
Abstract
Key message
We provide a transcriptional profile of coffee rust interaction and identified putative up regulated resistant genes
Abstract
Coffee rust disease, caused by the fungus Hemileia vastatrix, is one of the major diseases in coffee throughout the world. The use of resistant cultivars is considered to be the most effective control strategy for this disease. To identify candidate genes related to different mechanism defense in coffee, we present a time-course comparative gene expression profile of Caturra (susceptible) and Híbrido de Timor (HdT, resistant) in response to H. vastatrix race XXXIII infection. The main objectives were to obtain a global overview of transcriptome in both interaction, compatible and incompatible, and, specially, analyze up-regulated HdT specific genes with inducible resistant and defense signaling pathways. Using both Coffea canephora as a reference genome and de novo assembly, we obtained 43,159 transcripts. At early infection events (12 and 24 h after infection), HdT responded to the attack of H. vastatrix with a larger number of up-regulated genes than Caturra, which was related to prehaustorial resistance. The genes found in HdT at early hours were involved in receptor-like kinases, response ion fluxes, production of reactive oxygen species, protein phosphorylation, ethylene biosynthesis and callose deposition. We selected 13 up-regulated HdT-exclusive genes to validate by real-time qPCR, which most of them confirmed their higher expression in HdT than in Caturra at early stage of infection. These genes have the potential to assist the development of new coffee rust control strategies. Collectively, our results provide understanding of expression profiles in coffee—H. vastatrix interaction over a time course in susceptible and resistant coffee plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coffee is a worldwide-appreciated beverage and an important commodity for several countries. Coffea arabica and Coffea canephora are the two most economically important species (Davis et al. 2011). C. arabica, a natural alotetraploid (2n = 4x = 44) hybrid between the two diploids species C. canephora and C. eugenioides (Lashermes et al. 1999), accounts for 70% of the world production (Davis et al. 2011). One of the major problems in coffee production is the coffee rust disease, which can cause yield losses up to 50% (Zambolim 2016). The causal agent of this disease, Hemileia vastatrix, is a biotrophic fungus, which it is entirely dependent on the cells of living plants for its growth and reproduction. The fungus produces several specialized structures to colonize the plant. After spore germination, the germ tube differentiates to apressoria, which, in turn, became penetration hyphae at the stomata. This structure will reach the substomatic chamber where it will differentiate to haustoria that will invade adjacent cells altering plant metabolism to meet its nutritional needs and, thus, completing the fungus lifecycle. This mode of interaction involves a prolonged and effective suppression of the host immune system and, at the same time, the induction of host-specific genes for its establishment as biotrophic fungus (Schulze-Lefert and Panstruga 2003; Voegele and Mendgen 2003; Guerra-Guimarães et al. 2015). Therefore, in coffee—H. vastatrix pathosystem, identification of genes involved in the plant defense mechanism triggered by the presence of the pathogen is indispensable to find new defense genes to develop resistant cultivars.
Plant defense against pathogens starts with the recognition of pathogen-associated molecular patterns (PAMPs) by transmembrane proteins named pattern recognition receptors, PRRs (Jones and Dangl 2006). This step activates the PAMP-triggered imunity (PTI). Pathogen can overcome this initial defense by secreting small extracellular proteins called effectors (De Wit et al. 2009). However, plants can recognize these effectors direct or indirectly by a resistance (R) protein, and induce a second defense phase, named Effector-triggered immunity (ETI), which is more efficient than PTI (De Wit et al. 2009). Each R gene recognize a specific effector (called Avr gene, for Avirulent) (Flor 1942), therefore, plant immunity is related to the presence of specific alleles of R genes. Some pathogens have evolved their effectors proteins either by losing an Avr gene, or diversifying its genes into different effector protein and thus, it can suppress ETI and successfully colonize the host (Jones and Dangl 2006).
Cytological and biochemical studies have shown coffee activates some defense mechanisms when attacked by H. vastatrix. One such mechanism is the hypersensitive response (HR), a type of ETI, which is associated with the deposition of callose, phenolic compounds (flavonoid and chlorogenic acid), and cell wall lignification (Silva et al. 2002). Other types of responses involve enzymes such as lipoxygenase and peroxidase that stimulate the pathway of phenylpropanoids. PR proteins (pathogen related proteins), as β-1,3-glucanase and chitinase, are also found in association with resistance of some coffee cultivars to H. vastatrix race II (Silva et al. 2002; Fernandez et al. 2012). Molecular methodologies as suppression subtractive hybridization (SSH), 454 pyrosequencing and RT-qPCR have been used to identify several genes putatively involved in host resistance. Some of the expressed sequence tags (ESTs) found in these studies encode for proteins involved in resistance, stresses, defense and signal transduction pathways (i.e. chitinases, beta-1, 3 glucanases, PR10, lipoxygenase type-AP2 and WRKY transcription factors). Also, activity of oxidative enzymes (lipoxygenase, peroxidase and superoxide dismutase), phenylalanine ammonia lyase, chitinase and glucanase were detected in the resistance reaction to H. vastatrix race II (Fernandez et al. 2004, 2012; Ganesh et al. 2006; Diniz et al. 2012). On the other hand, coffee resistance against H. vastatrix is governed by at least nine major dominant genes (S H 1–S H 9) that have the corresponding virulence genes (v1–v9) in the pathogen (Rodrigues et al. 1975; Bettencourt and Rodrigues 1988; Várzea and Marques 2005). However, resistance breakdown by virulence factor v5, which is present in the race II of H. vastatrix and at least thirty other races, has been reported in most commercial coffee cultivars with S H 5 gene (Rodrigues et al. 1993).
Until 2005, more than 50 races of H. vastatrix had been broadly described worldwide (Várzea and Marques 2005; Zambolim 2016), in Brazil 15 races were identified: I, II, III, VII, X, XIII, XV, XVI, XVII, XXI, XXII, XXIII, XXIV, XXV or XXXI, and XXXVII (Cabral et al. 2009; Capucho et al. 2012). The H. vastatrix races recently discovered have been able to infect derivatives of Híbrido de Timor (HdT), a natural hybrid originated from C. arabica and C. canephora crossing (Bettencourt 1973; Rodrigues et al. 2004). HdT is the main source of resistance used in breeding programs throughout the world (Gichuru et al. 2012; Zambolim 2016). The presence of such rust virulent races in the field represents a serious risk to coffee production worldwide. An example is the cultivar Oeiras MG 6851, originated from a crossing between C. arabica cv. Caturra (CIFC 19/1) and HdT (CIFC 832/1) and released as an important rust resistant cultivar (Pereira et al. 2000). Twelve years after its release, the resistance was broken by race XXXIII of H. vastatrix (Capucho et al. 2012). Thus, to study the dynamics of the interaction between the pathogen and the plant through transcriptome profiling will help to understand the supplanting of plant resistance by new physiological races of the fungus.
Despite all the efforts so far, there is limited information available on the transcriptome analysis to reveal which defense genes are involved in response to H. vastatrix infection. Transcriptome profiling studies are essential to fully understand the biological pathways that are activated in various physiological conditions or stages of an organism development (Wang et al. 2009; Ozsolak and Milos 2010). Expression profiling of host and pathogen can provide a new understanding of this interaction, and allow the identification of virulence genes in the pathogen or defense pathways in the host cells (Westermann et al. 2012; Boyd et al. 2013). Here, we report a time-course high throughput transcriptome analysis of a susceptible and resistant genotypes challenged by H. vastatrix infection. In this approach, we could identify genes related to both compatible (susceptible) and incompatible (resistant) interactions that are important for disease development and resistance. This knowledge will help in better understanding the molecular basis of resistance to specific H. vastatrix races and develop new coffee rust control strategies.
