
Abstract
Key message
This piece of the submission is being sent via mail.
Abstract
Leaf senescence is essential for the nutrient economy of crops and is executed by so-called senescence-associated genes (SAGs). Here we explored the monocot C4 model crop Sorghum bicolor for a holistic picture of SAG profiles by RNA-seq. Leaf samples were collected at four stages during developmental senescence, and in total, 3396 SAGs were identified, predominantly enriched in GO categories of metabolic processes and catalytic activities. These genes were enriched in 13 KEGG pathways, wherein flavonoid and phenylpropanoid biosynthesis and phenylalanine metabolism were overrepresented. Seven regions on Chromosomes 1, 4, 5 and 7 contained SAG ‘hotspots’ of duplicated genes or members of cupin superfamily involved in manganese ion binding and nutrient reservoir activity. Forty-eight expression clusters were identified, and the candidate orthologues of the known important senescence transcription factors such as ORE1, EIN3 and WRKY53 showed “SAG” expression patterns, implicating their possible roles in regulating sorghum leaf senescence. Comparison of developmental senescence with salt- and dark- induced senescence allowed for the identification of 507 common SAGs, 1996 developmental specific SAGs as well as 176 potential markers for monitoring senescence in sorghum. Taken together, these data provide valuable resources for comparative genomics analyses of leaf senescence and potential targets for the manipulation of genetic improvement of Sorghum bicolor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Senescence is the final step of leaf development and was proposed to be selected through evolution to improve nutrient economy and the survival rate of plants (Leopold 1961). It has been noted for long that leaf senescence and crop yield are highly correlated, especially in cereals (Long et al. 2006; Richards 2000; Wu et al. 2012; Yang and Zhang 2006). Indeed, nutrient remobilisation could well explain the senescence syndrome (Bleecker 1998; Masclaux-Daubresse et al. 2008). However, it seems that leaf senescence has bi-directional effects on crop production. Early leaf senescence could decrease crop yields in general but increase pre-anthesis nitrogen use efficiency under low nitrogen condition (Gregersen et al. 2008). On the other hand, delaying leaf senescence may lower the nitrogen use efficiency although it may increase the final yield (Masclaux-Daubresse and Chardon 2011). Hence, fine-tuning the onset and progression of leaf senescence is one of the most important pre-requisites to achieve efficient crop production.
At the molecular level, the senescence syndrome is reflected by the changes in the expression of Senescence-Associated Genes (SAGs), suggesting that leaf senescence requires transcriptional adaptation. Indeed, both transcription and translation inhibitors effectively block the senescence programme (Noodén 1988). As a matter of fact, the up-regulation of SAGs is one of the hallmarks of leaf senescence (Zhou and Gan 2010). Senescence is a complex process which involves thousands of SAGs with diverse functions and pathways (Breeze et al. 2011; Buchanan-Wollaston et al. 2005; Van der Graaff et al. 2006). Genome-wide studies in Arabidopsis have shown that SAGs generally fall into four major categories: regulation, macromolecule degradation, nutrient mobilisation and transportation, as well as stress responses (Breeze et al. 2011). Some important genes belonging to these four categories have been cloned and functionally characterized, such as AtNAP (Guo and Gan 2006), ORE1 (Balazadeh et al. 2010; Kim et al. 2009; Rauf et al. 2013), WRKY53 (Miao et al. 2004; Zentgraf et al. 2010), ORE9 (Woo et al. 2001), LOX1 (He et al. 2002; Li et al. 2012), SAG101 (Chen et al. 2015; He and Gan 2002) and NTL4 (Lee et al. 2012). To date, transcriptome profiling of SAGs has been conducted in a limited number of crops. A survey of developmental leaf senescence in cotton (Gossypium hirsutum) showed that 3624 genes were differentially expressed and abundant transcription factors were involved (Lin et al. 2015). A study on leaf senescence in switchgrass (Panicum virgatum) revealed the enhancement of transport processes and potential roles of the NAC transcription factors in nutrient remobilisation (Palmer et al. 2015). During the developmental leaf senescence of maize (Zea mays), 4552 differentially expressed genes were identified and placed into 18 functional categories (Zhang et al. 2014b). These studies, together with those in wheat (Triticum aestivum) (Gregersen and Holm 2007), rice (Oryza sativa) (Liu et al. 2008) and Medicago truncatula (De Michele et al. 2009), further implicate that SAGs primarily consist of genes with function in gene expression regulation, macromolecule metabolism, nutrient mobilisation and transportation, and stress responses. Nonetheless, more crops, especially major cereals, need to be examined to have a holistic picture of the patterns and functional categories of genes involved in leaf senescence.
As one of the most important multipurpose crops, sorghum (Sorghum bicolor) is a major source of food, feed, fibre and fuel. For the underdeveloped regions or low income countries of the world, sorghum remains the primary source of energy and nutrition (FAO 1995). Varieties with high stem juice and sugar content, high biomass and high tolerance to stresses, known as sweet sorghum, are natural variants of the common grain sorghum and considered as an ideal biofuel crop for marginal land (Calvino and Messing 2012). In sorghum, it has been shown that the stay-green trait, or delayed leaf senescence, can substantially improve the post-flowering drought-tolerance (Diourte et al. 1995; Rosenow et al. 1983; Xu et al. 2000). And so far, four major QTLs named Stg1, Stg2, Stg3, and Stg4 as well as many additional minor QTLs have been identified and shown to condition the stay-green trait (Anami et al. 2015b; Crasta et al. 1999; Haussmann et al. 2002; Sanchez et al. 2002; Subudhi et al. 2000; Tao et al. 2000; Xu et al. 2000). However, the knowledge of molecular mechanisms controlling leaf senescence is still limited. Development of NGS technologies and the release of a complete genome sequence of sorghum (Paterson et al. 2009) provide an opportunity to investigate the senescence process at the whole genome scale. In addition, RNA-seq was employed to annotate the transcriptome of sorghum’s responses to osmotic stress and abscisic acid (Dugas et al. 2011). It was also revealed that pathogen infection could remarkably change the expression of particular paralogues which potentially encode components in the sorghum specific metabolic network (Mizuno et al. 2012). Putative key genes in the sorghum-Bipolaris sorghicola interaction were identified by simultaneous transcriptome analysis and de novo assembly (Yazawa et al. 2013). RNA-seq was also used to find aerial-shoot and rhizome specific expressed genes in Sorghum propinquum, the wild relative of sorghum (Zhang et al. 2014a).
Here, we employed the RNA-seq technology to examine dynamics of gene expression during developmental leaf senescence in sorghum. We identified 3396 SAGs by analysing the SAG expression profiles of a typical Chinese sorghum (kaoliang, Ji2731) with prominent senescence phenotype. Ji2731 has been used in many hybrid breeding programmes in China and was recently re-sequenced (Zheng et al. 2011). The results of detailed analyses of the SAGs gave us a global picture of biological processes and metabolic pathways involved and the key regulatory components of the senescence process in sorghum. We also compared the SAG profile of developmental leaf senescence with those induced by salt and darkness and defined a common set of senescence associated marker genes for sorghum.
Materials and methods
Plant material and growth conditions
The sorghum line used for the experiment is a typical Chinese sorghum (kaoliang, Ji2731), which does not have juice accumulation in the stem but has high grain yield, good seedling establishment and a short growth period (Zheng et al. 2011). In a collection of 46 sorghum accessions, Ji2731 showed a prominent senescence phenotype and was the most sensitive line to NaCl treatment (Wang et al. 2014). Seeds were first surface-sterilised, treated with 75 % ethanol for 1-min and 3 % NaClO solution for 15-min, and transferred to Petri dishes with saturated moist filter paper and let for germination for 7 days prior to transplanting into soil or hydroponic culture solutions.
For developmental senescence and dark-induced senescence, young seedlings were transferred to 20-cm-diameter pots in a potting mixture with commercially available peat moss substrates and growing media (PINDSTRUP SPHAGNUM, Denmark) and garden soil (1:1, v:v) in a greenhouse where temperature was set at 24 °C and photoperiod 10 h/14 h (day/night). Watering was applied when necessary and Hoagland solution applied equally in each pot every 2 weeks so that plant growth would not be limited by nutrients.
For salt-induced senescence, young seedlings were cultured with deionised water for 1 day, 1/4 Hogland solution for 2 days, 1/2 Hogland solution for 3 days, and then 1/2 Hogland solution for every 2 days until 10 days. Afterwards, seedlings were transferred to 200 mM NaCl solution as salt treatment.
