proteins (17.6 kDa class II heat shock protein, heat shock cognate protein 70-1, DNAJ heat shock family protein, heat shock protein 101 and two HSP20-like chaperones superfamily proteins) were only downregulated in response to D. rosae (Fig. 4).
Discussion
Fungal phytopathogens successfully colonize their hosts by manipulating the host defense mechanisms in different ways. To characterize the specific infection strategies based on the resulting transcriptomic changes in the early stages of infection with the hemibiotrophic D. rosae and the obligate biotrophic P. pannosa, we used next-generation sequencing based on the MACE (massive analysis of cDNA ends) approach.
A notable advantage of MACE compared to conventional RNA-Seq technology is that only one read is produced for each cDNA molecule, which allows a precise quantification, especially transcripts expressed at low levels. The sequencing (6–30 million reads per library) and mapping (76.3–92.9%) results (Supplementary Table 1) are comparable to those of other studies that applied this technique, e.g., Hradilová et al. (2017), applied the MACE technique to pea with an output of 8 to 15 million reads per library, and 12.3– 21.7 million reads per library were generated from the RNA of apple roots (Weiß et al. 2017).
The majority of significantly differentially expressed genes indicated by the MACE technique could be validated using RT-qPCR. Factoring that all three biological repeat experiments in the MACE analysis were derived from independent inoculation experiments and that the validation included three additional independent inoculation experiments, which introduced more biological and technical variability then conventional repeats, the correlation coefficient of 0.82 is surprisingly high. However, this might be due to the stringent conditions we applied to identify the differentially expressed genes. However, the MACE data might still indicate effects, which have been caused by outliers in one biological repeat. Thus, for a meaningful interpretation of the data, all the repeats have to be considered separately, not just the mean fold changes.
The selected infection method using detached leaves offers particularly high standardization and control options. Furthermore, in past experiments the detached leaf assay was always highly correlated to experiments conducted on potted resistant and susceptible plants. However, we cannot exclude effects of the experimental procedure on the overall gene expression levels in our analyses in both controls and inoculated samples.
Common response to both pathogens
The transcriptomic changes of the susceptible rose variety PC in response to both D. rosae and P. pannosa indicate a general pathogen response, characterized by the upregulation of PR10 genes, major allergens (Pru av1, Pru ar1, and Mal d 1) that belong to this class of PR-genes (Liu and Ekramoddoullah 2006) and chitinases. In addition, the GO terms enriched in the set of upregulated genes at 24 and 72 hpi indicating that a defense response to the pathogens was in progress.
The upregulation of the PR genes is a strong indicator of pathogen recognition (Bowles 1990). In particular, PR10 genes seem to play a central role in the response of the Rosaceae. Studies of the Rosaceae model plant Fragaria infected by pathogens with different lifestyles, such as the necrotrophic fungus Botrytis cinerea (González et al. 2013), the obligate biotrophic powdery mildew fungus Podosphaera aphanis (Jambagi and Dunwell 2015) and the hemibiotrophic oomycete Phytophthora cactorum (Toljamo et al. 2016), showed a strong upregulation of PR10 genes or major allergens. Studies on apple (Malus) infected with Venturia inaequalis (apple scab) (Poupard et al. 2003; Cova et al. 2017) and treated with fungal elicitors (Pühringer et al. 2000) also showed an upregulation of the PR10 genes. The biological functions of the PR10 gene family are not completely known, but, among other activities, antifungal activity has been described (Flores et al. 2002). The role of chitinases in the defense response of plants has been analyzed extensively in different systems. They block hyphal growth, trigger other defense mechanisms through the release of elicitors and play a role in the so-called “hypersensitive response” (Grover 2012).
Consistent with the upregulation of the PR genes, the transcriptional activation of other defense related genes was observed as well. Heat shock factor (Hsf) 4 (also known as HsfB1) and HsfB3 were observed to be upregulated in response to both pathogens. Another example is the upregulation of MYB108, a transcription factor whose expression is induced through Bortrytis infection (Mengiste et al. 2003).
These quick and conserved responses linked to both pathosystems might indicate the involvement of a PTI reaction elicited by chitin or the penetration of the cuticle. Since we considered compatible interactions in this study, it can be concluded that the two fungi were indeed detected, but reactions initiated were not sufficient for resistance. For resistance, both the right timing and the level of defense are of paramount importance. A susceptible interaction may be caused by an inadequate response of the plant or even by direct manipulation of the defense mechanisms by the pathogen. In the next section, we discuss the present regulations against the background of a successful colonization and seek to highlight origins for the increase in susceptibility.
For example, MYC2 and MYC3 are key factors involved in defense reactions linked to the jasmonic acid pathway (Kazan and Manners 2013), and the triple mutant myc2 myc3 myc4 cannot perform several JA-mediated defense responses against bacteria and insects (Fernández-Calvo et al. 2011). Their observed downregulation at 72 hpi is consistent with an unregulated JA-mediated pathogen response. In addition, MYC2 seems to play a role during flavonoid biosynthesis and the JA pathway in the more closely related Malus × domestica (An et al. 2016).
Zinc finger proteins comprise a large and abundant family of proteins that function in many aspects of plant growth and development as well as in defense response (Yu et al. 2016; Ciftci-Yilmaz and Mittler 2008). The downregulation of the C2H2-type zinc finger family proteins and zinc finger (CCCH type) helicase family protein in response to both pathogens is consistent with the analyzed susceptible interaction. In addition, they provide starting points for factors that support a successful pathogen attack.