Materials and methods
Plant inoculation, library preparation and RNA sequencing
Two coffee genotypes were used in this study: C. arabica cv. caturra vermelho CIFC 19/1 (susceptible) and Híbrido de Timor CIFC 832/1 (resistant). They are the parents of cultivar Oeiras, the one that have the resistance supplant by the race XXXIII of H. vastatrix. Plants (juvenile leaves from second pair) were inoculated with spores of H. vastatrix race XXXIII and sampled at 0, 12, 24, 96 h after inoculation (hai) and 17 days after inoculation (dai), totaling 10 different samples (5 time points for each of the 2 genotypes). Total RNA was extracted with RNeasy Plant Mini Kit (Qiagen) and cDNA synthesis was performed using the Mint-2 cDNA Synthesis kit (Evrogen), according to the manufacturer’s instructions. After this step, the samples were normalized with TRIMMER – cDNA Normalization kit (Evrogen), to reduce the abundant transcripts, especially rRNAs. RNA sequencing was performed using MiSeq platform (Illumina) at University of North Carolina Chape Hill, NC, USA, with TruSeq DNA Sample Preparation protocol (Illumina). This sequencing produced 2x250pb paired-end reads for each genotype and time sampled, and these ten libraries were sequenced in 10 MiSeq runs, each run comprising all libraries in a multiplex reaction, using a different barcode for each library.
Quality assessment and overlapping paired-end reads
Read quality was assessed with FastQC software version 0.11.3 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The strategy used to improve read quality and length was overlapping the forward (R1) and reverse (R2) reads using the PEAR software (Zhang et al. 2014), with T-30 parameter. Reads which did not overlap (unassembled) were filtered using Clean Solexa software, to remove low-quality regions (Q < 20). Finally, Q > 20 assembled and unassembled reads were concatenated and submitted to transcriptome assembly as single-end reads (Fig. 1).

Pipeline of filtering raw reads by quality using the PEAR and Clean Solexa software. R1 Read forward, R2 read reverse and strategy used for de novo assembly
Transcriptome assembly
The ten libraries were mapped against a reference genome. Despite C. arabica genome is not available to date, C. canephora had its genome recently made available (Denoeud et al. 2014). Since C. canephora is an ancestral of C. arabica, we used C. canephora as a reference genome. An index file of C. canephora was created with Bowtie2 version 2.2.5 software, and mapped against the set of reads with each library using Tophat2 version 2.0.13 (Trapnell et al. 2009). The assembly of each library was created with Cufflinks (Trapnell et al. 2010), pooled together with Cuffmerge software and annotated with Cuffcompare. For this latter process, we used the C. canephora GFF3 and CDS (Coding DNA Sequence) files, which contains structural and functional genes. The set of annotated transcripts was analyzed using RSEM (RNA-Seq by Expectation Maximization) version 1.2.20 software (Li and Dewey 2011) to estimate the abundance of genes. The Fasta, GFF3 and CDS files of C. canephora genome were downloaded at http://www.coffee-genome.org/coffeacanephora.
The next step was to get reads that mapped against H. vastatrix and subtracted from unmapped reads against C. canephora, using a partial genome of H. vastatrix (http://www.bioinformatics.cenicafe.org/index.php/wiki/CoffeeRustHybridDraftAssembly_Contigs). Additionally, the unmapped reads against H. vastatrix were subtracted from the mapped C. canephora (Fig. 1). The purpose of this strategy was to get reads unique to C. arabica. Based on the result, we produced a de novo assembly using Trinity software (Grabherr et al. 2011; Haas et al. 2013). Subsequently, the obtained transcripts were analyzed with the Transdecoder (cut ORFs > 250 pb) and compared with the non-redundant databases (-NR-NCBI) (e-value 1e−5) by BLASTX. In addition, transcripts were submitted to BLASTX against the annotated genomes of Arabidopsis thaliana, Vitis vinifera and Solanum lycopersicum (http://www.phytozome.net). Similarly, the set of annotated transcripts obtained by de novo methodology was analyzed using RSEM (RNA-Seq by Expectation Maximization) version 1.2.20 software (Li and Dewey 2011) to estimate the abundance of genes.
Identification and annotation of differentially expressed genes
Read counting and normalization (FPKM, fragments per kilobase of exon per million mapped reads) obtained in previous steps were used to analyze differential expression of genes using RSEM software two statistical packages (DESeq, Anders and Huber 2012 and EDGER; Robinson et al. 2010) were used to identify differentially expressed genes at time points 12 hai, 24 hai, 96 hai and 17 dai compared to 0 hai (control) for each genotype separately. The list of differentially expressed genes was filtered by Log 2 fold change ≥ 0.5 and Log 2 fold change ≤ − 0.5, with q value of 0.1. To assess the variability between samples, PCA plot was performed using the statistical R package, with reads count table from RSEM. Cluster analysis was performed using Cluster 3.0 software (De Hoon et al. 2004), starting from normalized read counts. Functional annotation related to biological process, molecular function and extracellular component was performed using BLAST2GO (Conesa et al. 2005).
Candidate gene selection and validation by real time qPCR
Only differentially expressed genes identified by both statistical packages (DESeq and EDGER) were used for quantitative real time PCR validation. Initially, the up- and down-regulated genes within each genotype and at all contrasting times were identified (Fig. 2). Within the intersection of each group, a new intersection between the up-regulated and down-regulated genes was done in susceptible and resistant cultivars for all the time points monitored. Then with the unique genes of the HdT (resistant) in each time, a last intersection was separated in order to select important genes related to plant defense (Fig. 2).

Pipeline strategy to select candidate genes. HDT Híbrido de Timor, hai hours after inoculation, dai days after inoculation
The qPCR was conducted to compare the expression pattern of some selected genes during compatible (Caturra—H. vastatrix) and incompatible (Híbrido de Timor—H. vastatrix) interactions. Primers flanking the sequence of each candidate gene were designed (Table 1) with Primer-BLAST software of NCBI (available at: http://www.ncbi.nlm.nih.gov/tools/primer-blast/) with the following parameters: Amplicon length between 90 and 150 bp, primer size: 20 ± 2 bp, annealing temperature (Ta) between 55 and 60 °C, GC content of ± 50%. Reference genes used were GADPH (glyceraldehyde-3-phosphate dehydrogenase), UBQ10 (polyubiquitin 10) and S24 (ribosomal protein) (Cruz et al. 2009), see Table 1. Primer efficiency was tested by developing a standard curve of five dilution points of cDNA (1:5), and primer efficiency (E) was calculated from the slope (a) of standard curve [E = 10(−1/a) − 1] with the Cts obtained for each dilution.