Leaf sampling
We chose the sixth leaves (counted from the bottom of the plant) of individual plants for developmental and dark-induced senescence. In sorghum, leaf senescence follows the typical pattern of monocots with the initiation of yellowing from the tip of the leaf blade and subsequent progression inward to leaf sheath. For developmental senescence, leaves were harvested at four stages, which were arbitrarily defined as mature (Mature), early senescence (ES), middle senescence (MS) and late senescence (LS). The four stages were defined according to the yellowing ratio of the leaf blade area; the Mature stage was defined when a leaf reached full expansion and showed no signs of yellowing, the ES leaves were those with 10 % yellowing of the leaf blade, the MS leaves with yellowing areas between 10 and 25 %, and the LS leaves with yellowing areas above 50 %. Under our growth conditions, the sixth leaves of sorghum reached full maturity at about 54 days after sowing (DAS) when the eleventh leaves were about to emerge. The sixth leaves reached ES, MS and LS at about 61 DAS, 70 DAS and 73 DAS, respectively. For dark-induced senescence, we cut the middle part of a fully expanded leaf (54 DAS) for dark treatment. Samples were harvested every second days until the sixth day when the leaf yellow ratio was over 50 %. Fully expanded leaves before dark treatment were set as D0, and the rest were sequentially set as D2, D4, and D6. For salt-induced senescence, the fully expanded fourth leaves (23 DAS) prior to salt treatment were harvested as S0. Leaves samples from plants following 2-, 4- or 5-day’s salt treatment were harvested and designated as S2, S4, S5, respectively. All experiments were repeated at least twice.
Chlorophyll content measurement
Chlorophyll of leaf samples was extracted by 80 % acetone solution with 24-hour dark-incubation at room temperature. Absorbance of light with wave length 663 and 645 nm was measured with spectrophotometer (MAPADA, Shanghai) and recorded as A663 and A645, respectively. Contents of chlorophyll a (Ca), chlorophyll b (Cb) and total chlorophyll (Ct) were calculated by the formulae below:
Ca (mg·L−1) = 12.7A663 − 2.69A645, Cb (mg·L−1) = 22.9A645 − 4.68A663, Ct (mg·L−1) = Ca + Cb = 20.2A645 + 8.02A663. Content of chlorophyll in leaf samples (CL) was calculated by the following formula
C represents Ca, Cb or Ct (mg·L−1), V represents the volume of extracted solution (mL) and W represents weight of leaf sample (g).
RNA extraction, pooling and sequencing
Leaf samples from 10 plants in one replication were mixed for RNA extraction. RNA was extracted using the RNApre pure Plant Kit (TIANGEN, Beijing) according to the Manufacturer’s recommendations. RNA was subsequently quantified by NANODROP 1000 spectrophotometer (Thermo, USA). For each time-point, RNA from two replications was combined as one RNA pool and RNA pools of Mature, ES, MS, LS, D0, D6, S0 and S5 were used for RNA-seq. Libraries of each RNA pool (one library for each pool) were constructed with Illumina Truseq RNA Sample Prep Kits (Illumina, Santiago). Insertion length of the fragments were about 130–150 bp. Pair-end sequencing was performed on an Illumina HiSeq2000 sequencer. The length of raw reads was 101 bp. The whole set of original sequence data in fastq format has been deposited in the NCBI Short Read Archive under the study accession SRP054999.
Identification of differentially expressed genes (DEGs)
Adapter contained reads were firstly deleted from raw reads using the fastx_clipper command in the FASTX-Toolkit (v0.0.13). The parameters for fastx_clipper in use are “-C -Q 33 -n -M 33”. Using an in-house Perl script, low quality reads were cut or filtered. The filtering rules were set as follows: the maximum percentage of bases of which the quality score <20 was set as 20 %, the maximum percentage of bases of which the quality score <13 was set as 10 % and the minimum average quality score was set as 20. The last base of a read that could not fulfill the above conditions was deleted to make reevaluation for the read. And the procedure was repeated until the read fulfilled the conditions or otherwise was deleted when the length was <50. After the filtering steps, the remaining single reads were deleted to get clean paired reads.
A published pipeline (Trapnell et al. 2012) was used for DEGs identification. We made index for sorghum genome sequence with Bowtie2 (v2.0.2), mapped the clean reads to the sorghum reference genome with Tophat (v2.0.6) and identified DEGs with Cuffdiff (v2.0.1), a part of the Cufflinks package (Trapnell et al. 2010), based on the annotation. Within the result file, genes assigned as significant were identified as DEGs. Sequences of sorghum genome (Paterson et al. 2009) and genes annotation files were downloaded from Ensembl Plants (release 16) (Kersey et al. 2014).
Semi-quantitative RT-PCR (RT-PCR) validation and quantitative real-time PCR (qPCR) test
RNA samples used for semi-quantitative RT-PCR (RT-PCR) validation were the same as used for sequencing. ReverTraAce kit (Toyobo) was used to synthesize first strand cDNA from 1 mg of total RNA in a 10-mL reaction volume. For quantitative real-time PCR (qPCR), leaf samples were collected before dark-treatment (green at the Mature stage) and after dark-treatment (25 and 50 % yellowing). Total RNA and cDNA was prepared as mentioned above. Quantitative real-time PCR analysis was performed with an Eco system (Illumina) using SYBR Green Realtime PCR Master Mix (Toyobo). The relative expression levels were quantified using the comparative CT method (2−∆∆Ct) (Livak and Schmittgen 2001). The Pearson correlation coefficients of the expression patterns of selected differentially expressed TFs between qPCR and developmental transcriptome were calculated using the SPSS software.
Clustering of DEGs expression patterns
The expression value of each gene was evaluated by Cufflinks using FPKM value. We took the logarithm (base 10) for each FPKM value. Formatted data were used for clustering. We used a time series-clustering software SplineCluster (Heard et al. 2006) to cluster the formatted data of the identified DEGs. The parameter priorprecision was set as 0.001 because it generates the most divided clusters.
GO enrichment analysis
We performed GO enrichment analysis with the agriGO toolkit (Du et al. 2010). Sorghum GO annotation in Biomart database of Ensembl plants (Kinsella et al. 2011) was used as customised reference. We selected hypergeometric test as the statistical test method and Yekutieli (FDR under dependency) as the multi-test adjustment method, or otherwise described in the text. The significance level was set at 0.05 and the minimum number of mapping entries was set at 5. Plant GO slim was used for GO enrichment analysis of all SAGs and complete GO was used for enriched GO comparison analysis for sorghum and Arabidopsis.
Pathway enrichment analysis
Pathway enrichment analysis was performed using the KOBAS 2.0 platform (Xie et al. 2011). First, we annotated query genes for KEGG genes in the format of Entrez ID of sorghum genes. Subsequently, we identified enriched pathways of query genes using Hypergeometric test with a False Discovery Rate (FDR) threshold 0.05 using the Benjamini and Hochberg procedure (Benjamini and Hochberg 1995). During the identification step, the KEGG pathway database was used (Kanehisa 2002). Entrez ID of sorghum genes were downloaded from Biomart of Ensembl Plants (Kinsella et al. 2011).
Transcription factors (TFs) and transcription factor binding site (TFBS) enrichment analysis
Transcription factor annotation in sorghum genome was acquired from the plant transcription factor database (PlantTFDB v2.0) (Zhang et al. 2011). TFs with data sources not from JGI (Joint Genome Institute) were filtered. A total of 1761 TFs belonging to 54 families were analysed. Numbers of TFs in each comparison were recorded for Fisher’s Exact Test (Fisher test function in the R package). Numbers of TFs in each family in sorghum genome were used as background. The p values were corrected for the number of clusters tested using FDR. Corrected p values <0.05 were considered significantly enriched.
Sequences in length of 2 kb up-stream of individual DEGs were extracted as promoters. TFBSs data set was obtained from the database of Plant cis-acting regulatory DNA elements (PLACE) (Higo et al. 1999). One WRKY53 binding site (Miao et al. 2004), two NAC binding sites (Olsen et al. 2005) and six EIN3 binding sites (Chang et al. 2013) were added to the TFBS data set. We detected each TFBS in promoters using an in-house Perl script. And the numbers of genes that contain the TFBS in their 2 kb promoters in each cluster were recorded for Fishers’ Exact Test and the p values were adjusted using the FDR method in R. FDR threshold was set to 0.05.
Results
Identification of differentially expressed genes during sorghum developmental leaf senescence by RNA-seq
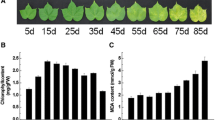
Prior to the large-scale RNA-seq work, we performed pilot experiments to assess visual leaf senescence in sorghum and optimised leaf sampling for RNA-seq. Leaf yellowing is a reliable visible symptom to measure the onset and progression of leaf senescence (Ougham et al. 2008; Wu et al. 2012). In this study, the stage when a leaf is fully expanded was defined as the Mature stage (Mature). We then arbitrarily divided the senescence process into three stages, Early Senescence (ES), Middle Senescence (MS) and Late Senescence (LS) according to the yellowing ratio of the leaf blade (Fig. 1a). Each senescence stage exhibited the expected decline of chlorophyll content (Fig. 1b). These results indicated that the samples could reflect the progression of leaf senescence and be used for the identification of SAGs.