Plant hormones are an important part of the signaling cascade in response to biotic stress (e.g. Yang et al. 2015). A downregulated or disturbed auxin pathway may enhance susceptibility, as in the case for Arabidopsis mutants and their defense response to necrotrophic fungi (Llorente et al. 2008). Bouzroud et al. (2018) describe auxin responsive factors (ARFs) as part of biotic and abiotic stress signaling in Solanum. The role of ethylene as a defense regulator has been well established, primarily due to its function as a signaling molecule in the defense against necrotrophic pathogens in combination with jasmonic acid. Although there are major contradictions in the ethylene-related expression patterns, which could indicate a disturbed signal transmission, our observation of the upregulated ethylene responsive ABR1 genes indicates a negative impact on the abscisic acid (ABA) signaling pathway which might also contribute to susceptibility (Pandey et al. 2005).
NDR1, which interacts with RPM1 Interacting Protein4 (RIN4) to control the activation of disease resistance signaling by the CC-NB-LRR class of resistance proteins, was downregulated in both pathosystems. It was shown that NDR1 in combination with RIN4 are needed to activate a number of disease resistance pathways against bacterial and fungal pathogens in Arabidopsis (Century et al. 1995; Day et al. 2006). Two NDR1-like sequences identified in soybean (GmNDR1a, b) were also shown to be required for resistance to Pseudomonas syringae (Selote et al. 2013).
The network of WRKY transcription factors is involved in many stress responses, and some are key factors in the defense response (Phukan et al. 2016). The downregulated WRKY33, although only weakly downregulated in the interaction with P. pannosa, provides further evidence of a repression of the defense responses. This was reported several times, including such pathogens as Botrytis and Alternaria as well as Pseudomonas syringae in Arabidopsis and Fusarium in tomato (Birkenbihl and Somssich 2011; Buscaill and Rivas 2014; Garner et al. 2016; Rushton et al. 2010; Aamir et al. 2018). WRKY75 is a particularly interesting transcription factor, because at 72 hpi it was exclusively expressed in the inoculated samples. It is notable that this transcription factor was initially described only in the context of phosphate stress (Devaiah et al. 2007). However, descriptions connected with defense reaction could also be found in recent years (Encinas-Villarejo et al. 2009; Jiang et al. 2014).
Changes in the transcriptome specific to the interaction with P. pannosa
In contrast to the D. rosae infection the upregulation of PR10 and chitinase genes, and thus the reaction to P. pannosa occurred only at 72 hpi instead of 24 hpi. In addition, the expression levels were often lower and no specific group of genes related to common defense functions was found to be upregulated exclusively in the reaction to P. pannosa. One of the reasons for the low number of significantly regulated genes at 24 hpi and the decreased expression levels could be that the inoculation density was lower for P. pannosa than for D. rosae. In addition, the infection zone was restricted to the epidermis with fewer penetration sites. Either fewer transcriptional changes were initiated as response or genes with low expression remained below our stringent criteria to select the DEGs. In addition to differences in the upregulation of the PR genes a gene for a heat shock protein (Hsp) 90.1 is upregulated specifically in response to P. pannosa. In wheat, Hsp90 genes were found to be essential for resistance to the stripe rust fungus (Wang et al. 2011, 2015).
A specific reaction to P. pannosa infection is that the genes involved in the light reaction of photosynthesis were downregulated more strongly than those in response to D. rosae, which is surprising, considering that P. pannosa only infects epidermal cells, which are not photosynthetically active, while D. rosae infects also infects the cell layers below the epidermis. However, the downregulation of the photosynthic genes is often observed in plant-pathogen interactions (e.g., Cremer et al. 2013; Milli et al. 2012; Balan et al. 2018). This reaction may be explained by a pathogen-induced source-sink transition of infected leaf tissues (Berger et al. 2007). In addition to their role to nourish pathogens, sugar transporters and sugar signaling have a large impact on the defense response of plants as described by Bezrutczyk et al. (2018). Interestingly, Morkunas and Ratajczak (2014) propose an increased resistance due to the high sugar levels in different pathosystems. For rice, it is well described that defective OsSWEET13 or 14 genes, bidirectional sugar transporters, lead to the resistance of rice against Xanthomonas oryzae pv. oryzae (e.g., Zhou et al. 2015; Blanvillain-Baufumé et al. 2017). In rose leaves infected with P. pannosa sugar transporters were upregulated, a first indication of the manipulation of the host metabolism by the fungus. However, further studies are needed to determine the influence of P. pannosa on photosynthesis and sugar metabolism.
Another process strongly regulated in response to P. pannosa and contributing to susceptibility was the cell wall modification process. Different factors, such as expansins, TBLs or FLAs, were downregulated in response to the pathogen. They are known to be involved in modification processes such as cell wall loosening, plant cell expansion and secondary cell wall formation (Cosgrove 2000; Schindelman et al. 2001; Johnson et al. 2003; Van Sandt et al. 2007; Bischoff et al. 2010). The WAT1 genes might be of particular interest. Two WAT1 gene homologs were expressed in our samples, and one was downregulated with a generally low expression level also in the control leaves, while the other, which had a much higher expression level, was exclusively downregulated in response to P. pannosa at 72 hpi. WAT1 was identified as an essential factor for secondary cell wall formation by loss-of-function mutations in Arabidopsis (Ranocha et al. 2010). In addition, wat1 mutants showed a dwarf phenotype and downregulation of almost all the genes in the lignin biosynthetic pathway. The downregulation of the highly expressed WAT1 gene exclusively observed in response to P. pannosa might be an important factor leading to the observed differences in cell wall formation and the lignin pathway. Since it is not regulated in the interaction with D. rosae it constitutes one of the differences in the suppression of the host response that contrasts with the infection strategy of the two pathogens. Interestingly, wat1 mutants also showed increased tissue-specific resistance to vascular pathogens (Denancé et al. 2013).