Using a different set of plants of those used for transcriptome study, total RNA was extracted from leaves of Caturra and Híbrido de Timor genotypes infected with H. vastatrix at 0, 12, 24 and 96 hai. Three biological and three technical replicates were performed for each cDNA sample. Reverse transcription was conducted using the ImProm-II™ Reverse Transcriptase system (Promega, Madison, USA), with 1 μg of total RNA. The qPCR reactions were carried out in 7500 Real Time PCR System (Applied Biosystems, Thermo Fisher Scientific, USA), in final volume of 10 μl with 50 ng/μl of cDNA and 100 nΜ of forward and reverse primers in 1x GoTaq® qPCR Master Mix (Promega, Madison, USA) as final concentrations. Reaction parameters were 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min and melting curve stage was set to default conditions. The expression levels of the candidate genes were calculated using qBase software (Hellemans et al. 2007). All statistical analyses were carried out using the GraphPad Prism (version 5; GraphPad Software Inc.; La Jolla, CA, USA). To determine significant differences between control and treated samples, the one-way ANOVA with Dunnet test (p < 0.05) was performed. Means were compared using the Tukey test (p < 0.05).
Results and discussion
Read quality filtering, mapping and assembly
Transcriptome of coffee during compatible (Caturra) and incompatible (HdT) interactions with H. vastatrix race XXXIII was analyzed, using various bioinformatics tools. Firstly, a total of 103,031,664 2X250 pair-end reads were generated from ten libraries sequenced by the Illumina Miseq platform (see Table 2). The overlap between paired reads was performed producing 70,450,936 overlapped reads. This approach generated assembled (overlapped) and unassembled (R1 and R2) 300-bp reads, which were concatenated generating a total of 110,135,873 single-end reads (Fig. 1; Table 2). As such, the quality and average length of number of reads was increased and improving the chance to obtain good transcriptome assembly.
Since C. arabica reference genome has not made available to date, we used a genome from a closely related species, i.e. C. canephora, as a reference for read mapping. In the susceptible genotype, read mapping against C. canephora genome showed a decrease of alignment along the infection time course, starting from 48% (0 hai) and reducing to 33% (17 dai) (Fig. 3a). In contrast, resistant library alignment was almost constant at all time points (Fig. 3b). On the contrary, when mapping was made against H. vastatrix partial genome (Cristancho et al. 2014), the mapping of reads in the susceptible genotype during the early hours of infection (12 and 24) was 3%, then started to increase at 96 hai, up to 8% in the 17 dai library (Fig. 3a). This final library showed a high percentage of reads mapped against H. vastatrix genome due to increased fungus biomass at this time point. Approximately 20 days after the start of the infection process, hyphae forming a large mycelial density, appearing on the outside through the stomata, a uredosporic serum in a “bouquet” occurs in the area of penetration (Silva et al. 2006). In resistant genotype the effect was the opposite; the amount of the fungus reads aligned began to decrease from 12 hai and onwards (Fig. 3b).

Percentage of reads mapping against Coffea canephora (green) and Hemileia vastatrix (yellow) reference genomes during infection course. a Susceptible libraries, b resistant libraries
The transcriptome assembly using C. canephora reference genome produced 28,119 contigs with an N50 value of 1626 (Table 3). Approximately 50% of the total reads of each HdT’ libraries mapped to C. canephora genome, which is higher than observed for the susceptible libraries (Fig. 3). This higher mapping percentage in the resistant libraries is probably due to HdT origin, since it is a natural hybrid between Coffea arabica and C. canephora (Bettencourt 1973; Rodrigues et al. 2004), therefore a larger part of its genome derives from C. canephora. As a complement to the strategy using C. canephora as a reference, and in order to increase the number of contigs to have a greater coverage of the transcriptome, de novo assembly of unaligment reads was adopted in this study. We obtained a total of 15,040 contigs with N50 of 1317 (Table 3). A total of 43,159 contigs were produced using C. canephora genome reference and de novo assembly. This number is much greater than reported by Fernandez et al. (2012) in a transcriptome analysis of C. arabica CIFC H147/1 (resistant) during the interaction with H. vastatrix using the 454-pyrosequencing platform, the only published study reporting transcriptome profiling of this pathosystem. The authors produced 352,146 reads assembled into 13,951 contigs with an average size of 631 bp. The functions of the majority of the genes were related to caffeine or chlorogenic acid biosynthesis, and genes associated to plant defense response such as WRKY transcription factor, pathogenesis-related (PR) proteins (1,3-b-glucanases, PR1b, PR-5 of the thaumatin-like protein family and chitinases). Similar genes were founded in our experiment; however, other HdT exclusive genes such as NAC, MYB, Beta-glucosidase, F-box protein and peroxidase were found involved in defense mechanisms at early hours (12 and 24) of H. vastatrix infection.
In addition to obtaining a greater number of contigs, we analyzed both compatible and incompatible coffee—H. vastatrix interaction with the aim of providing a better understanding of gene expression profile. Moreover, here, we used Illumina sequencing which has less low error rate, compared to 454-pyrosequencing platform (Loman et al. 2012). Recently, other transcriptome studies of coffee, however not involving plant-pathogen interactions, showed similar number of contigs assembled as in this present study. Leaf and fruit transcriptome analysis of Coffea eugenioides produced 36,935 contigs using Illumina HiSeq platform (Yuyama et al. 2016). Also Mofatto et al. (2016) obtained a total of 41,512 contigs from C. arabica transcriptome, comparing the molecular responses to drought in two commercial cultivars using 454—pyrosequencing and Sanger platforms. The expression profiles (read counts) in all libraries were submitted for PCA analysis in order to evaluate the intrinsic variation between libraries. A distinction between the group of resistant and susceptible genotype libraries was observed when C. canephora was used as a reference for transcriptome assembly (Fig. 4). The results from de novo assembly (data not shown) are similar to the result elucidated by C. canephora assembly. The PCA clearly revealed that the greatest changes in host transcriptome were caused by the fungal infection following the interaction process. In particular, in susceptible genotype 17 dai library was well separated from all other libraries.

Principal component analysis (PCA) of susceptible and resistant genotype libraries during the interaction with H. vastatrix
Profiling of differentially expressed genes (DEGs)
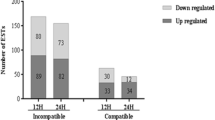
Plants are in a constant struggle with pathogens, and use different defense mechanisms to preserve their integrity, such as PTI and ETI. In coffee—H. vastatrix interaction, the pathogen establishes a biotrophic relation with the host within the first hours after infection (< 24 hai). Two main groups of expression levels can be observed from the heatmap (Fig. 5a). The first group correspond to the early infection events (12 and 24 hai) and HdT responded to the attack of H. vastatrix with larger number of up-regulated genes than Caturra, which is indicated as a higher red intensity in heatmap (Fig. 5a) and total number of up-regulated genes (Fig. 5b), which shows around 500 more up-regulated genes than in Caturra. In the second group, both genotypes had similar pattern expression during 96 hai. However, at 17 dai the expression of some genes was increased in resistant genotypes unlike in susceptible genotypes. The markedly up-regulation of genes in HdT may reflect the exploitation of cellular resources and / or the activation of defense responses (Grenville-Briggs and West 2005). In fact, it has been reported that coffee plants with complete resistance to H. vastatrix, fungus growth ceases in the early stages of infection where there is a disruption of the cytoplasmic contents of the fungal infection structures (Diniz et al. 2012). Therefore, this overall up-regulation of transcription might be a direct consequence of fungus attack and it is responsible for a coordinate and effective defense.