Characterisation of developmental leaf senescence in sorghum. The four stages were arbitrary defined based on the visible yellowing and chlorophyll content of leaves. a, b Show representative photos and chlorophyll content of the four stages of leaf senescence, respectively. ES early senescence, MS middle senescence, LS late senescence
RNA samples from the four stages, one RNA pool for each stage, were then used for RNA-seq library construction and Illumina HiSeq 2000 sequencing. The sorghum genome was annotated to contain 34568 genes (v1.4) (Paterson et al. 2009). In the four developmental senescence stages, 24976 genes were detected (FPKM > 0, FPKM: fragments per kilobase of transcript sequence per millions base pairs). The expression levels of genes at the four developmental stages were pair-wisely compared and hence six comparisons obtained. In total, 4293 differentially expressed genes were identified, including 2351 genes with up-regulated expression only, 897 with down-regulated expression only, and 1045 genes with either up-down or down-up regulated expression (Table 1). Primers for 23 DEGs, including 20 up-regulated genes and three down-regulated genes (ESM Dataset1-s1), were designed and the expression of these genes were examined by semiquantitative reverse transcription polymerase chain reaction (RT-PCR). Twenty-one of the 23 genes showed similar expression patterns in both the RT-PCR and the transcriptome analyses (ESM Dataset1-s2), suggesting a high quality of the RNA-seq.
Associated with the dysfunction of a cell during leaf senescence, many cellular processes are suppressed, as envisaged by the discovery of genes with down-regulated expression. In this study, 897 down-regulated genes were found and were enriched in 12 GO categories of cellular component groups, including plastid, thylakoid, cytoplasmic part, intracellular membrane-bounded organelle and intracellular organelle part (ESM Dataset2-s1), confirming the notion that most down-regulated genes were photosynthesis-associated. In the KEGG pathway analysis, we did not find any particular enrichment (corrected p values <0.05) of genes, although in total there were 95 pathways involved (ESM Dataset2-s2). Furthermore, forty-six transcription factors were found to be down-regulated including members from bHLH, bZIP, MYB_related, MYB and WRKY families (ESM Dataset2-s3). The importance of these TFs during sorghum leaf senescence requires further functional analysis.
Due to the observation that both transcription and translation inhibitors effectively inhibit the leaf senescence programme (Noodén 1988), up-regulated genes during senescence process are considered as SAGs (Zhou and Gan 2010). Hence, the remaining part of this report focuses only on the analysis of the 3396 up-regulated genes, including 2351 genes only up-regulated and 1045 genes up-down and/or down-up regulated.
Profiles of SAGs during developmental leaf senescence in sorghum
As shown in Fig. 2, Gene Ontology (GO) analysis using plant GO slim of the AgriGO platform (Du et al. 2010) showed the enrichment of SAGs involved in 15 biological processes in terms of abiotic/biotic stress responses, endogenous/external stimuli, the metabolism of carbohydrates and secondary metabolites to catabolic processes. In the molecular function aspect, genes were enriched in three categories: catalytic activity, transferase activity and transporter activity. In the cellular component aspect, genes were enriched in six categories involved in extracellular region, vacuole, plastid and plasma membranes. Categories of “Metabolic process” and “Catalytic activity” were not shown in the figure for the reason that over 40 % genes were assigned to these two categories. Details of enriched SAGs could be found in supplementary material online (ESM Dataset3-s1).

Overrepresented GO categories of 3396 SAGs analysed by plant GO slim. The X axis shows the three major categories analysed. The overrepresented GO terms in each category are arranged in an increased order of FDR values (hypergeometric test, FDR < 0.05). The Y axis indicates the number of SAGs in each term
We further analysed the SAGs in detail and assigned them into four major groups (Fig. 3). In the regulation related group, genes with protein kinase activities accounted for the biggest share and were followed by genes with transcription factor activities. In the macromolecule degradation group, genes involved in carbohydrate metabolic process occupied the largest portion, genes involved in metal ion binding in the transportation related group, and the response to stress in the stress-related group. For comparison, we included the analysis of SAGs in Arabidopsis (Breeze et al. 2011) and maize (Zhang et al. 2014b). It was found that the category of responses to stress was constantly overrepresented in sorghum, Arabidopsis and maize (Fig. 3). Furthermore, SAGs were enriched in GO categories of surfur metabolic process, carbohydrate metabolic process, response to stress and secondary metabolic process both in sorghum and maize (Fig. 3). SAGs in sorghum were also enriched in the cellular catabolic processes for amino acids, nucleotides and nucleic acids, as well as the flavonoid metabolic process (Fig. 3).

Comparison of the percentages of SAGs in senescence related GO categories among sorghum, maize and Arabidopsis. GO categories were assigned in four groups of regulation, macromolecule degradation, transportation and stress related. Blue, green and red stars represent overrepresented GO categories in sorghum, maize and Arabidopsis, respectively. SAGs of Arabidopsis and maize were obtained from Breeze et al. (2011) and Zhang et al. (2014b)
Pathway enrichment analysis using KOBAS 2.0 (Xie et al. 2011) showed that sorghum SAGs were enriched in 13 pathways (Table 2, ESM Dataset3-s2), mainly in those related to the biosynthesis of secondary metabolites including flavonoids, phenylpropanoids, phenylalanines, flavones and flavonols. Pathways related to nitrogen metabolism were also overrepresented, including tyrosine metabolism, tropane, piperidine and pyridine alkaloid biosynthesis and cysteine and methionine metabolism pathways. Interestingly, only one pathway, the phenylalanine metabolism, was overrepresented in maize SAGs (Zhang et al. 2014b). In Arabidopsis, SAGs (Breeze et al. 2011) were enriched in seven pathways, including plant hormone signal transduction, ubiquitin mediated proteolysis, alpha-Linolenic acid metabolism, indole alkaloid biosynthesis (ESM Dataset3-s3). It appears that more distinctive pathways and genes are employed for leaf senescence amongst these species.
SAGs are overrepresented on seven regions on chromosomes 1, 4, 5 and 7
To investigate the distribution of SAGs among chromosomes and chromosomal regions, we analysed the density of all sorghum genes and SAGs on individual chromosomes using a 500-kb slide window (Fig. 4a, b).

Distributions of all genes and SAGs in the ten chromosomes of the sorghum genome. The window size was 500 kb. a Distribution of all genes. b Distribution of all SAGs. c–e Distribution of SAGs in stages from Mature to ES, MS and LS, respectively. Red stars indicate the SAGs enriched regions following fisher’s exact test with FDR < 0.05
Four regions were identified with condensed SAGs on Chromosomes 1, 5 and 7, as indicated by red stars in Fig. 4b. Genes in these ‘hotspots’ prone to contain duplicated genes or members of a superfamily (ESM Dataset4-s1). Within the 19 SAGs residing from 53.5 to 54 Mb on Chromosome 1, six copies of the GSTU18 and two copies of the GSTU17 were found. GO annotation suggests that GSTU18 may have glutathione transferase activity. GSTU17 was reported to play a negative role in drought and salt stress tolerance in Arabidopsis (Chen et al. 2012b). More interestingly, all nine SAGs of 12 genes from 49 MB to 49.5 Mb on Chromosome 5, were copies of the CHS (chalcone synthase gene). CHS is a key enzyme involved in the biosynthesis of flavonoids, and is required for the accumulation of purple anthocyanins in leaves and stems (Brown et al. 2001; Feinbaum and Ausubel 1988). Six CHS genes concomitantly increased their expression levels at the ES stage and three at the MS stage, and expression of all the nine genes were increased throughout the senescence process. Furthermore, within the 12 SAGs between 60.5–61 Mb on Chromosome 5, five of them were copies of the Bowman–Birk type bran trypsin inhibitor precursor gene (BBTI13) and two were copies of O-methyltransferase family genes. Finally, eight of the 10 SAGs between 7–7.5 Mb on Chromosome 7 were cupins superfamily genes and six of them were members of RmlC-like cupins superfamily genes. GO annotation showed that RmlC-like cupins superfamily genes had manganese ion binding and nutrient reservoir activity.
We next examined whether there are any ‘temporal hotspots’ for SAGs, to understand the co-expression of SAGs. Figure 4c–e show the SAGs rich regions at each stage. Hotspots of all SAGs on chromosomes 1 and 5 (60.5–61 Mb) showed temporal changes while hotspots on chromosomes 5 (49–49.5 Mb) and 7 did not. The hotspot with the CHS gene family on chromosome 5 (49–49.5 Mb) was consistently identified throughout the three stages while hotspot on Chromosome 7 was not identified in separated stages. Furthermore, three more hotspots were identified at MS and LS stages, containing six PAL (phenylalanine ammonia-lyase) genes and four PYR (pyrabactin resistance) like genes (Fig. 4c–e, ESM Dataset4-s2-s4). Upregulation of PAL and CHS might suggests the activation of the synthesis pathways derived from phenylalanine.
Forty-eight co-expression clusters were identified
To investigate the global co-expression patterns of the SAGs, we clustered 3396 SAGs using SplineCluster (Heard et al. 2006) and obtained 48 clusters. As shown in the heatmap (Fig. 5a), these 48 clusters were assigned to three major groups. Group I genes, including Clusters 1–9, represent those with relatively high basal levels of expression and were enriched in categories of responses to stimuli and stress as well as cellular nitrogen compound metabolic process. Group II genes, including Cluster 10–27, represent SAGs with intermediate expression levels and were enriched in categories of transmembrane transport activities, thiamin and derivative biosynthetic process, regulation of abscisic acid mediated signaling pathway and responses to reactive oxygen species. Group III genes, including cluster 28–48, represent SAGs with low expression levels and were enriched in categories of hormone metabolic process, glucan biosynthetic process, lipid metabolic process and nutrient reservoir activity. Categories of oxidoreductase activity and electron carrier activity were overrepresented in all the three groups.