Many obligate biotrophs and hemibiotrophs induce host responses via the SA pathway in which one of the key features is the transcriptional activation of the PR1 genes. We observed the downregulation of a PR1 gene after P. pannosa infection at both time points (Supplementary Table 4) and together with a lack of regulation of other the SA related genes, such as EDS1 and PAD4 homologs, this points to a suppression of part of the SA-based defense in the P. pannosa-rose pathosystem (van Loon et al. 2006; Derksen et al. 2013).
Surprisingly, we observed a large number of DEGs at the starting point at 0 hpi. This might be a temporary effect due to our inoculation method. After 24 h, the number of DEGs dropped to a similar level at the start point of the D. rosae inoculation before dramatically rising again. Of the total of 131 upregulated genes only one (a putative RNA polymerase) was still upregulated at 24 hpi and of the 142 downregulated, 16 still showed a significantly lower expression. During P. pannosa inoculation (see “Material and Methods”) the detached rose leaves lay for 30–60 min in an open inoculation box, which may have caused a generalized stress response and, in particular, osmotic stress. For example, the two aquaporin genes TIP1-2 and PIP2-1 were upregulated compared to the control, and PIP2-1 was upregulated in both the control and D. rosae-inoculated leaves.
Changes in the transcriptome specific to the interaction with D. rosae
The transcriptomic changes specific to the interaction with D. rosae include the upregulation of many genes encoding enzymes in the lignin biosynthetic and flavonoid pathways. The synthesis of lignin or lignin-like phenolic polymers is a common phenomenon in response to pathogens. For instance, Chinese cabbage plants infected with the necrotrophic bacterium Erwinia carotovora accumulated high levels of lignin monomers, and 12 genes involved in lignin biosynthesis were upregulated (Zhang et al. 2007). Infection with the fungus Sclerotinia sclerotium led to the lignification of Camelina sativa cells and was correlated with upregulation of the CCR genes (Eynck et al. 2012). In addition, the defense response of the model plant Arabidopsis thaliana infected with Pseudomonas syringae was accompanied by the upregulation of the CAD genes. Loss-of-function mutations of these genes resulted in plants with a reduced resistance to the pathogen (Tronchet et al. 2010). Lignin has different functions in plant defense. As part of the secondary cell wall, it provides a physical barrier against the entry of the pathogen. Simultaneously, it prevents the spread of pathogen toxins and enzymes into the neighboring cells and the transfer of water and nutrients from the host to the pathogen. In addition, cell wall components can have signaling functions in defense (Miedes et al. 2014).
In addition, to the lignin biosynthetic pathway, genes encoding almost all enzymes of the flavonoid pathway were upregulated specifically in response to D. rosae. Products of the flavonoid pathway have various functions in plants. For example, they act as chemical messengers that interact with insects and microbes and function as pigments to attract pollinators or protect against UV light. More importantly, many phytoalexins are synthesized by this pathway (Piasecka et al. 2015; Falcone Ferreyra et al. 2012). In Fragaria, catechin and catechin-derived proanthocyanidins have been shown to be involved in the defense response to Alternaria alternata and B. cinerea in infected leaves and fruits, respectively (Yamamoto et al. 2000; Puhl and Treutter 2008). In addition, a recent transcriptomic study of roots infected with P. cactorum showed an upregulation of the flavonoid pathway genes leading to these products (Toljamo et al. 2016). Interestingly, in our data, FLS genes were highly upregulated, indicating that the synthesis of flavonols is more important in the response to D. rosae than in the response to P. pannosa. In plants, flavonols function as antioxidants during high light conditions and as detoxifying agents against reactive oxygen species (ROS), which might explain their role in plant defense (Pollastri and Tattini 2011). Metabolic analyses focusing on this group of metabolites might be needed to confirm the induction of this pathway in the rose–D. rosae interaction.
Different SA response genes were exclusively upregulated in response to D. rosae. PAD4, EDS1 and SAG101 cooperate to stimulate the production of SA and are essential for SA-mediated pathogen responses (Zhou et al. 1998; Feys et al. 2005; Rietz et al. 2011), which are typically characterized by the upregulation of the PR1, PR2 and PR5 genes. PR1 and different thaumatins (PR5) were also exclusively upregulated in response to D. rosae, except for one PR5 paralog that was also upregulated in response to P. pannosa. This could be an indication that the additional D. rosae-specific stress responses are regulated by the SA-mediated signaling pathway, which is either suppressed during the interaction with P. pannosa or not upregulated in the P. pannosa pathosystem because D. rosae is recognized differently and signaling and desired resistance is attempted via other pathways. Interestingly, it is described, e.g. for tomato (Rahman et al. 2012), that the SA pathways may promote necrotrophic disease development, possibly by antagonizing JA. That could also be helpful for the later stages of the D. rosae infection. In addition, Cui et al. (2018) proposed that EDS1/PAD4 suppresses MYC2 and therefore the JA pathway, which could explain the stronger downregulation of the leaves infected with D. rosae that was observed.
All of these reactions could be based on a partial ETI reaction, in which specific effectors of D. rosae are detected. We know from previous experiments that there are more than 20 genes in the rose genome that are similar to the resistance-mediating muRdr1A gene of the Rdr1-locus (Terefe-Ayana et al. 2011) but do not mediate a full resistance. However, they might function to activate a partial ETI reaction, leading to the differences in the reaction of roses to the pathogens observed.