Expression profile in Híbrido de Timor (resistant) and Caturra (susceptible) inoculated with H. vastatrix. a Hierarchical clustering according to the changes in expression level along the time course. Cluster analysis was performed using Cluster 3.0 software. Each column represents the Log2 fold change in transcript levels in coffee at the indicated times. Up-regulated and down-regulated genes are shown in red and green, respectively. The intensity color scale indicates the level of expression. b, c Total up and down-regulated genes, respectively
Generally, sets of genes in plants are induced against a biotic or abiotic stress condition. This response may be early or late, but the first response is key to induce signaling cascades for expression of genes involved in defense mechanisms (Kumar and Kirti 2011). For the majority of pathosystems, all this process starts with the recognition or perception, signal transduction, followed by the activation of defense genes (Oliveira et al. 2016). By inducing a hierarchical network of genes that regulate the expression of resistance genes, the advancing pathogen within the plant is suppressed (Kushalappa et al. 2016). Here, we found several DEGs encoding receptors in the transcriptome of HdT such as receptor-like kinases (RLKs) and receptor-like proteins (RLPs) in the initial stage of infection (data not shown). These receptors could be related to pathogen perception, such as PAMPs recognition by PRRs, which leads to a signaling cascade that alerts the plant to the presence of a pathogen and induces PTI (Ishiga et al. 2013). This PTI response frequently involves ion fluxes, the production of reactive oxygen species, protein phosphorylation, ethylene biosynthesis, and callose deposition (Boller and Felix 2009). Linked to such functions during the early hours of HdT-H. vastatrix interaction, we found genes such as calcium-transporting, peroxidase, ethylene-responsive transcription and callose synthase.
Additionally, we confirmed that the early response of HdT to the H. vastatrix attack was related to pre-haustorial resistance as suggested (Heath 1977; Mellersh and Heath 2003; Niks and Rubiales 2002; Freitas et al. 2014). Usually the haustoria formation of H. vastatrix begins after 24 hai in the host. Although, in coffee is also possible to found post haustorial resistance, the type of response depend of the coffee genotype (Silva et al. 2006). The pre-haustorial response was also observed in the barley-Puccinia graminis f. sp. tritici pathosystem, during this interaction the resistance gene RPG1 was expressed a few minutes after inoculation with stem rust races (Nirmala et al. 2010). More recently, in the same pathosystem it was observed early responses to avirulent stem rust races in wheat carrying resistance genes Sr5 and Sr36 involved callose deposition in stomatal guard cells (Wang et al. 2015). It is possible that the genes identified here as induced in early hours in HdT may trigger a rapid and efficient response for the plant defense, and these mechanisms may involve pre-haustorial resistance in coffee-H. vastatrix interaction, in the same way found for barley and wheat pre-haustorial defense to P. graminis. Niks and Rubiales (2002) suggest that combining different resistance mechanisms acting at different stages of the infection process could provide multiple barriers that are not easily overcome by simple race-type changes of the pathogen. In addition, different members of a host plant species contain different R gene complements. Rust infections can therefore lead to either a resistant or a susceptible outcome, depending upon the plant and pathogen genotypes involved (Bettgenhaeuser et al. 2014).
In the case of a group of genes in susceptible plant, more up-regulated genes at 12 hai (Fig. 5a) may be associated with basal resistance of the plant (Jones and Dangl 2006). In the following time points, however, the level of gene expression was suppressed (Fig. 5a). Note that in all time points, HdT showed a higher gene expression, which can be noticed by the color intensity in heatmap (Fig. 5a). This shows that transcription programming in the resistant genotype is completely different from susceptible genotype. On the other hand, the number of down-regulated genes is comparable in both genotypes (Fig. 5b). Plant–pathogen interactions are complex processes that trigger a series of molecular responses at several expression levels. While resistant plants initiate responses in incompatible interactions, susceptible plants can also launch a series of basal defense responses in compatible interactions. Although they present similar expression profiles, defense gene induction in compatible interactions occurs later than that in incompatible interactions (Balaji et al. 2008; Lara-Ávila et al. 2012).
BLAST2GO analysis was performed for genes up-regulated at 12 and 24 hai in both resistant and susceptible genotypes (Fig. S1). These time points are when actual biotrophic interactions are assumed to occur. The result presented for the biological process category, the two genotypes showed the existence of up-regulated genes associated with programmed cell death but at different time points (12 hai in susceptible and 24 hai in resistant genotype). It could be inferred that programmed cell death is a delayed response to efficiently deter the ingression and establishment of the invading pathogen. A microscopic study analyzing the same pathosystem that we used (HdT CIFC 832/1 and Caturra CIFC 19/1 inoculated with H. vastatrix race XXXIII) revealed 30% of HR sites in both genotypes at 12 hai. However, this same study showed an increase HR to 70% in HdT at 24 hai and maintained 30% in Caturra (Freitas et al. 2014). On the other hand, for the molecular function category for the majority of the genes (70%) were represented by genes involved in catalytic activity in resistant genotype. Of remarkable difference that can be noticed is the higher proportion of up-regulated genes predicted as cell wall component under the cellular component category in resistant genotype as part of structural resistance.
On the other hand, analysis of differentially expressed genes using C. canephora genome reference, represented using venn diagrams in all time courses between Caturra and HdT, allowed to identify one set of up-regulated genes unique to HdT for each time of infection (Fig. 6a–d), the results from de novo assembly (data not shown) are similar to the result elucidated. However, within each time it was possible to observe that both genotypes have some genes in common (Fig. 6a–d), some of which could be associated with basal resistance in both genotypes, considering that Caturra does not show vertical resistance against H. vastatrix (Silva et al. 2006). Basal resistance is a kind of defense response mechanism common in plants when they are attacked by biotrophic fungi (Niks et al. 2015), and that is initiated during the early phases of pathogen infection (Gill et al. 2015). Finally, one more intersection of this pool of genes allowed us to choose 13 genes exclusively up-regulated in HdT, which showed importance within the plant defense mechanisms. This selection was made from 219, 129 and 118 genes corresponding to 12, 24 and 96 h after infection in HdT, respectively (Fig. 6e).

Number of up-regulated genes (DEGs) of the C. canephora assembly in Caturra and HdT in response to H. vastatrix infection. a Venn diagram displaying the number of up-regulated DEGs between Caturra (Cat) and HdT at 12 hai. b Up-regulated DEGs between Cat and HdT at 24 hai. c Up-regulated DEGs between Cat and HdT at 96 hai. d Up-regulated DEGs between Cat and HdT at 17 dai. e Up-regulated DEGs exclusive to HdT at all times investigated
The qPCR analysis of 13 different types of genes related to plant defense mechanisms were conducted at 0, 12, 24 and 96 hai in susceptible and resistant coffee genotypes. Expression pattern in qPCR was similar to transcriptome analysis for most of the studied genes (Fig. S2). A similar correlation between qPCR and transcriptome profiling was reported by Rubio et al. (2015). Both analyzes have their own strengths and limitations, the different expression levels between Transcriptome and qPCR could be caused by the bioinformatics process performed in the Transcriptome analysis, which includes several factors that can affect the reproducibility of quantitative expression profiles, including alignment choices, estimation of transcript expression, etc. (Labaj et al. 2011). The first group of analyzed genes was transcription factors (Putative basic helix-loop-helix bHLH DNA-binding superfamily protein, and Ethylene-responsive transcription factor 1B), which showed early expression in HdT in contrast to Caturra (Fig. 7a, b). The bHLH (Fig. 7a) presented a high peak expression at 12 hai in the incompatible interaction. The results suggest that this gene may be involved in regulating the expression of some resistance genes or defense mechanisms only in resistant genotype. Kundu et al. (2015) evidenced that the expression profiling of transcription factor of this same family (bHLH) may play a complementary and/or overlapping role in enhancing expression of downstream components of the defense pathway.