SplineCluster analysis of 3396 developmental SAGs. a Heat map of the expression level of 3396 SAGs clustered into 48 clusters. Green color represents low expressed genes, while red color and black color represent highly expressed genes and intermediate genes, respectively. Base 10 logarithm of FPKM value 0 was set as −2 to pass through the software. b Forty-eight clusters for base 10 logarithm of FPKM value of the 3396 SAGs. The same color of cluster number indicates that those clusters are in the same group
Enriched GO terms can also be found in single clusters. Genes in Clusters 2, 8, 25 and 26, were enriched in chloroplast related processes; genes in Clusters 1, 6, and 41 in categories responsive to stress and stimuli; and genes in Clusters 5, 13, and 22 in transferase activities (Fig. 5b).
Candidate senescence regulating transcription factors and their target SAGs in sorghum
Transcription factors(TFs) in SAGs As shown in Table 3, a total of 222 TFs distributed in 32 TF families were identified as SAGs in sorghum. Five TFs families were statistically overrepresented (Fishers’ Exact Test) during sorghum leaf senescence, including NAC, ERF, WRKY, HSF and CO-like families. The NAC family SAGs were distributed in 11 clusters, mainly in cluster 13 and cluster 6, and were found to be omnipresent throughout all senescence stages. The ERF family SAGs were expressed at lower levels than those of NAC family SAGs and distributed in 12 clusters, primarily in cluster 32 and cluster 13. Their expression mainly increased after the MS stage. The WRKY family SAGs, most of which were expressed at intermediate levels, were distributed in 13 clusters primarily prior to the MS stage. The HSF family SAGs were distributed in 9 clusters and their expression increased in the ES stage. Four of the seven CO-like family SAGs were down-regulated at the ES stage. At the MS stage, their expression increased but not higher than those at the Mature Stage. NAC, ERF and WRKY families were prominently overrepresented of all TFs, and hence these families and their candidate targets were surveyed and presented below.
ORE1 homologues and candidate targets NAC domain transcription factors consist of a large portion of SAGs in leaf senescence (Andersson et al. 2004; Breeze et al. 2011; Buchanan-Wollaston et al. 2005; Guo et al. 2004; Lin and Wu 2004). In Arabidopsis, ORE1 (AT5G39610) was demonstrated to participate in the network of ethylene-regulated and age-dependent senescence (Kim et al. 2009). In sorghum, we found 16 genes whose protein sequences had 40.4–53.3 % similarity comparing with ORE1 in Arabidopsis. Nine of them were not expressed. Six of them increased expression and were identified as SAGs (Fig. 6a) and these six genes were selected as homologous genes of ORE1. Among these six genes, Sb01g036590 possesses of highest protein similarity (53.3 %) to ORE1 and was much higher expressed than the others (Fig. 6a). In 2010, Balazadeh et al. identified 170 target genes of ANAC092/ORE1 in Arabidopsis (Balazadeh et al. 2010). We searched for the homologues of these targets in sorghum, and 64 genes among them were identified as SAGs. Of these 64 genes, 60 contained at least one NAC binding motif (Olsen et al. 2005) in their 2 kb promoter sequences. However, different expression patterns of the 60 candidate target SAGs were found (Fig. 6b).

The expression patterns of representative TFs and their candidate targets. The expression patterns of ORE1, EIN3, WRKY53 homologues are shown in a, c, e, and their corresponding targets in b, d, f, respectively. The expression values of candidate targets are represented by the average expression values of candidate targets of SAGs in the corresponding clusters. And only five mostly SAGs containing clusters are shown
EIN3 homologues and candidate targets The role of the phytohormone ethylene in leaf senescence has been illustrated for long time (Abeles et al. 1988; Chen et al. 2012a; Jing et al. 2002; Patterson and Bleecker 2004; Stead et al. 2003). As a crucial regulator of ethylene signaling, EIN3 directly modifies expression of ethylene response factors (ERFs), the downstream regulatory factors of ethylene pathway which influence ethylene responses and stress responses (An et al. 2010; Konishi and Yanagisawa 2008; Yoo et al. 2008; Zhong et al. 2009; Zhu et al. 2011). There are one EIN3 gene (Sb01g036740) and five EIN3-like genes in sorghum (Table 4). Sb01g036740 was expressed at much higher level than the others (Fig. 6c). In Arabidopsis, Chang et al. employed a ChIP-Seq experiment and identified 1314 candidate targets of EIN3 (Chang et al. 2013). Among homologues of these 1314 genes in sorghum, 305 genes with different expression patterns were identified as SAGs, of which 175 contained at least one EIN3 binding motif in their 2 kb promoter sequences (Fig. 6d).
WRKY53 homologues and candidate targets WRKY53 is a well characterized senescence-regulating transcription factors and functions at the early stage of leaf senescence (Hinderhofer and Zentgraf 2001). There are four WRKY53 homologous genes in sorghum (Table 4). Sb02g022290, Sb05g001220 and Sb10g004000 were annotated as ATWRKY53 (AT4G23810) but with weak similarity to ATWRKY53. Sb09g015900 was annotated as ATWRKY33 (AT2G38470) and OsWRKY53 (LOC_Os05g27730). Sb09g015900 has much higher similarity with OsWRKY53 (76.6 %) than ATWRKY33 (42.9 %) and Sb09g015900 had the highest expression level (Fig. 6e). We propose that Sb09g015900 might be the WRKY53 homologue in sorghum. Of the 63 candidate targets identified in Arabidopsis (Miao et al. 2004), 19 homologous genes (based on the annotation) were identified as SAGs by this study, of which 12 contained at least one WRKY53 binding motif (Miao et al. 2004) in their 2 kb promoter sequences (Table 4). The expression level of most of these twelve genes were increased in the ES stage and kept being increasd to the LS stage (Fig. 6f).
We surveyed the distribution of transcription factor binding sites (TFBSs) in the promoter regions in the 48 clusters. Sequences of TFBSs were downloaded from the PLACE database (Higo et al. 1999) and examined for the binding sites of EIN3 (Chang et al. 2013), ANAC092/ORE1 (Olsen et al. 2005) and WRKY53 (Miao et al. 2004) individually. Enrichment patterns of 12 TFBSs in each cluster are shown in Fig. 7. In Cluster 3, overrepresented TFBSs including ABREATCONSENSUS, CACGTGMOTIF, GBOXLERBCS and IRO2OS contain the core sequence (“-ACGTG-”) of G-box, which are involved in response to ABA and binding with TFs in the bHLH family. Two additional TFBSs were overrepresented in Cluster 3, including MYBPZM and E2FCONSENSUS. MYBPZM is a TFBS identified in maize and recognised by a Myb homologue encoded by the maize P gene, which directly activated a flavonoid biosynthetic gene subset to control phlobaphene pigmentation in maize floral organs (Grotewold et al. 1994). E2FCONSENSUS is a TFBS recognised by TFs in the E2F family (Vandepoele et al. 2005), which control the temporal expression of genes that are needed for multiple processes during the cell cycle (van den Heuvel and Dyson 2008). In Cluster 6, overrepresented TFBSs including ACGTABOX, ACGTCBOX and CACGTGMOTIF contain the core sequence “-ACGT-”, which is the main binding site of TFs in the bZIP family (Izawa et al. 1993). CGCGBOXAT was also overrepresented in Cluster 6. It is a target of AtSR1 and involved in multiple signaling pathways including ethylene signaling, abscisic acid signaling, and light signal perception (Yang and Poovaiah 2002). TFBSs including CGCGBOXAT and an EIN3 de novo motif (Chang et al. 2013) were overrepresented in Cluster 16, revealing that some ethylene response genes might be enriched Cluster 16. A NAC TFBS (NAC_2) and a WRKY53 TFBS (WRKY53_target) were overrepresented in Cluster 35. This implies that candidate targets of NAC and WRKY TFs might be also enriched in Cluster35 and a cross talk of NAC family genes and WRKY family genes might exist.