An indication of the specific interference of black spot with host resistance is the strong downregulation of WRKY33 and 53 known to have a function in resistance (Birkenbihl and Somssich 2011). In particular the downregulation of WRKY 33 and WRKY 53 transcription factors related to resistance and the upregulation of WRKYs 27, 40, 50 and 51 as repressors of resistance is interesting, since it indicates major differences in how both pathogens interfere with the SA-inducible host resistance response.
In addition, the specific downregulation of six heat shock proteins in response to D. rosae (17.6 kDa class II heat shock protein, heat shock cognate protein 70-1, DNAJ heat shock family protein, heat shock protein 101 and two HSP20-like chaperones superfamily proteins) can be reconciled with the current susceptible interaction, since their regulation might may decrease the strength or the timing of the defense response (Park and Seo 2015; Lee et al. 2012).
Conclusions
With this first analysis of the rose defense transcriptome, we showed contrasting responses of the host to two fungal pathogens, the hemibiotrophic D. rosae and the biotrophic P. pannosa displaying different lifestyles. In addition to a common response to both pathogens, characterized by an upregulation of the PR10 genes and chitinases, processes such as photosynthesis and cell wall modification were primarily downregulated in response to P. pannosa, while the secondary metabolism in form of the phenylpropanoid and flavonoid pathway was primarily upregulated in response to D. rosae. Surprisingly, PR1 and components of the SA-pathway were exclusively upregulated in response to the hemibiotrophic D. rosae and not, as often observed in other biotrophic systems, also in the interaction with P. pannosa. In contrast, the transcriptional regulation of some factors known to interfere with host resistances was distinct for both pathogens. This information is an important first step to understand the response to both rose pathogens and revealed many processes, which merit analysis in more detail.
References
Aamir M, Singh VK, Dubey MK, Kashyap SP, Zehra A, Upadhyay RS, Singh S (2018) Structural and functional dissection of differentially expressed tomato WRKY transcripts in host defense response against the vascular wilt pathogen (Fusarium oxysporum f. sp. lycopersici). PLoS ONE 13(4):e0193922. https://doi.org/10.1371/journal.pone.0193922
An JP, Li HH, Song LQ, Su L, Liu X, You CX, Wang XF, Hoa YJ (2016) The molecular cloning and functional characterization of MdMYC2, a bHLH transcription factor in apple. Plant Physiol Biochem 108:24–31. https://doi.org/10.1016/j.plaphy.2016.06.032
Aronescu A (1934) Diplocarpon rosae: from spore germination to haustorium formation. Bull Torr Bot Club 61:291–329. https://doi.org/10.2307/2480882
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. https://doi.org/10.1038/75556
Balan B, Marra FP, Caruso T, Martinelli F (2018) Transcriptomic responses to biotic stresses in Malus x domestica: a meta-analysis study. Sci Rep 8(1):1970. https://doi.org/10.1038/s41598-018-19348-4
Bent AF, Mackey D (2007) Elicitors, effectors, and R genes: the new paradigm and a lifetime supply of questions. Annu Rev Phytopathol 45:399–436. https://doi.org/10.1146/annurev.phyto.45.062806.094427
Berger S, Sinha AK, Roitsch T (2007) Plant physiology meets phytopathology: plant primary metabolism and plant-pathogen interactions. J Exp Bot 58:4019–4026. https://doi.org/10.1093/jxb/erm298
Bezrutczyk M, Yang J, Eom JS, Prior M, Sosso D, Hartwig et al (2018) Sugar flux and signaling in plant-microbe interactions. Plant J 93(4):675–685. https://doi.org/10.1111/tpj.13775
Birkenbihl RP, Somssich IE (2011) Transcriptional plant responses critical for resistance towards necrotrophic pathogens. Front Plant Sci 2:76. https://doi.org/10.3389/fpls.2011.00076
Bischoff V, Nita S, Neumetzler L, Schindelasch D, Urbain A, Eshed R et al (2010) TRICHOME BIREFRINGENCE and its homolog AT5G01360 encode plant-specific DUF231 proteins required for cellulose biosynthesis in Arabidopsis. Plant Physiol 153(2):590–602. https://doi.org/10.1104/pp.110.153320
Blanvillain-Baufumé S, Reschke M, Solé M, Auguy F, Doucoure H, Szurek B et al (2017) Targeted promoter editing for rice resistance to Xanthomonas oryzae pv. oryzae reveals differential activities for SWEET14-inducing TAL effectors. Plant Biotechnol J 15(3):306–317. https://doi.org/10.1111/pbi.12613
Borges AF, Ferreira RB, Monteiro S (2013) Transcriptomic changes following the compatible interaction Vitis vinifera-Erysiphe necator. Paving the way towards an enantioselective role in plant defence modulation. Plant Physiol Biochem 68:71–80. https://doi.org/10.1016/j.plaphy.2013.03.024
Bouzroud S, Gouiaa S, Hu N, Bernadac A, Mila I, Bendaou N et al (2018) Auxin response factors (ARFs) are potential mediators of auxin action in tomato response to biotic and abiotic stress (Solanum lycopersicum). PLoS ONE 13(2):e0193517. https://doi.org/10.1371/journal.pone.0193517