Validation of candidate genes by qPCR. Y-axis represent the relative expression at various times after H. vastatrix inoculation in susceptible and resistant coffee genotypes. Error bars = SEM, n = 3 independent biological replicates. Red asterisk (*) shows significantly up-or down-regulated relative to uninoculated samples (0 hai) within interaction as defined by one-way ANOVA followed by Dunnett test (p < 0.05); black asterisk (*) shows significant difference in expression level between the same hai between interactions defined by Tukey test (p < 0.05)
The second transcription factor, Ethylene-responsive transcription factor 1B (ERF) presented two expression peaks (at 12 and 24 hai) in resistant genotype and also significantly higher when compared to susceptible genotype (Fig. 7b). Usually, the transcription factors are activated by external stimuli and subsequently regulate the expression of genes involved in plant defense. A number of ERF genes confer tolerance to various biotic stresses when expressed in different plants under different conditions (Licausi et al. 2013). There is evidence that ERFs induce transcription resistance genes, pathogenesis-related (PR) genes, osmotin, chitinase and b-1,3-glucanase (Licausi et al. 2013). This kind of gene has been selected through evolution to regulate a series of stress-response pathways and could be used in genetic engineering for the breeding of plants with specific traits related to stress tolerance (Licausi et al. 2013). Ganesh et al. (2006) evaluated the expression of three defense related genes during coffee—H. vastatrix interaction, including a transcription factor. These genes showed that they were transiently induced during early stages (12–24 hai) of pathogen infection. They reported that two of the three genes (CaR111, gene encoding a protein of unknown function and CaWRKY1, a transcription factor) were up-regulated in incompatible samples, which suggest their involvement in defense mechanism.
The group of genes related to recognition of pathogen elicitor proteins [Serine-threonine protein-receptor-like kinase SD1-8 and Probable putative protein receptor-like kinase (At5g39020 homolog)], exhibited a constant expression in HdT during infection process while the expression in Caturra was delayed (24 hai) (Fig. 7c, d). Such behavior are associated with signal perception through their extracellular domain and propagate the signal through the intracellular kinase domain to activate R genes (Afzal et al. 2008). A similar expression profiling was found with the resistance gene group: Putative late blight resistance protein homolog R1A-6, NB-ARC domain-containing disease resistance protein and Putative disease resistance protein RGA1 (Fig. 7e–g). These genes group are most likely involved in the activation of plant defense mechanisms and their expression during all time course, is possibly associated with controlling the differentiation of several fungal structures. In coffee, Diniz et al. (2012) evaluated the expression of CaRLK, which is involved in recognition, signaling and defense, showed that this gene was activated in the early events of the HdT 832/2—H. vastatrix interaction. CaRLK showed expression peak at 6 to 12 hai, a period when appressoria and penetration hypha differentiate. According to this report, the second highest expression peak was when the anchors and haustoria mother cells differentiate (21–24 hai). In the present work, RLK was continuously expressed along all the time courses studied in resistant genotype unlike in the susceptible genotype in which the expression was delayed. The result suggests that this gene is one of the key defense components which is in accordance to the work of (Diniz et al. 2012).
In the case of Putative disease resistance-response (dirigent-like protein) family protein, it presented a unique and high relative expression at 96 hai (Fig. 7h). This might be a case of an R-genes that is highly genotype specific (Narusaka et al. 2013) and downstream activated by signaling cascades (Hammond-Kosack and Jones 1997). The PR protein showed high expression in both genotypes at 24 hai, however the susceptible was higher than resistant genotype (Fig. 7i), which suggests that in general these types of genes are important in the defense of plants against pathogens, mainly in susceptible plants. Several types of PR proteins in resistant and susceptible wheat genotypes were also reported as up-regulated (Xin et al. 2012). The PR protein expression is often triggered by pathogen infection, limiting the pathogen progression (Silva et al. 2006).
Another group of genes analyzed by qPCR were Premnaspiridione oxygenase, Putative Ankyrin repeat-containing protein At3g12360, Cationic peroxidase and Putative E3 ubiquitin-protein ligase PUB24. The first gene encodes solavetivone, a potent antifungal phytoalexin (Takahashi et al. 2007). Generally, phytoalexins are organic compounds, which possess antimicrobial or repellent activities (Jeandet et al. 2013). Their production starts a few hours after the pathogen or pest attack (Pedras et al. 2011). The results found here are in agreement with the literature, since in the resistant genotype Premnaspiridione oxygenase up-regulation starts at 12 hai while in the susceptible genotype it only starts at 24 hai (Fig. 7j). The putative ankyrin repeat-containing protein followed a similar expression pattern (Fig. 7k). Functional characterization of Ankyrin repeats proteins across species have indicated a conserved role for them in protection against pathogen and disease resistance, as promoter of Systemic Acquired Resistance (SAR) (Sharma and Pandey 2016). This protein is important for transducing the Salicylic acid (SA) signal. In some R–avr-mediated interactions, SA is required for the R gene-dependent host programmed cell death (called the hypersensitive response, HR) and/or for disease resistance (Lu 2003). Perhaps, the induction of Ankyrin repeat-containing protein in the resistant plant, within the first hours after infection, promotes the production of SA, important compound to trigger HR.
The Cationic peroxidase gene is related with the accumulation of lignin-like compounds and reduction in pathogen multiplication in leaves and onset of the HR. We found this gene up-regulated at 12 hai in HdT and at 24 hai in Caturra (Fig. 7l). Its rice homologue is also induced at 24 hai during incompatible interaction with Xanthomonas oryzae pv oryzae pathogen (Young et al. 1995) and during compatible interaction at 48 hai. The early expression of cationic peroxidase in HdT may be involved in most important responses in coffee HR (Silva et al. 2002). Putative E3 ubiquitin-protein ligase PUB24 is related to different positive and negative functions in different steps of plant defense such as regulation of RLKs (Duplan and Rivas 2014), involved in recognition of pathogen elicitor proteins (Kenn 1999; Ebel 1997; Shibuya and Minami 2001; Peck 2003). Wang et al. (2006) demonstrated that the interaction of E3 Ubligase gene with a RLKs type protein triggered cascades of defense gene activation in rice plant against Xanthomonas oryzae pv oryzae. Here, we found a high gene expression of Putative E3 ubiquitin-protein ligase PUB24 at 12 hai (Fig. 7m) in the resistant plant at the same time of an up-regulation of an Serine-threonine protein-receptor-like kinase SD1-8 and Probable putative protein receptor-like kinase (At5g39020 homolog) genes (Fig. 7c, d). Therefore, due to their expression pattern, it is reasonable to hypothesize that these three genes might be involved in a defense mechanism similar to that found in rice against X. oryzae pv oryzae, where coffee E3 ubiquitin-ligase acts together with these two receptor kinases to activate the signaling cascade leading to defense response.