Statistical analysis of 12 Trancsription factor binding site (TFBS) in the promoter regions (2 kb upperstream of ATG) of SAGs in each cluster. Colored backgrounds represent enriched TFBS with fdr < 0.05 (Fisher’s exact test). From yellow to red, the darker the color indicates that the FDR value is smaller
Identification of SAGs during stress-induced senescence in sorghum
To have a holistic view of sorghum SAGs and to identify a subset of SAGs specific for developmental leaf senescence, we also surveyed SAGs induced by dark and salt treatment. As shown in Fig. 8, typical senescence symptoms were observed. We then performed RNA-seq on samples collected before dark treatment (D0) and after 6-day dark treatment (D6) for dark-induced senescence and before salt treatment (S0) and after 5-day salt treatment (S5) for salt-induced senescence. Following the pipelines used for developmental senescence analysis, we acquired the data sets of differentially expressed genes during dark- and salt-induced senescence. In the end, 1444 and 1264 up-regulated genes were found for these two treatments, respectively. In dark-induced senescence, the up-regulated genes were enriched in GO categories of carbohydrate metabolic process, response to external/abiotic/extracellular stimulus, cellular amino acid and derivative metabolic process, lipid metabolic process, cell communication, vacuole and extracellular region part (Fig. 9a). On the other hand, the up-regulated genes in salt-induced senescence, were enriched in GO categories of response to abiotic/extracellular/endogenous stimulus, response to stress, cell communication and transcription factor activity (Fig. 9b). KEGG pathway enrichment analysis showed that two pathways including flavonoid biosynthesis and biosynthesis of secondary metabolites were overrepresented in dark-induced genes, while three pathways including glutathione metabolism, beta-Alanine metabolism and taurine and hypotaurine metabolism were overrepresented in salt-induced genes. Compared with developmental senescence, more dark-induced genes were enriched in the similar GO categories and KEGG pathways than salt-induced genes. Flavonoid biosynthesis pathway was overrepresented in both developmental and dark-induced leaf senescence, but not in salt-induced one. Among dark-induced genes, 122 genes were identified as transcription factors, mainly in ERF (21), NAC (17), bHLH (12), MYB (12), WRKY (10), bZIP (9), and Trihelix (8), while among salt-induced genes, 106 genes were identified as transcription factors, mainly in NAC (18), ERF (12), bZIP (8), MYB (8), WRKY (8) and bHLH (6). ERF family and Trihelix family were overrepresented in dark-induced genes, while NAC family was overrepresented in salt-induced genes. The main distributed TF families in dark- and salt-induced genes showed high similarity when compared with the TFs in developmental SAGs.

Characterisation of dark- and salt-induced leaf senescence in sorghum. a, b Show representative photos and chlorophyll content after 0-day, 2-day, 4-day and 6-day dark treatment, respectively. c, d Show representative photos and chlorophyll content after 0-day, 2-day, 4-day and 5-day salt treatment, respectively

Overrepresented GO categories of induced SAGs. a Shows the overrepresented GO categories in up-regulated genes of dark-induced senescence. b Shows the overrepresented GO categories in up-regulated genes of salt-induced senescence. GO enrichment analysis was performed in AgriGO platform using plant GO slim
Potential senescence marker genes identified by comparing developmental and induced senescence
One of the main goals in senescence study is to define molecular markers to monitor the onset and progression of the process. We define marker genes as those with relatively abundant expression levels and should show increasing expression levels along the progression of leaf senescence. The second consideration for marker genes is the timing of up-regulation, hence, genes showing up-regulation at ES, MS and LS should be sought for to represent the onset of leaf senescence at various stages. Furthermore, comparison of SAG profiles of developmental and induced senescence could lead to the identification of SAGs common to the core processes of senescence or specific to developmental senescence. We initially found that 507 up-regulated genes were shared by the three conditions (ESM Fig. S1, ESM Dataset5-s1), we then examined their expression profiles and found 12, 28 and 32 marker genes for monitoring senescence at early, middle and late stages, respectively (Table 5). These genes are proposed to be the core marker genes common to developmental and induced senescence. Among the core marker genes, 15 genes were mapped on KEGG pathways, the majority of which belong to biosynthesis pathways of secondary metabolites (seven genes) and pathway of alanine, aspartate and glutamate metabolism. MLS (Sb06g020720) and a decarboxylase coding gene (Sb07g003040) which were mapped to several KEGG pathways were expressed slightly at the Mature stage and up-regulated throughout the whole senescence stages. Five cytochrome P450 family genes were proposed to be the core marker genes, of which CYP81D4 (Sb03g040280) and CYP71B34 (Sb07g000510) for early senescence, CYP72B1 (Sb10g023380) and CYP71B35 (Sb06g020210) for middle senescence, and CYP704A2 (Sb01g031080) for late senescence. All the five cytochrome P450 family genes were expressed at relatively low levels at the Mature stage and substantially increased the expression levels at the LS stage, suggesting cytochrome P450 genes play important roles during senescence. In plants, a wide range of biosynthetic processes, leading to plant hormones and defensive compounds involve Cytochrome P450. Thus, Cytochrome P450 genes could be important for senescence in sorghum.
Similarly, we identified 1996 genes specifically up-regulated during developmental senescence (ESM Fig. S1, ESM Dataset5-s2) by a three-way comparison of the profiles of the up-regulated genes during developmental and induced senescence. Further, by analysing their expression patterns, we identified 104 marker genes specific for developmental senescence in sorghum (Table 6). Interestingly, the majority of those genes (96 genes) showed late up-regulation patterns, and only limited numbers of middle senescence marker genes (eight genes) were found. Among the developmental senescence specific marker genes, 22 genes were mapped on KEGG pathways, most of which belong to biosynthesis pathways of secondary metabolites (nine genes) and pathways of amino sugar and nucleotide sugar metabolism (three Chitinase A genes). Two WRKY family genes were identified as developmental senescence marker genes, in which Sb04g034440 was the middle senescence marker gene and Sb03g026280 was the late senescence marker gene. One NAC family gene (Sb01g006410) was identified as middle senescence marker gene. Two ARF family genes (Sb04g003240, Sb04g026610) and one ERF family gene (Sb10g001620) were identified as late senescence marker genes.
To test the usefulness of these marker genes in sorghum leaf senescence, we surveyed the expression patterns of 17 differentially expressed TFs in dark-induced senescence. The expression of 10 genes, including CBF2 (Sb02g030300), bZIP21 (Sb03g040530), WRKY51 (Sb03g032800), BIM1 (Sb02g027460), ANAC081 (Sb03g037940), EDF2 (Sb03g031860), WIP4 (Sb03g040490), BHLH92 (Sb03g036100), LUX (Sb03g039610) and GT2 (Sb04g033390), were increased both in transcriptome of dark-treatment and qPCR. While the expression of four genes including HEC1 (Sb02g026530), bZIP23 (Sb03g037300), Sb03g040160, and RL6 (Sb09g029560) were decreased both in transcriptome of dark-treatment and qPCR. Amongst the ten up-regulated genes, ANAC081, EDF2, and WIP4 were also identified as SAGs during developmental senescence, while GT2 was identified as one of the core SAGs. The expression of 12 differentially expressed TFs in dark-induced stages (Green, 25 % yellowing and 50 % yellowing) and developmental stages (Mature, MS and LS) had a high correlation (Fig. 10).

qPCR test of selected differentially expressed TFs dark-induced senescence. The histograms represent the relative intensity of qPCR from independent biological replicates (left y-axis); the lines represent the FPKM value of development transcriptome (right y-axis). r Indicates the pearson correlation coefficient value. DGE represents digital gene expression
Discussion
SAGs are the drivers of the complex process of leaf senescence, a very important stage for leaf development and influences crop production. In order to have a holistic picture of events in plant senescence, transcriptomic analyses of leaf senescence are required in more plants, particularly in major crops. Sorghum is a multipurpose crop and is a fail-proof food supply for arid and semi-arid low income countries (Anami et al. 2015a). Delayed leaf senescence or functional stay-green is an important trait for the survival of sorghum in harsh drought environments. Nonetheless, the molecular mechanism to understand this remarkable trait is still not well addressed. We present here a comprehensive characterisation of the transcriptome of sorghum leaves during developmental senescence.
Characteristics of leaf senescence transcriptome in sorghum
In all four developmental senescence stages, 24976 genes consisting of about 72.3 % of whole sorghum genome were detected (FPKM > 0). Furthermore, numbers of detected genes and genes expressed at different regions of expression values did not show much difference in the four stages (ESM Fig. S2). Otherwise, numbers of genes expressed at low level (1 < FPKM < 10) accounted for the vast majority of expressed genes in each stages and the trends were similar among four stages. This implicates that numbers of expressed genes were relatively stable throughout developmental processes of sorghum leaves. In sorghum, about 9.8 % genes of whole genome were identified as SAGs, while about 9.7 % in Arabidopsis (Breeze et al. 2011) and 9.6 %, in maize, respectively. Nonetheless, although similar proportions of the genomes were involved in the senescence process, the overrepresented pathways of the SAGs were rather different in these species.
Seven SAG condensed regions were identified in sorghum and four regions were found specifically for SAGs in different senescence stages. The SAGs in six condensed regions are paralogs of GSTU18, CHS, PAL, PYL6 and BBTI13 or genes of the RmlC-like cupin superfamily. GSTs were proposed to play a protective role under various stress conditions by reducing endogenous plant toxins which accumulate when oxidative stress increasing (Marrs 1996; Sappl et al. 2009). In plants, expression of GST could be induced by phytohormones and various stresses (Deng et al. 2007; Marrs 1996; Moons 2003). The hotspot of GSTU18 might indicate increased endogenous oxidative stress and active phytohormones signaling pathways. It has been reported that six of the nine duplicated CHS genes on chromosome 5 were up-regulated in sorghum leaves infected by Bipolaris sorghicola while several PAL genes were highly expressed but not differentially expressed after pathogen infection (Mizuno et al. 2012). Duplication of CHS genes and PAL genes reveals the importance of secondary metabolites in sorghum. These genes in the hotspot regions showed similar expression patterns during leaf senescence. Gene duplication is widely recongnised as one of the primary driving forces in the evolution of genomes and genetic systems (Engel et al. 1970; Gu et al. 2003). On the aspect of evolution, duplicated SAGs and their divergent expression patterns during senescence may facilitate plants adaptation to various environments or increase the efficiencies of the relevant processes.