Bowles DJ (1990) Defense-related proteins in higher plants. Annu Rev Biochem 59:873–907. https://doi.org/10.1146/annurev.bi.59.070190.004301
Buscaill P, Rivas S (2014) Transcriptional control of plant defence responses. Curr Opin Plant Biol 20:35–46. https://doi.org/10.1016/j.pbi.2014.04.004
Century KS, Holub EB, Staskawicz BJ (1995) NDR1, a locus of Arabidopsis thaliana that is required for disease resistance to both a bacterial and a fungal pathogen. Proc Natl Acad Sci USA 92:6597–6601. https://doi.org/10.1073/pnas.92.14.6597
Ciftci-Yilmaz S, Mittler R (2008) The zinc finger network of plants. Cell Mol Life Sci 65(7–8):1150–1160. https://doi.org/10.1007/s00018-007-7473-4
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. https://doi.org/10.1093/bioinformatics/bti610
Cosgrove DJ (2000) Loosening of plant cell walls by expansins. Nature 407:321–326. https://doi.org/10.1038/35030000
Cova V, Paris R, Toller C, Patocchi A, Velasco R, Komjanc M (2017) Apple genes involved in the response to Venturia inaequalis and salicylic acid treatment. Sci Hortic 226:157–172. https://doi.org/10.1016/j.scienta.2017.08.043
Coyier DL (1983) Control of rose powdery mildew in the greenhouse and field. Plant Dis 67:919–923
Cui H, Qui J, Zhou Y, Bhandari DD, Zhao C, Bautor J, Parker JE (2018) Antagonism of transcription factor MYC2 by EDS1/PAD4 complexes bolsters salicylic acid defense in arabidopsis effector-triggered immunity. Mol Plant 11(8):1053–1066. https://doi.org/10.1016/j.molp.2018.05.007
Davies DR (1980) Rapid propagation of roses in vitro. Sci Hortic 13:385–389. https://doi.org/10.1016/0304-4238(80)90097-7
Day B, Dahlbeck D, Staskawicz BJ (2006) NDR1 Interaction with RIN4 mediates the differential activation of multiple disease resistance pathways in Arabidopsis. Plant Cell 18:2782–2791. https://doi.org/10.1105/tpc.106.044693
de Cremer K, Mathys J, Vos C, Froenicke L, Michelmore RW, Cammue, BPA et al (2013) RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis cinerea. Plant Cell Environ 36:1992–2007. https://doi.org/10.1111/pce.12106
Debener T, Byrne DH (2014) Disease resistance breeding in rose: current status and potential of biotechnological tools. Plant Sci 228:107–117. https://doi.org/10.1016/j.plantsci.2014.04.005
Debener T, Drewes-Alvarez R, Rockstroh K (1998) Identification of five physiological races of blackspot, Diplocarpon rosae, Wolf on roses. Plant Breed 1998:267–270. https://doi.org/10.1111/j.1439-0523.1998.tb01937.x
Denancé N, Ranocha P, Oria N, Barlet X, Rivière MP, Yadeta KA et al (2013) Arabidopsis wat1 (walls are thin1)-mediated resistance to the bacterial vascular pathogen, Ralstonia solanacearum, is accompanied by cross-regulation of salicylic acid and tryptophan metabolism. Plant J 73(2):225–239. https://doi.org/10.1111/tpj.12027
Derksen H, Rampitsch C, Daayf F (2013) Signaling cross-talk in plant disease resistance. Plant Sci 207:79–87. https://doi.org/10.1016/j.plantsci.2013.03.004
Devaiah BN, Karthikeyan AS, Raghothama KG (2007) WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol 143(4):1789–1801. https://doi.org/10.1104/pp.106.093971
Dohm A, Ludwig C, Nehring K, Debener T (2001) Somatic embryogenesis in roses. Acta Hortic 547:341–347. https://doi.org/10.17660/ActaHortic.2001.547.40
Encinas-Villarejo S, Maldonado AM, Amil-Ruiz F, de los Santos B, Romero F, Pliego-Alfaro F et al (2009) Evidence for a positive regulatory role of strawberry (Fragaria x ananassa) Fa WRKY1 and Arabidopsis At WRKY75 proteins in resistance. J Exp Bot 60:3043–3065. https://doi.org/10.1093/jxb/erp152
Eynck C, Seguin-Swartz G, Clarke WE, Parkin IA (2012) Monolignol biosynthesis is associated with resistance to Sclerotinia sclerotiorum in Camelina sativa. Mol Plant Pathol 13:887–899. https://doi.org/10.1111/j.1364-3703.2012.00798.x
Falcone Ferreyra ML, Rius SP, Casati P (2012) Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front Plant Sci 3:222. https://doi.org/10.3389/fpls.2012.00222
Fernández-Calvo P, Chini A, Fernández-Barbeo G, Chico JM, Gimenez-Ibanez S, Geerinck J et al (2011) The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 23(2):701–715. https://doi.org/10.1105/tpc.110.080788
Feys BJ, Wiermer M, Bhat RA, Moisan LJ, Medina-Escobar N, Neu C et al (2005) Arabidopsis SENESCENCE-ASSOCIATED GENE101 stabilizes and signals within an ENHANCED DISEASE SUSCEPTIBILITY1 complex in plant innate immunity. Plant Cell 17:2601–2613. https://doi.org/10.1105/tpc.105.033910
Flores T, Alape-Giron A, Flores-Diaz M, Flores HE (2002) Ocatin. A novel tuber storage protein from the Andean tuber crop oca with antibacterial and antifungal activities. Plant Physiol 128:1291–1302. https://doi.org/10.1104/pp.010541
Gachomo EW, Dehne H-W, Steiner U (2006) Microscopic evidence for the hemibiotrophic nature of Diplocarpon rosae, cause of black spot disease of rose. Physiol Mol Plant Pathol 69:86–92. https://doi.org/10.1016/j.pmpp.2007.02.002