Taken together, the qPCR expression analysis between resistant and susceptible genotypes confirmed that genes such as putative basic helix-loop-helix bHLH DNA-binding superfamily protein, ethylene-responsive transcription factor 1B, putative disease resistance protein RGA1, putative disease resistance-response (dirigent-like protein) family protein, and premnaspiridione oxygenase showed higher expression at early stage upon challenge with the biotrophic pathogen H. vastatrix in resistant plant. In contrast, no such up-regulation in early hours was detected in susceptible genotype. These genes could represent potential candidates for different biotechnological and effective management tools for operational application in new strategies for controlling coffee rust. In general, the regulation of gene expression is a dynamic process. We confirmed that early expression patterns of up-regulated genes in HdT are directly related to prehaustorial resistance. This transcriptome study paves a way for more detailed work on coffee resistance to H. vastatrix, which will be important for future development of plants with certain adaptive characteristics to coffee rust.
Change history
17 October 2019
All the transcriptome sequencing data mentioned in the original article is publicly available at the National Center of Biotechnology Information (NCBI).
17 October 2019
All the transcriptome sequencing data mentioned in the original article is publicly available at the National Center of Biotechnology Information (NCBI).
References
Afzal AJ, Wood AJ, Lightfoot DA (2008) Plant receptor-like serine threonine kinases: roles in signaling and plant defense. Mol Plant Microbe Interact 21:507–517. doi:10.1094/MPMI-21-5-0507
Anders S, Huber W (2012) Differential expression of RNA-Seq data at the gene level—the DESeq package. EMBL 11:R106. doi:10.1186/gb-2010-11-10-r106
Balaji V, Mayrose M, Sherf O, Jacob-Hirsch J, Eichenlaub R, Iraki N, Manulis-Sasson S, Rechavi G, Barash I, Sessa G (2008) Tomato transcriptional changes in response to Clavibacter michiganensis subsp. michiganensis reveal a role for ethylene in disease development. Plant Physiol 146:1797–1809. doi:10.1104/pp.107.115188
Bettencourt AJ (1973) Considerações gerais sobre o “Hibrido de Timor.” Circ. No 23 20
Bettencourt AJ, Rodrigues C (1988) Principles and practice of coffee breeding for resistance to rust and other diseases. Elsevier Appl Sci 3:199–234
Bettgenhaeuser J, Gilbert B, Ayliffe M, Moscou MJ (2014) Nonhost resistance to rust pathogens—a continuation of continua. Front Plant Sci 5:664. doi:10.3389/fpls.2014.00664
Boller T, Felix G (2009) A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol 60:379–406. doi:10.1146/annurev.arplant.57.032905.105346
Boyd LA, Ridout C, O’Sullivan DM, Leach JE, Leung H (2013) Plant-pathogen interactions: disease resistance in modern agriculture. Trends Genet 29:233–240. doi:10.1016/j.tig.2012.10.011
Cabral PGC, Zambolim EM, Zambolim L, Lelis TP, Capucho AS, Caixeta ET (2009) Identification of a new race of Hemileia vastatrix in Brazil. Australas Plant Dis Notes 4:129–130
Capucho AS, Zambolim EM, Freitas RL, Haddad F, Caixeta ET, Zambolim L (2012) Identification of race XXXIII of Hemileia vastatrix on Coffea arabica Catimor derivatives in Brazil. Australas Plant Dis Notes 7:189–191. doi:10.1007/s13314-012-0081-7
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. doi:10.1093/bioinformatics/bti610
Cristancho MA, Botero-Rozo DO, Giraldo W, Tabima J, Riaño-Pachón DM, Escobar C, Rozo Y, Rivera LF, Durán A, Restrepo S, Eilam T, Anikster Y, Gaitán AL (2014) Annotation of a hybrid partial genome of the coffee rust (Hemileia vastatrix) contributes to the gene repertoire catalog of the Pucciniales. Front Plant Sci 5:594. doi:10.3389/fpls.2014.00594
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LMG, Romano E, Grossi-de-Sá MF, Vaslin M, Alves-Ferreira M (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Mol Breed 23:607–616. doi:10.1007/s11032-009-9259-x
Davis AP, Tosh J, Ruch N, Fay MF (2011) Growing coffee: Psilanthus (Rubiaceae) subsumed on the basis of molecular and morphological data; implications for the size, morphology, distribution and evolutionary history of Coffea. Bot J Linn Soc 167:357–377. doi:10.1111/j.1095-8339.2011.01177.x
De Hoon MJL, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20:1453–1454. doi:10.1093/bioinformatics/bth078
De Wit PJGM, Mehrabi R, Van Den Burg HA, Stergiopoulos I (2009) Fungal effector proteins: past, present and future. Mol Plant Pathol 10:735–747. doi:10.1111/j.1364-3703.2009.00591.x
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G, Aury J-M, Bento P, Bernard M, Bocs S, Campa C, Cenci A, Combes M-C, Crouzillat D, Da Silva C, Daddiego L, De Bellis F, Dussert S, Garsmeur O, Gayraud T, Guignon V, Jahn K, Jamilloux V, Joet T, Labadie K, Lan T, Leclercq J, Lepelley M, Leroy T, Li L-T, Librado P, Lopez L, Munoz A, Noel B, Pallavicini A, Perrotta G, Poncet V, Pot D, Priyono, Rigoreau M, Rouard M, Rozas J, Tranchant-Dubreuil C, VanBuren R, Zhang Q, Andrade AC, Argout X, Bertrand B, de Kochko A, Graziosi G, Henry RJ, Jayarama, Ming R, Nagai C, Rounsley S, Sankoff D, Giuliano G, Albert VA, Wincker P, Lashermes P (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184. doi:10.1126/science.1255274
Diniz I, Talhinhas P, Azinheira HG, Várzea V, Medeira C, Maia I, Petitot AS, Nicole M, Fernandez D, do Céu Silva M (2012) Cellular and molecular analyses of coffee resistance to Hemileia vastatrix and nonhost resistance to Uromyces vignae in the resistance-donor genotype HDT832/2. Eur J Plant Pathol 133:141–157. doi:10.1007/s10658-011-9925-9
Duplan V, Rivas S (2014) E3 ubiquitin-ligases and their target proteins during the regulation of plant innate immunity. Front Plant Sci. doi:10.3389/fpls.2014.00042
Ebel J, Scheel D (1997) Signals in host-parasite interactions. In: Carrol GC, Tudzynski P (eds) The Mycota, vol V. Springer, Berlin, pp 85–105
Fernandez D, Santos P, Agostini C, Bon MC, Petitot AS, Silva MC, Guerra-Guimarães L, Ribeiro A, Argout X, Nicole M (2004) Coffee (Coffea arabica L.) genes early expressed during infection by the rust fungus (Hemileia vastatrix). Mol Plant Pathol 5:527–536. doi:10.1111/J.1364-3703.2004.00250.X
Fernandez D, Tisserant E, Talhinhas P, Azinheira H, Vieira A, Petitot AS, Loureiro A, Poulain J, da Silva C, do Céu Silva M, Duplessis S (2012) 454-pyrosequencing of Coffea arabica leaves infected by the rust fungus Hemileia vastatrix reveals in planta-expressed pathogen-secreted proteins and plant functions in a late compatible plant-rust interaction. Mol Plant Pathol 13:17–37. doi:10.1111/j.1364-3703.2011.00723.x