Transcription factors and their candidate targets
Transcriptional regulation of leaf senescence is achieved through multiple members of transcription factor families. In this study, 222 transcription factors were identified as SAGs, of which 199 TFs exhibited low expression levels. It suggests that most TFs does not need to keep high expression level to function during senescence. NAC, ERF and WRKY families were overrepresented in the senescence regulatory TFs in sorghum, further confirming their importance as global leaf senescence regulators. Interestingly, CO-like family was identified to be significantly down-regulated during late senescence in Arabidopsis (Breeze et al. 2011), but in our research, CO-like family members are significantly up-regulated. Furthermore, 233 TFs were identified as DEGs in leaf senescence transcriptome in maize (Zhang et al. 2014b). Among those TFs, 184 were up-regulated during leaf senescence, such as the MYB, WRKY, bHLH, HS, NAC, AP2/EREBP, CO-like and bZIP families. In cotton (Gossypium hirsutum L.), WRKY and C3H family were significantly up-regulated at early senescence, NAC family showed altered expression at various times during senescence, and bHLH, GRAS, MYB, DBP and AP2-EREBP TFs were also up-regulated during the early stages (Lin et al. 2015).
Using Arabidopsis as the reference, we selected the candidate targets of ORE1, EIN3 and WRKY53 in sorghum. However, when searching for binding motifs of TFs in 2-kb promoter regions, we could not find the binding motifs in all the candidate targets. This may suggest that it is not sufficient to identify targets just by searching the homologues but the promoter should also be considered. Surprisingly, our analysis showed that the promoter regions of about 33.1 % of the SAGs contain the binding motifs for ORE1, 56.3 % for EIN3, and 60.9 % for WRKY53. It suggested that the majority of the SAGs could be regulated by these three TF families, although not all the predicted binding motifs are authenticated. Furthermore, 403 SAGs (occupied about 11.9 % of all SAGs) concomitantly contained the ORE1, WRKY53 and EIN3 binding motifs (ESM Fig. S3), which implied cross-talks among transcription factors and complex regulation of leaf senescence.
Expression pattern of SAGs and potential marker genes for sorghum leaf senescence
The leaf senescence database (Li et al. 2014) indicates that 11 rice SAGs and 91 Arabidopsis SAGs promote senescence and could be potential senescence marker genes. NYE1 was identified to promote senescence both in rice (LOC_Os09g36200) and Arabidopsis (AT4G22920). The sorghum homologue (Sb02g030830) also showed an increased expression pattern during leaf senescence. In addition, five genes including LOX2, PHT5, FAAH, ACS2, PAO, which were identified to promote senescence in Arabidopsis, also increased expression during sorghum senescence. LOX2 is a gene that encodes a key enzyme in the jasmonate (JA) biosynthetic pathway (Schommer et al. 2008), and JA biosynthesis was reported to be up-regulated in senescing leaves (Seltmann et al. 2010). PHT5 was reported to influences the interaction between phosphate homeostasis and ethylene signaling (Nagarajan et al. 2011), while ACS2 gene encodes a key enzyme in ethylene biosynthetic pathway. These shared marker genes might suggest the important role of hormone signaling and biosynthesis and their cross-talks with other metabolic pathways during leaf senescence. Three homologues of SAG12 in sorghum, including Sb01g007820, Sb02g011230 and Sb04g003305, were expressed in developmental senescence. Sb01g007820, which is the best hit of See1 (GRMZM2G045706) gene in maize, was identified as SAG. Sb01g007820 was up-regulated form Mature to ES stage and MS to LS stage and also in the comparison of Mature to LS stage. One ERD1/SAG15 homologue (Sb04g021410) was identified as SAG during developmental senescence. Expression of Sb04g021410 was increased through all stages and was significantly higher than Mature stage. One SAG21 homologue (Sb03g012940) started to be expressed at ES stage and was significantly up-regulated at MS and LS stages when compared Mature stage.
Abbreviations
- cDNA:
-
Complementary DNA
- DEGs:
-
Differentially expressed genes
- DGE:
-
Digital gene expression
- ES:
-
Early senescence
- FDR:
-
False discovery rate
- FPKM:
-
Fragment per kilo base per million mapped fragment
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LS:
-
Late senescence
- MS:
-
Middle senescence
- NGS:
-
Next-generation sequencing
- QTL:
-
Quantitative trait locus
- SAGs:
-
Senescence-associated genes
- TF:
-
Transcription factor
- TFBS:
-
Transcription factor binding site
References
Abeles FB, Dunn LJ, Morgens P, Callahan A, Dinterman RE, Schmidt J (1988) Induction of 33-kD and 60-kD Peroxidases during Ethylene-induced senescence of cucumber cotyledons. Plant Physiol 87:609–615
An FY, Zhao QO, Ji YS, Li WY, Jiang ZQ, Yu XC, Zhang C, Han Y, He WR, Liu YD, Zhang SQ, Ecker JR, Guo HW (2010) Ethylene-induced stabilization of ETHYLENE INSENSITIVE3 and EIN3-LIKE1 Is Mediated by Proteasomal Degradation of EIN3 Binding F-Box 1 and 2 that requires EIN2 in Arabidopsis. Plant Cell 22:2384–2401
Anami SE, Zhang L-M, Xia Y, Zhang Y-M, Liu Z-Q, Jing H-C (2015a) Sweet sorghum ideotypes: genetic improvement of the biofuel syndrome. Food Energ Secur 4(3):159–177
Anami SE, Zhang LM, Xia Y, Zhang YM, Liu ZQ, Jing HC (2015b) Sweet sorghum ideotypes: genetic improvement of stress tolerance. Food Energ Secur 4:3–24
Andersson A, Keskitalo J, Sjodin A, Bhalerao R, Sterky F, Wissel K, Tandre K, Aspeborg H, Moyle R, Ohmiya Y, Bhalerao R, Brunner A, Gustafsson P, Karlsson J, Lundeberg J, Nilsson O, Sandberg G, Strauss S, Sundberg B, Uhlen M, Jansson S, Nilsson P (2004) A transcriptional timetable of autumn senescence Genome Biol 5(4):R24
Balazadeh S, Siddiqui H, Allu AD, Matallana-Ramirez LP, Caldana C, Mehrnia M, Zanor MI, Kohler B, Mueller-Roeber B (2010) A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J 62:250–264
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate – a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300
Bleecker AB (1998) The evolutionary basis of leaf senescence: method to the madness? Curr Opin Plant Biol 1:73–78
Breeze E, Harrison E, McHattie S, Hughes L, Hickman R, Hill C, Kiddle S, Kim YS, Penfold CA, Jenkins D, Zhang CJ, Morris K, Jenner C, Jackson S, Thomas B, Tabrett A, Legaie R, Moore JD, Wild DL, Ott S, Rand D, Beynon J, Denby K, Mead A, Buchanan-Wollaston V (2011) High-Resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 23:873–894
Brown DE, Rashotte AM, Murphy AS, Normanly J, Tague BW, Peer WA, Taiz L, Muday GK (2001) Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis. Plant Physiol 126:524–535
Buchanan-Wollaston V, Page T, Harrison E, Breeze E, Lim PO, Nam HG, Lin JF, Wu SH, Swidzinski J, Ishizaki K, Leaver CJ (2005) Comparative transcriptome analysis reveals significant differences in gene expression and signalling pathways between developmental and dark/starvation-induced senescence in Arabidopsis. Plant J 42:567–585
Calvino M, Messing J (2012) Sweet sorghum as a model system for bioenergy crops. Curr Opin Biotechnol 23:323–329
Chang KN, Zhong S, Weirauch MT, Hon G, Pelizzola M, Li H, Huang SS, Schmitz RJ, Urich MA, Kuo D, Nery JR, Qiao H, Yang A, Jamali A, Chen H, Ideker T, Ren B, Bar-Joseph Z, Hughes TR, Ecker JR (2013) Temporal transcriptional response to ethylene gas drives growth hormone cross-regulation in Arabidopsis. ELife 2:e00675
Chen G-H, Liu C-P, Chen S-CG, Wang L-C (2012a) Role of ARABIDOPSIS A-FIFTEEN in regulating leaf senescence involves response to reactive oxygen species and is dependent on ETHYLENE INSENSITIVE2. J Exp Bot 63:275–292
Chen JH, Jiang HW, Hsieh EJ, Chen HY, Chien CT, Hsieh HL, Lin TP (2012b) Drought and salt stress tolerance of an Arabidopsis Glutathione S-Transferase U17 knockout mutant are attributed to the combined effect of Glutathione and Abscisic Acid. Plant Physiol 158:340–351