Garner CM, Kim SH, Spears BJ, Gassmann W (2016) Express yourself: transcriptional regulation of plant innate immunity. Semin Cell Dev Biol 56:150–162. https://doi.org/10.1016/j.semcdb.2016.05.002
González G, Fuentes L, Moya-León MA, Sandoval C, Herrera R (2013) Characterization of two PR genes from Fragaria chiloensis in response to Botrytis cinerea infection: a comparison with Fragaria x ananassa. Physiol Mol Plant Pathol 82:73–80. https://doi.org/10.1016/j.pmpp.2013.02.001
Grover A (2012) Plant chitinases: genetic diversity and physiological roles. Crit Rev Plant Sci 31:57–73. https://doi.org/10.1080/07352689.2011.616043
Guidarelli M, Carbone F, Mourgues F, Perrotta G, Rosati C, Bertolini P, Baraldi E (2011) Colletotrichum acutatum interactions with unripe and ripe strawberry fruits and differential responses at histological and transcriptional levels. Plant Pathol 60(4):685–697. https://doi.org/10.1111/j.1365-3059.2010.02423.x
Hibrand Saint-Oyant L, Ruttink T, Hamama L, Kirov I, Lakhwani D, Zhou NN et al (2018) A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat Plants. https://doi.org/10.1038/s41477-018-0166-1
Horbach R, Navarro-Quesada AR, Knogge W, Deising HB (2011) When and how to kill a plant cell: infection strategies of plant pathogenic fungi. J Plant Physiol 168:51–62. https://doi.org/10.1016/j.jplph.2010.06.014
Hradilová I, Trněný O, Válková M, Cechová M, Janská A, Prokešová L et al (2017) A combined comparative transcriptomic, metabolomic, and anatomical analyses of two key domestication traits: pod dehiscence and seed dormancy in pea (Pisum sp.). Front Plant Sci 8:542. https://doi.org/10.3389/fpls.2017.00542
Jambagi S, Dunwell JM (2015) Global transcriptome analysis and identification of differentially expressed genes after infection of Fragaria vesca with powdery mildew (Podosphaera aphanis). Transcriptomics 03. https://doi.org/10.4172/2329-8936.1000106
Jiang Y, Duan Y, Yin J, Ye S, Zhu J, Zhang F et al (2014) Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J Exp Bot 65:6629–6644. https://doi.org/10.1093/jxb/eru381
Johnson KL, Jones BJ, Bacic A, Schultz CJ (2003) The fasciclin-like arabinogalactan proteins of arabidopsis. A multigene family of putative cell adhesion molecules. Plant Physiol 133:1911–1925. https://doi.org/10.1104/pp.103.031237
Jones JD, Dangl JL (2006) The plant immune system. Nature 444:323–329. https://doi.org/10.1038/nature05286
Kahl G, Molina C, Rotter B, Juengling R, Frank A, Krezdorn N et al (2012) Reduced representation sequencing of plant stress transcriptomes. J Plant Biochem Biotechnol 21:119–127. https://doi.org/10.1007/s13562-012-0129-y
Kazan K, Manners JM (2013) MYC2: the master in action. Mol Plant 6(3):686–703. https://doi.org/10.1093/mp/sss128
Klie M, Debener T (2011) Identification of superior reference genes for data normalisation of expression studies via quantitative PCR in hybrid roses (Rosa hybrida). BMC Res Notes 4:518. https://doi.org/10.1186/1756-0500-4-518
Kolde R (2018) pheatmap: Pretty Heatmaps. R package version 1.0.10. https://CRAN.R-project.org/package=pheatmap
Lee JH, Yun HS, Kwon C (2012) Molecular communications between plant heat shock responses and disease resistance. Mol Cells 34(2):109–116. https://doi.org/10.1007/s10059-012-0121-3
Linde M, Debener T (2003) Isolation and identification of eight races of powdery mildew of roses (Podosphaera pannosa) (Wallr. Fr.) de Bary and the genetic analysis of the resistance gene Rpp1. Theor Appl Genet 107:256–262. https://doi.org/10.1007/s00122-003-1240-1
Linde M, Shishkoff N (2003) Fungi: powdery mildew. In: Roberts A, Debener T, Gudin S (eds) Encyclopedia of rose science. Elsevier, Academic Press, Oxford, pp 158–165
Liu J-J, Ekramoddoullah AKM (2006) The family 10 of plant pathogenesis-related proteins: their structure, regulation, and function in response to biotic and abiotic stresses. Physiol Mol Plant Pathol 68:3–13. https://doi.org/10.1016/j.pmpp.2006.06.004
Llorente F, Muskett P, Sánchez-Vallet A, López G, Sánchez-Rodríquez C, Jordá L, Parker J, Molina A (2008) Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol Plant 1(3):469–509. https://doi.org/10.1093/mp/ssn025
Mengiste T, Chen X, Salmeron J, Dietrich R (2003) The BOTRYTIS SUSCEPTIBLE1 gene encodes an R2R3MYB transcription factor protein that is required for biotic and abiotic stress responses in Arabidopsis. Plant Cell 15(11):2551–2565. https://doi.org/10.1105/tpc.014167
Menz I, Straube J, Linde M, Debener T (2017) The TNL gene Rdr1 confers broad-spectrum resistance to Diplocarpon rosae. Mol Plant Pathol. https://doi.org/10.1111/mpp.12589
Miedes E, Vanholme R, Boerjan W, Molina A (2014) The role of the secondary cell wall in plant resistance to pathogens. Front Plant Sci 5:358. https://doi.org/10.3389/fpls.2014.00358
Milli A, Cecconi D, Bortesi L, Persi A, Rinalducci S, Zamboni A et al (2012) Proteomic analysis of the compatible interaction between Vitis vinifera and Plasmopara viticola. J Proteom 75:1284–1302. https://doi.org/10.1016/j.jprot.2011.11.006