Flor H (1942) Inheritance of pathogenicity in Melampsora lini. Phytopathology 32:653–669
Freitas RL, Zambolim E, Silva MC, Caixeta ET, Lelis DT, Zambolim L, Sakiyama N (2014) Cytological evaluation of the infection process of Hemileia vastatrix (race XXXIII) in resistant and susceptible coffee. In: Proceedings of the 25th international conference on coffee science (ASIC), pp 42–46. Armenia, Colombia
Ganesh D, Petitot AS, Silva MC, Alary R, Lecouls AC, Fernandez D (2006) Monitoring of the early molecular resistance responses of coffee (Coffea arabica L.) to the rust fungus (Hemileia vastatrix) using real-time quantitative RT-PCR. Plant Sci 170:1045–1051. doi:10.1016/j.plantsci.2005.12.009
Gichuru EK, Ithiru JM, Silva MC, Pereira AP, Varzea VMP (2012) Additional physiological races of coffee leaf rust (Hemileia vastatrix) identified in Kenya. Trop Plant Pathol 37:424–427. doi:10.1590/S1982-56762012000600008
Gill US, Lee S, Mysore KS (2015) Host versus nonhost resistance: distinct wars with similar arsenals. Phytopathology 105:580–587. doi:10.1094/PHYTO-11-14-0298-RVW
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi:10.1038/nbt.1883
Grenville-Briggs LJ, West P van (2005) The biotrophic stages of oomycete–plant interactions. 57:217–243. doi:10.1016/S0065-2164(05)57007-2
Guerra-Guimarães L, Tenente R, Pinheiro C, Chaves I, Silva M, do C, Cardoso FMH, Planchon S, Barros DR, Renaut J, Ricardo CP (2015) Proteomic analysis of apoplastic fluid of Coffea arabica leaves highlights novel biomarkers for resistance against Hemileia vastatrix. Front Plant Sci 6:1–16. doi:10.3389/fpls.2015.00478
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. doi:10.1038/nprot.2013.084
Hammond-Kosack KE, Jones JD (1997) Plant disease resistance genes. Annu Rev Plant Physiol Plant Mol Biol 48:575–607
Heath MC (1977) A comparative study of non-host interactions with rust fungi. Physiol Plant Pathol 10:73–88. doi:10.1016/0048-4059(77)90009-1
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. doi:10.1186/gb-2007-8-2-r19
Ishiga Y, Upplapapti SR, Mysore KS (2013) Expression analysis reveals a role for hydrophobic or epicuticular wax signals in pre-penetration structure formation of Phakopsora pachyrhizi. Plant Signal Behav 8:e26959. doi:10.4161/psb.26959
Jeandet P, Clement C, Courot E, Cordelier S (2013) Modulation of phytoalexin biosynthesis in engineered plants for disease resistance. Int J Mol Sci 14:14136–14170
Jones JDG, Dangl JL (2006) The plant immune system. Nature 444:323–329. doi:10.1038/nature05286
Kenn NT (1999) Plant disease resistance: progress in basic understanding and practical application. In: Advances in botanical research. Academic Press, London, pp 291–328
Kumar KRR, Kirti PB (2011) Differential gene expression in Arachis diogoi upon interaction with peanut late leaf spot pathogen, Phaeoisariopsis personata and characterization of a pathogen induced cyclophilin. Plant Mol Biol 75:497–513. doi:10.1007/s11103-011-9747-3
Kundu A, Patel A, Paul S, Pal A (2015) Transcript Dynamics at Early Stages of Molecular Interactions of MYMIV with Resistant and Susceptible Genotypes of the Leguminous Host, Vigna mungo. PLoS ONE 10:e0124687. doi:10.1371/journal.pone.0124687
Kushalappa AC, Yogendra KN, Karre S (2016) Plant innate immune response: qualitative and quantitative resistance. CRC Crit Rev Plant Sci 35:38–55. doi:10.1080/07352689.2016.1148980
Labaj PP, Leparc GG, Linggi BE, Markillie LM, Wiley HS, Kreil DP (2011) Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics 27:i383–i391. doi:10.1093/bioinformatics/btr247
Lara-Ávila JP, Isordia-Jasso MI, Castillo-Collazo R, Simpson J, Alpuche-Solís ÁG (2012) Gene expression analysis during interaction of tomato and related wild species with Clavibacter michiganensis subsp. michiganensis. Plant Mol Biol Report 30:498–511. doi:10.1007/s11105-011-0348-8
Lashermes P, Combes M-C, Robert J, Trouslot P, D’Hont A, Anthony F, Charrier A (1999) Molecular characterisation and origin of the Coffea arabica L. genome. Mol Gen Genet MGG 261:259–266. doi:10.1007/s004380050965
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323. doi:10.1186/1471-2105-12-323
Licausi F, Ohme-Takagi M, Perata P (2013) APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol 199:639–649
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ (2012) Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 30:434–439. doi:10.1038/nbt.2198
Lu H (2003) ACD6, a novel ankyrin protein, is a regulator and an effector of salicylic acid signaling in the Arabidopsis defense response. Plant Cell Online 15:2408–2420. doi:10.1105/tpc.015412
Mellersh DG, Heath MC (2003) An investigation into the involvement of defense signaling pathways in components of the nonhost resistance of Arabidopsis thaliana to rust fungi also reveals a model system for studying rust fungal compatibility. Mol Plant-Microbe Interact 16:398–404. doi:10.1094/MPMI.2003.16.5.398
Mofatto LS, Carneiro F, de A, Vieira, Duarte NG, Vidal KE, Alekcevetch RO, Cotta JC, Verdeil MG, Lapeyre-Montes J-L, Lartaud F, Leroy M, De Bellis T, Pot F, Rodrigues D, Carazzolle GC, Pereira MF, Andrade GAG, Marraccini AC P (2016) Identification of candidate genes for drought tolerance in coffee by high-throughput sequencing in the shoot apex of different Coffea arabica cultivars. BMC Plant Biol 16:94. doi:10.1186/s12870-016-0777-5
Narusaka M, Kubo Y, Hatakeyama K, Imamura J, Ezura H, Nanasato Y, Tabei Y, Takano Y, Shirasu K NY (2013) Interfamily transfer of dual NB-LRR genes confers resistance to multiple pathogens. PLoS ONE 8:e55954. doi:10.1371/journal.pone.0055954
Niks RE, Rubiales D (2002) Potentially durable resistance mechanisms in plants to specialised fungal pathogens. Euphytica 124:201–216. doi:10.1023/A:1015634617334
Niks RE, Qi X, Marcel TC (2015) quantitative resistance to biotrophic filamentous plant pathogens: concepts, misconceptions, and mechanisms. Annu Rev Phytopathol 53:445–470. doi:10.1146/annurev-phyto-080614-115928
Nirmala J, Drader T, Chen X, Steffenson B, Kleinhofs A (2010) Stem rust spores elicit rapid RPG1 phosphorylation. Mol Plant-Microbe Interact 23:1635–1642. doi:10.1094/MPMI-06-10-0136
Oliveira MDM, Varanda CMR, Félix MRF (2016) Induced resistance during the interaction pathogen x plant and the use of resistance inducers. Phytochem Lett 15:152–158. doi:10.1016/j.phytol.2015.12.011
Ozsolak F, Milos P (2010) RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12:87–98