Chen QF, Xu L, Tan WJ, Chen L, Qi H, Xie LJ, Chen MX, Liu BY, Yu LJ, Yao N, Zhang JH, Shu W, Xiao S (2015) Disruption of the Arabidopsis defense regulator Genes SAG101, EDS1, and PAD4 confers enhanced freezing tolerance. Mol Plant 8:1536–1549
Crasta OR, Xu WW, Rosenow DT, Mullet J, Nguyen HT (1999) Mapping of post-flowering drought resistance traits in grain sorghum: association between QTLs influencing premature senescence and maturity. Mol Gen Genet 262:579–588
De Michele R, Formentin E, Todesco M, Toppo S, Carimi F, Zottini M, Barizza E, Ferrarini A, Delledonne M, Fontana P, Lo Schiavo F (2009) Transcriptome analysis of Medicago truncatula leaf senescence: similarities and differences in metabolic and transcriptional regulations as compared with Arabidopsis, nodule senescence and nitric oxide signalling. New Phytol 181:563–575
Deng Z, Zhang X, Tang W, Oses-Prieto JA, Suzuki N, Gendron JM, Chen H, Guan S, Chalkley RJ, Peterman TK, Burlingame AL, Wang ZY (2007) A proteomics study of brassinosteroid response in Arabidopsis. Mol Cell Proteomics 6:2058–2071
Diourte M, Starr JL, Jeger MJ, Stack JP, Rosenow DT (1995) Charcoal Rot (Macrophomina-Phaseolina) Resistance and the effects of water-stress on disease development in Sorghum. Plant Pathol 44:196–202
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70
Dugas DV, Monaco MK, Olsen A, Klein RR, Kumari S, Ware D, Klein PE (2011) Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genomics 12:514–534
Engel W, Hof JO, Wolf U (1970) [Gene duplication by polyploid evolution: the isoenzyme of the sorbitol dehydrogenase in herring- and salmon-like fishes (Isospondyli)]. Humangenetik 9:157–163
FAO R (1995) Sorghum and millets in human nutrition. FAO Food Nutrition Series 27:16–19
Feinbaum RL, Ausubel FM (1988) Transcriptional regulation of the Arabidopsis thaliana chalcone synthase gene. Mol Cell Biol 8:1985–1992
Gregersen PL, Holm PB (2007) Transcriptome analysis of senescence in the flag leaf of wheat (Triticum aestivum L.) Plant Biotechnol J 5:192–206
Gregersen PL, Holm PB, Krupinska K (2008) Leaf senescence and nutrient remobilisation in barley and wheat. Plant Biol 10:37–49
Grotewold E, Drummond BJ, Bowen B, Peterson T (1994) The myb-homologous P gene controls phlobaphene pigmentation in maize floral organs by directly activating a flavonoid biosynthetic gene subset. Cell 76:543–553
Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li WH (2003) Role of duplicate genes in genetic robustness against null mutations. Nature 421:63–66
Guo YF, Gan SS (2006) AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J 46:601–612
Guo Y, Cai Z, Gan S (2004) Transcriptome of Arabidopsis leaf senescence. Plant Cell Environ 27:521–549
Haussmann BIG, Mahalakshmi V, Reddy BVS, Seetharama N, Hash CT, Geiger HH (2002) QTL mapping of stay-green in two sorghum recombinant inbred populations. Theor Appl Genet 106:133–142
He Y, Gan S (2002) A gene encoding an acyl hydrolase is involved in leaf senescence in Arabidopsis. Plant Cell 14:805–815
He Y, Fukushige H, Hildebrand DF, Gan S (2002) Evidence supporting a role of jasmonic acid in Arabidopsis leaf senescence. Plant Physiol 128:876–884
Heard NA, Holmes CC, Stephens DA (2006) A Quantitative Study of Gene Regulation Involved in the Immune Response of Anopheline Mosquitoes. J Amer Statistical Assoc 101:18–29
Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27:297–300
Hinderhofer K, Zentgraf U (2001) Identification of a transcription factor specifically expressed at the onset of leaf senescence. Planta 213:469–473
Izawa T, Foster R, Chua NH (1993) Plant bZIP protein DNA binding specificity. J Mol Biol 230:1131–1144
Jing HC, Sturre MJG, Hille J, Dijkwel PP (2002) Arabidopsis onset of leaf death mutants identify a regulatory pathway controlling leaf senescence. Plant J 32:51–63
Kanehisa M (2002) The KEGG database. Novart Found Symp 247:91–103
Kersey PJ, Allen JE, Christensen M, Davis P, Falin LJ, Grabmueller C, Hughes DST, Humphrey J, Kerhornou A, Khobova J, Langridge N, McDowall MD, Maheswari U, Maslen G, Nuhn M, Ong CK, Paulini M, Pedro H, Toneva I, Tuli MA, Walts B, Williams G, Wilson D, Youens-Clark K, Monaco MK, Stein J, Wei XH, Ware D, Bolser DM, Howe KL, Kulesha E, Lawson D, Staines DM (2014) Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res 42:D546–D552
Kim JH, Woo HR, Kim J, Lim PO, Lee IC, Choi SH, Hwang D, Nam HG (2009) Trifurcate Feed-Forward Regulation of Age-Dependent Cell Death Involving miR164 in Arabidopsis. Science 323:1053–1057
Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, Kersey P, Flicek P (2011) Ensembl BioMarts: a hub for data retrieval across taxonomic space Database-Oxford
Konishi M, Yanagisawa S (2008) Ethylene signaling in Arabidopsis involves feedback regulation via the elaborate control of EBF2 expression by EIN3. Plant J 55:821–831
Lee S, Seo PJ, Lee HJ, Park CM (2012) A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J 70:831–844
Leopold AC (1961) Senescence in plant development. Science 134:1727–1732
Li Z, Peng J, Wen X, Guo H (2012) Gene network analysis and functional studies of senescence-associated genes reveal novel regulators of Arabidopsis leaf senescence. J Integr Plant Biol 54:526–539
Li Z, Zhao Y, Liu X, Peng J, Guo H, Luo J (2014) LSD 2.0: an update of the leaf senescence database. Nucleic Acids Res 42:D1200–D1205
Lin JF, Wu SH (2004) Molecular events in senescing Arabidopsis leaves. Plant J 39:612–628
Lin M, Pang C, Fan S, Song M, Wei H, Yu S (2015) Global analysis of the Gossypium hirsutum L. Transcriptome during leaf senescence by RNA-Seq. BMC Plant Biol 15:43
Liu L, Zhou Y, Zhou G, Ye RJ, Zhao L, Li XH, Lin YJ (2008) Identification of early senescence-associated genes in rice flag leaves. Plant Mol Biol 67:37–55
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Long SP, Zhu XG, Naidu SL, Ort DR (2006) Can improvement in photosynthesis increase crop yields? Plant Cell Environ 29:315–330
Marrs KA (1996) The Functions and Regulation of Glutathione S-Transferases in Plants. Annu Rev Plant Physiol Plant Mol Biol 47:127–158
Masclaux-Daubresse C, Chardon F (2011) Exploring nitrogen remobilization for seed filling using natural variation in Arabidopsis thaliana. J Exp Bot 62:2131–2142
Masclaux-Daubresse C, Reisdorf-Cren M, Orsel M (2008) Leaf nitrogen remobilisation for plant development and grain filling Plant Biol (Stuttg) 10(Suppl 1):23–36
Miao Y, Laun T, Zimmermann P, Zentgraf U (2004) Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol Biol 55:853–867
Mizuno H, Kawahigashi H, Kawahara Y, Kanamori H, Ogata J, Minami H, Itoh T, Matsumoto T (2012) Global transcriptome analysis reveals distinct expression among duplicated genes during sorghum-Bipolaris sorghicola interaction. BMC Plant Biol 12:121
Moons A (2003) Osgstu3 and osgtu4, encoding tau class glutathione S-transferases, are heavy metal- and hypoxic stress-induced and differentially salt stress-responsive in rice roots. FEBS Lett 553:427–432
Nagarajan VK, Jain A, Poling MD, Lewis AJ, Raghothama KG, Smith AP (2011) Arabidopsis Pht1;5 Mobilizes Phosphate between Source and Sink Organs and Influences the Interaction between Phosphate Homeostasis and Ethylene Signaling. Plant Physiol 156:1149–1163
Noodén LD (1988) 1-The Phenomena of Senescence and Aging. In: Noodén LD, Leopold AC (eds) Senescence and Aging in Plants. Academic Press, pp 1–50
Olsen AN, Ernst HA, Lo Leggio L, Skriver K (2005) DNA-binding specificity and molecular functions of NAC transcription factors. Plant Sci 169:785–797
Ougham H, Hortensteiner S, Armstead I, Donnison I, King I, Thomas H, Mur L (2008) The control of chlorophyll catabolism and the status of yellowing as a biomarker of leaf senescence. Plant Biol 10:4–14