Morkunas I, Ratajczak L (2014) The role of sugar signaling in plant defense responses against fungal pathogens. Acta Physiol Plant 36:1607–1619. https://doi.org/10.1007/s11738-014-1559-z
Pandey GK, Grant JJ, Cheong YH, Kim BG, Li L, Luan S (2005) ABR1, an APETALA2-domain transcription factor that functions as a repressor of ABA response in Arabidopsis. Plant Physiol 139(3):1185–1193. https://doi.org/10.1104/pp.105.066324
Park CJ, Seo YS (2015) Heat shock proteins: a review of the molecular chaperones for plant immunity. Plant Pathol J 31(4):323–333. https://doi.org/10.5423/PPJ.RW.08.2015.0150
Phukan UJ, Jeena GS, Shukla RK (2016) WRKY transcription factors: molecular regulation and stress responses in plants. Front Plant Sci 7:2645. https://doi.org/10.3389/fpls.2016.00760
Piasecka A, Jedrzejczak-Rey N, Bednarek P (2015) Secondary metabolites in plant innate immunity: conserved function of divergent chemicals. New Phytol 206:948–964. https://doi.org/10.1111/nph.13325
Pollastri S, Tattini M (2011) Flavonols: old compounds for old roles. Ann Bot 108:1225–1233. https://doi.org/10.1093/aob/mcr234
Poupard P, Parisi L, Campion C, Ziadi S, Simoneau P (2003) A wound- and ethephon-inducible PR-10 gene subclass from apple is differentially expressed during infection with a compatible and an incompatible race of Venturia inaequalis. Physiol Mol Plant Pathol 62:3–12. https://doi.org/10.1016/S0885-5765(03)00008-0
Puhl I, Treutter D (2008) Ontogenetic variation of catechin biosynthesis as basis for infection and quiescence of Botrytis cinerea in developing strawberry fruits. J Plant Dis Protect 115:247–251. https://doi.org/10.1007/BF03356272
Pühringer H, Moll D, Hoffmann-Sommergruber K, Watillon B, Katinger H, da Câmara Machado ML (2000) The promoter of an apple Ypr10 gene, encoding the major allergen Mal d 1, is stress- and pathogen-inducible. Plant Sci 152:35–50. https://doi.org/10.1016/S0168-9452(99)00222-8
R Core Team. R: a language and environment for statistical computing. http://www.R-project.org/
Rahman TA, Oirdi ME, Gonzalez-Lamothe R, Bouarab K (2012) Necrotrophic pathogens use the salicylic acid signaling pathway to promote disease development in tomato. Mol Plant Microbe Interact 25(12):1584–1593. https://doi.org/10.1094/MPMI-07-12-0187-R
Ranocha P, Denancé N, Vanholme R, Freydier A, Martinez Y, Hoffmann L et al (2010) Walls are thin 1 (WAT1), an Arabidopsis homolog of Medicago truncatula NODULIN21, is a tonoplast-localized protein required for secondary wall formation in fibers. Plant J 63:469–483. https://doi.org/10.1111/j.1365-313X.2010.04256.x
Rietz S, Stamm A, Malonek S, Wagner S, Becker D, Medina-Escobar N et al (2011) Different roles of enhanced disease susceptibility1 (EDS1) bound to and dissociated from phytoalexin deficient4 (PAD4) in Arabidopsis immunity. New Phytol 191:107–119. https://doi.org/10.1111/j.1469-8137.2011.03675.x
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA (eds) Bioinformatics methods and protocols. Methods in molecular biology™, vol 132. Humana Press, Totowa. https://doi.org/10.1385/1-59259-192-2:365
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15(5):247–258. https://doi.org/10.1016/j.tplants.2010.02.006
Schindelman G, Morikami A, Jung J, Baskin TI, Carpita NC, Derbyshire P et al (2001) COBRA encodes a putative GPI-anchored protein, which is polarly localized and necessary for oriented cell expansion in Arabidopsis. Genes Dev 15:1115–1127. https://doi.org/10.1101/gad.879101
Selote D, Shine MB, Robin GP, Kachroo A (2013) Soybean NDR1-like proteins bind pathogen effectors and regulate resistance signaling. New Phytol 202:485–498. https://doi.org/10.1111/nph.12654
Silvia Sebastiani M, Bagnaresi P, Sestili S, Biselli C, Zechini A, Orrù L et al (2017) Transcriptome Analysis of the melon-Fusarium oxysporum f. sp. melonis race 1.2 pathosystem in susceptible and resistant plants. Front Plant Sci 8:362. https://doi.org/10.3389/fpls.2017.00362
Slusarenko AJ, Fraser RSS, van Loon LC (2000) Mechanisms of resistance to plant diseases. Springer, Dordrecht
Terefe-Ayana D, Yasmin A, Le TL, Kaufmann H, Biber A, Kühr A et al (2011) Mining disease-resistance genes in roses: functional and molecular characterization of the Rdr1 locus. Front Plant Sci 2:35. https://doi.org/10.3389/fpls.2011.00035
Toljamo A, Blande D, Kärenlampi S, Kokko H (2016) Reprogramming of strawberry (Fragaria vesca) root transcriptome in response to Phytophthora cactorum. PloS ONE 11:e0161078. https://doi.org/10.1371/journal.pone.0161078
Tronchet M, Balague C, Kroj T, Jouanin L, Roby D (2010) Cinnamyl alcohol dehydrogenases-C and D, key enzymes in lignin biosynthesis, play an essential role in disease resistance in Arabidopsis. Mol Plant Pathol 11:83–92. https://doi.org/10.1111/j.1364-3703.2009.00578.X
van Loon LC, Rep M, Pieterse CMJ (2006) Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44:135–162. https://doi.org/10.1146/annurev.phyto.44.070505.143425