Peck SC (2003) Early phosphorylation events in biotic stress. Curr Opin Plant Biol 6:334–338. doi:10.1016/S1369-5266(03)00056-6
Pedras MSC, Yaya EE, Glawischnig E (2011) The phytoalexins from cultivated and wild crucifers: chemistry and biology. Nat Prod Rep 28:1381. doi:10.1039/c1np00020a
Pereira AA, Zambolim L, Chaves GM, Sakiyama NS (2000) Cultivar de café resistente à Ferrugem: Oeiras-MG 6851. Rev Ceres 46:121–124
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi:10.1093/bioinformatics/btp616
Rodrigues CJ, Bettencourt AJ, Rijo L (1975) Races of the pathogen and resistance to coffee rust. Annu Rev Phytopathol 13:49–70. doi:10.1146/annurev.py.13.090175.000405
Rodrigues Jr CJ, Varzea VMP, Godinho IL, Palma S, Rato RC (1993) New physiologic races of Hemileia vastatrix. In: 15° Colloque Scientifique International sur le café. Montpellier, France, pp 318–321
Rodrigues Jr CJ, Gonçalves MM, Várzea VMP (2004) Importância do Híbrido de Timor para o território e para o melhoramento da cafeicultura mundial. Rev Ciências Agrárias 27:203–213
Rubio M, Rodríguez-Moreno L, Ballester AR, de Moura MC, Bonghi C, Candresse T, Martínez-Gómez P (2015) Analysis of gene expression changes in peach leaves in response to Plum pox virus infection using RNA-SEq. Mol Plant Pathol 16:164–176. doi:10.1111/mpp.12169
Schulze-Lefert P, Panstruga R (2003) Establishment of biotrophy by parasitic fungi and reprogramming of host cells for disease resistance. Annu Rev Phytopathol 41:641–667. doi:10.1146/annurev.phyto.41.061002.083300
Sharma M, Pandey GK (2016) Expansion and function of repeat domain proteins during stress and development in plants. Front Plant Sci 6:1218. doi:10.3389/fpls.2015.01218
Shibuya N, Minami E (2001) Oligosaccharide signalling for defence responses in plant. Physiol Mol Plant Pathol 59:223–233. doi:10.1006/pmpp.2001.0364
Silva MC, Nicole M, Guerra-GuimarÃes L, Rodrigues CJ (2002) Hypersensitive cell death and post-haustorial defence responses arrest the orange rust (Hemileia vastatrix) growth in resistant coffee leaves. Physiol Mol Plant Pathol 60:169–183. doi:10.1006/pmpp.2002.0389
Silva M do, Várzea C, Guerra-Guimarães V, Azinheira L, Fernandez HG, Petitot D, Bertrand A-S, Lashermes B, Nicole P M (2006) Coffee resistance to the main diseases: leaf rust and coffee berry disease. Brazilian J Plant Physiol 18:119–147. doi:10.1590/S1677-04202006000100010
Takahashi S, Yeo Y-S, Zhao Y, O’Maille PE, Greenhagen BT, Noel JP, Coates RM, Chappell J (2007) Functional characterization of premnaspirodiene oxygenase, a cytochrome P450 catalyzing regio- and stereo-specific hydroxylations of diverse sesquiterpene substrates. J Biol Chem 282:31744–31754. doi:10.1074/jbc.M703378200
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-SEq. Bioinformatics 25:1105–1111. doi:10.1093/bioinformatics/btp120
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi:10.1038/nbt.1621
Várzea VMP, Marques DV (2005) Resistance, population variability of Hemileia vastatrix vs. coffee durable. In: Zambolim L, Zambolim E, Várzea VMP (eds) Durable resistance to coffee leaf rust. UFV Press, Viçosa, pp 53–74
Voegele RT, Mendgen K (2003) Rust haustoria: nutrient uptake and beyond. New Phytol 159:93–100. doi:10.1046/j.1469-8137.2003.00761.x
Wang Y-S, Pi L-Y, Chen X, Chakrabarty PK, Jiang J, De Leon AL, Liu G-Z, Li L, Benny U, Oard J, Ronald PC, Song W-Y (2006) Rice XA21 binding protein 3 is a ubiquitin ligase required for full Xa21-mediated disease resistance. Plant Cell Online 18:3635–3646. doi:10.1105/tpc.106.046730
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi:10.1038/nrg2484
Wang X, McCallum BD, Fetch T, Bakkeren G, Saville BJ (2015) Sr36- and Sr5-mediated resistance response to Puccinia graminis f. sp. tritici is associated with callose deposition in wheat guard cells. Phytopathology 105:728–737. doi:10.1094/PHYTO-08-14-0213-R
Westermann AJ, Gorski SA, Vogel J (2012) Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630. doi:10.1038/nrmicro2852
Xin M, Wang X, Peng H, Yao Y, Xie C, Han Y, Ni Z, Sun Q (2012) Transcriptome comparison of susceptible and resistant wheat in response to powdery mildew infection. Genom Proteom Bioinform 10:94–106. doi:10.1016/j.gpb.2012.05.002
Young SA, Guo A, Guikema JA, White FF, Leach JE (1995) Rice cationic peroxidase accumulates in xylem vessels during incompatible interactions with Xanthomonas oryzae pv oryzae. Plant Physiol 107:1333–1341. doi:10.1104/pp.107.4.1333
Yuyama PM, Junior OR, Ivamoto ST, Domingues DS, Carazzolle MF, Pereira GA, Charmetant P, Leroy T, Pereira LP (2016) Transcriptome analysis in Coffea eugenioides, an Arabica coffee ancestor, reveals differentially expressed genes in leaves and fruits. Mol Genet Genom 291:323–336. doi:10.1007/s00438-015-1111-x
Zambolim L (2016) Current status and management of coffee leaf rust in Brazil. Trop Plant Pathol 41:1–8. doi:10.1007/s40858-016-0065-9
Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30:614–620. doi:10.1093/bioinformatics/btt593
Acknowledgements
We thank Jorge Badel for his helpful suggestions during the preparation of the manuscript. We are also grateful to CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) for the scholarship and Brazilian Coffee Research and Development Consortium (Consórcio Brasileiro de Pesquisa e Desenvolvimento do Café – CBP&D/Café), the Foundation for Research Support of the state of Minas Gerais (FAPEMIG), National Council of Scientific and Technological Development (CNPq), and National Institutes of Science and Technology of Coffee (INCT/Café) for the financial support, DTI (Diretoria de Tecnologia da Informação) of the Universidade Federal de Viçosa and LGE (Laborátorio de Genômica e Expressão) of the Universidade Estadual de Campinas for providing bioinformatics facilities.
Author information
Authors and Affiliations
Contributions
JCF, LCM and MFC analyzed the sequencing data for transcriptome assembly; RDLFL and EMZ developed the biological experiments for the sequencing; JCF and SSF developed the qPCR experiments; Wrote the manuscript draft: JCF, SSF and ETC edited and revised the manuscript; ETC and LZ formulated the idea.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Additional information
Juan Carlos Florez and Luciana Souto Mofatto have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Florez, J.C., Mofatto, L.S., do Livramento Freitas-Lopes, R. et al. High throughput transcriptome analysis of coffee reveals prehaustorial resistance in response to Hemileia vastatrix infection. Plant Mol Biol 95, 607–623 (2017). https://doi.org/10.1007/s11103-017-0676-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-017-0676-7