Palmer NA, Donze-Reiner T, Horvath D, Heng-Moss T, Waters B, Tobias C, Sarath G (2015) Switchgrass (Panicum virgatum L) flag leaf transcriptomes reveal molecular signatures of leaf development, senescence, and mineral dynamics. Funct Integr Genomics 15:1–16
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob ur R, Ware D, Westhoff P, Mayer KFX, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Patterson SE, Bleecker AB (2004) Ethylene-dependent and -independent processes associated with floral organ abscission in Arabidopsis. Plant Physiol 134:194–203
Rauf M, Arif M, Dortay H, Matallana-Ramirez LP, Waters MT, Gil Nam H, Lim PO, Mueller-Roeber B, Balazadeh S (2013) ORE1 balances leaf senescence against maintenance by antagonizing G2-like-mediated transcription. EMBO Rep 14:382–388
Richards RA (2000) Selectable traits to increase crop photosynthesis and yield of grain crops J Exp Bot 51 Spec No:447–458
Rosenow DT, Quisenberry JE, Wendt CW, Clark LE (1983) Drought tolerant sorghum and cotton germplasm. Agr Water Manage 7:207–222
Sanchez AC, Subudhi PK, Rosenow DT, Nguyen HT (2002) Mapping QTLs associated with drought resistance in sorghum (Sorghum bicolor L. Moench). Plant Mol Biol 48:713–726
Sappl PG, Carroll AJ, Clifton R, Lister R, Whelan J, Harvey Millar A, Singh KB (2009) The Arabidopsis glutathione transferase gene family displays complex stress regulation and co-silencing multiple genes results in altered metabolic sensitivity to oxidative stress. Plant J 58:53–68
Schommer C, Palatnik JF, Aggarwal P, Chetelat A, Cubas P, Farmer EE, Nath U, Weigel D (2008) Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol 6:e230
Seltmann MA, Stingl NE, Lautenschlaeger JK, Krischke M, Mueller MJ, Berger S (2010) Differential Impact of Lipoxygenase 2 and Jasmonates on Natural and Stress-Induced Senescence in Arabidopsis. Plant Physiol 152:1940–1950
Stead AD, Malcolm P, Buchanan-Wollaston V, Thomas B, Breeze E, Laarhoven LJ, Harren F, Rogers HJ, Wagstaff C (2003) Ethylene and Alstroemeria flowers—A grey area. Biol Biotech Plant Hormone Ethylene iii 349:340–344
Subudhi PK, Rosenow DT, Nguyen HT (2000) Quantitative trait loci for the stay green trait in sorghum (Soughum bicolor L. Moench): consistency across genetic backgrounds and environments. Theor Appl Genet 101:733–741
Tao YZ, Henzell RG, Jordan DR, Butler DG, Kelly AM, McIntyre CL (2000) Identification of genomic regions associated with stay green in sorghum by testing RILs in multiple environments. Theor Appl Genet 100:1225–1232
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation Nat. Biotech 28:511–515
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
van den Heuvel S, Dyson NJ (2008) Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 9:713–724
Van der Graaff E, Schwacke R, Schneider A, Desimone M, Flugge UI, Kunze R (2006) Transcription analysis of arabidopsis membrane transporters and hormone pathways during developmental and induced leaf senescence. Plant Physiol 141:776–792
Vandepoele K, Vlieghe K, Florquin K, Hennig L, Beemster GT, Gruissem W, Van de Peer Y, Inze D, De Veylder L (2005) Genome-wide identification of potential plant E2F target genes. Plant Physiol 139:316–328
Wang T-T, Ren Z-J, Liu Z-Q, Feng X, Guo R-Q, Li B-G, Li L-G, Jing H-C (2014) SbHKT1;4, a member of the high-affinity potassium transporter gene family from Sorghum bicolor, functions to maintain optimal Na+/K+ balance under Na+ stress. J Integr Plant Biol 56:315–332
Woo HR, Chung KM, Park JH, Oh SA, Ahn T, Hong SH, Jang SK, Nam HG (2001) ORE9, an F-box protein that regulates leaf senescence in Arabidopsis. Plant Cell 13:1779–1790
Wu XY, Kuai BK, Jia JZ, Jing HC (2012) Regulation of Leaf Senescence and Crop Genetic Improvement. J Integr Plant Biol 54:936–952
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322
Xu W, Subudhi PK, Crasta OR, Rosenow DT, Mullet JE, Nguyen HT (2000) Molecular mapping of QTLs conferring stay-green in grain sorghum (Sorghum bicolor L. Moench). Genome 43:461–469
Yang T, Poovaiah BW (2002) A calmodulin-binding/CGCG box DNA-binding protein family involved in multiple signaling pathways in plants. J Biol Chem 277:45049–45058
Yang JC, Zhang JH (2006) Grain filling of cereals under soil drying. New Phytol 169:223–236
Yazawa T, Kawahigashi H, Matsumoto T, Mizuno H (2013) Simultaneous transcriptome analysis of Sorghum and Bipolaris sorghicola by using RNA-seq in combination with de novo transcriptome assembly. PLoS One 8:e62460
Yoo SD, Cho YH, Tena G, Xiong Y, Sheen J (2008) Dual control of nuclear EIN3 by bifurcate MAPK cascades in C2H4 signalling. Nature 451:789–795
Zentgraf U, Laun T, Miao Y (2010) The complex regulation of WRKY53 during leaf senescence of Arabidopsis thaliana. Eur J Cell Biol 89:133–137
Zhang H, Jin J, Tang L, Zhao Y, Gu X, Gao G, Luo J (2011) PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res 39:D1114–D1117
Zhang T, Zhao X, Wang W, Huang L, Liu X, Zong Y, Zhu L, Yang D, Fu B, Li Z (2014a) Deep transcriptome sequencing of rhizome and aerial-shoot in Sorghum propinquum. Plant Mol Biol 84:315–327
Zhang WY, Xu YC, Li WL, Yang L, Yue X, Zhang XS, Zhao XY (2014b) Transcriptional analyses of natural leaf senescence in maize. PLoS One 9:e115617
Zheng LY, Guo XS, He B, Sun LJ, Peng Y, Dong SS, Liu TF, Jiang SY, Ramachandran S, Liu CM, Jing HC (2011) Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor) Genome Biol 12.
Zhong S, Zhao M, Shi T, Shi H, An F, Zhao Q, Guo H (2009) EIN3/EIL1 cooperate with PIF1 to prevent photo-oxidation and to promote greening of Arabidopsis seedlings. Proc Natl Acad Sci USA 106:21431–21436
Zhou C, Gan S (2010) Senescence. In: Pua EC, Davey MR (eds) Plant Dev Biol—Biotechnological Perspectives. Springer Berlin Heidelberg, pp 151–169
Zhu Z, An F, Feng Y, Li P, Xue L, A M, Jiang Z, Kim J-M, To TK, Li W, Zhang X, Yu Q, Dong Z, Chen W-Q, Seki M, Zhou J-M, Guo H (2011) Derepression of ethylene-stabilized transcription factors (EIN3/EIL1) mediates jasmonate and ethylene signaling synergy in Arabidopsis. Proc Natl Acad Sci USA 108(30):12539–12544
Acknowledgments
Members of Hai-Chun Jing’s lab are acknowledged for stimulating discussion on this project. This work was partially funded by grants to Hai-Chun Jing from National Natural Science Foundation of China (31271797, 30970252 and 31471570) and Ministry of Science and Technology of the People’s Republic of China (2013BAD22B01, 2015BAD15B03). We are also grateful to the handling editor and two anonymous reviewers for their critical comments and useful suggestions on a previous version of the manuscript.
Author contributions
Hai-Chun Jing designed and conceived the research, Wei-Juan Hu and Li-dong Wang performed the experiments, Xiao-yuan Wu and Hong Luo analysed the sequencing data, Yan Xia, Yi Zhao, Li-min Zhang, Jing-Chu Luo and Hai-Chun Jing assisted data analysing and gave thoughtful discussion. Xiao-Yuan Wu, Hong Luo and Hai-Chun Jing wrote the whole paper with feedback from Jing-Chu Luo. Xiao-Yuan Wu, Wei-Juan Hu, and Hong Luo contributed equally to the paper.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Xiao-Yuan Wu, Wei-Juan Hu and Hong Luo have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11103_2016_532_MOESM6_ESM.eps
ESM Fig. S1 Conditions shared and specific SAGs. A Venn diagram showing the numbers of SAGs shared by and specific to developmental, dark- and salt-induced senescence, respectively. DSAGs represents developmental specific SAGs (EPS 21034 KB)
11103_2016_532_MOESM8_ESM.eps
ESM Fig. S3 Number of SAGs contain three kinds of binding motifs. Candidate targets of transcription factors were identified by searching transcription factors binding sites in 2 kb promoter region of each sorghum gene (EPS 20871 KB)
Rights and permissions
About this article

Cite this article
Wu, XY., Hu, WJ., Luo, H. et al. Transcriptome profiling of developmental leaf senescence in sorghum (Sorghum bicolor). Plant Mol Biol 92, 555–580 (2016). https://doi.org/10.1007/s11103-016-0532-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-016-0532-1