van Sandt VS, Suslov D, Verbelen J-P, Vissenberg K (2007) Xyloglucan endotransglucosylase activity loosens a plant cell wall. Ann Bot 100:1467–1473. https://doi.org/10.1093/aob/mcm248
Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W (2010) Lignin biosynthesis and structure. Plant Physiol 153(3):895–905. https://doi.org/10.1104/pp.110.155119
Vidhyasekaran P (2015) Plant hormone signaling systems in plant innate immunity. 1st edn. Springer, Rotterdam. https://doi.org/10.1007/978-94-017-9285-1
von Malek B, Debener T (1998) Genetic analysis of resistance to blackspot (Diplocarpon rosae) in tetraploid roses. Theor Appl Genet 96:228–231. https://doi.org/10.1007/s001220050731
Wang GF, Wie X, Fan R, Zhou H, Wang X, Yu C et al (2011) Molecular analysis of common wheat genes encoding three types of cytosolic heat shock protein 90 (Hsp90): functional involvement of cytosolic Hsp90s in the control of wheat seedling growth and disease resistance. New Phytol 191(2):418–431. https://doi.org/10.1111/j.1469-8137.2011.03715.x
Wang GF, Fan R, Wang X, Wang D, Zhang X (2015) TaRAR1 and TaSGT1 associate with TaHsp90 to function in bread wheat (Triticum aestivum L.) seedling growth and stripe rust resistance. Plant Mol Biol 87(6):577–589. https://doi.org/10.1007/s11103-015-0298-x
Weiß S, Bartsch M, Winkelmann T (2017) Transcriptomic analysis of molecular responses in Malus domestica ‘M26’ roots affected by apple replant disease. Plant Mol Biol 94:303–318. https://doi.org/10.1007/s11103-017-0608-6
Winkel-Shirley B (2002) Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Biol 5(3):218–223. https://doi.org/10.1016/S1369-5266(02)00256-X
Yamamoto M, Nakatsuka S, Otani H, Kohmoto K, Nishimura S (2000) (+)-Catechin acts as an infection-inhibiting factor in strawberry leaf. Phytopathology 90:595–600. https://doi.org/10.1094/PHYTO.2000.90.6.595
Yang Y-X, Ahammed GJ, Wu C, Fan S-Y, Zhou Y-H (2015) Crosstalk among jasmonate, salicylate and ethylene signaling pathways in plant disease and immune responses. Curr Protein Pept Sci 16:450–461. https://doi.org/10.2174/1389203716666150330141638
Yu Y-H, Li X-Z, Wu Z-J, Chen D-X, Li G-R, Li X-Q, Zhang G-H (2016) VvZFP11, a Cys2His2-type zinc finger transcription factor, is involved in defense responses in Vitis vinifera. Biol Plant 60(2):292–298. https://doi.org/10.1007/s10535-016-0598-2 dio
Zhang S-H, Yang Q, Ma R-C (2007) Erwinia carotovora ssp. Carotovora infection induced “defense lignin” accumulation and lignin biosynthetic gene expression in chinese cabbage (Brassica rapa L. ssp. pekinensis). J Integr Plant Biol 49:993–1002. https://doi.org/10.1111/j.1672-9072.2007.00478.x
Zhou N, Tootle TL, Tsui F, Klessig DF, Glazebrook J (1998) PAD4 functions upstream from salicylic acid to control defense responses in Arabidopsis. Plant Cell 10:1021–1030. https://doi.org/10.1105/tpc.10.6.1021
Zhou J, Peng Z, Long J, Sosso D, Liu B, Eom JS et al (2015) Gene targeting by the TAL effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J Cell Mol Biol 82:632–643. https://doi.org/10.1111/tpj.12838
Zhu L, Ni W, Liu S, Cai B, Xing H, Wang S (2017) Transcriptomics analysis of apple leaves in response to Alternaria alternata apple pathotype infection. Front Plant Sci 20:8–22. https://doi.org/10.3389/fpls.2017.00022
Acknowledgements
We want to give special thanks to the Institut für Rebenzüchtung of the Julius Kühn-Institut (JKI) in Siebeldingen. In particular, we thank the group of Prof. Dr. Eva Zyprian for the support in performing high-throughput qPCR with the Biomark system. Additionally, we thank the DFG for the funding of this project in the background of the GRK 1798.
Funding
This work was funded by the DFG GRK 1798 “Signaling at the Plant-Soil Interface” (GRK1798/1).
Author information
Authors and Affiliations
Contributions
The contribution of the authors to this study is as follows: EN was involved in conceptualizing the project, performing and evaluating the experiments, interpreting the data and writing and reviewing the article. HD was involved in performing and evaluating the qPCR experiments, evaluating and interpreting the MACE data and writing and reviewing the article. IM was involved in performing and evaluating the qPCR experiments, evaluating and interpreting the MACE data and writing and reviewing the article. HK was involved conceptualizing the project, performing the experiments for the generation of the MACE data and writing and reviewing the article. ML was involved in writing and reviewing the article. TD was involved in conceptualizing and supervising the project, interpreting the data and writing and reviewing the article.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Neu, E., Domes, H.S., Menz, I. et al. Interaction of roses with a biotrophic and a hemibiotrophic leaf pathogen leads to differences in defense transcriptome activation. Plant Mol Biol 99, 299–316 (2019). https://doi.org/10.1007/s11103-018-00818-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-018-00818-2