
Abstract
Proteins are the cell’s functional entities. Rather than operating independently, they interact with other proteins. Capturing in vivo protein complexes is therefore crucial to gain understanding of the function of a protein in a cellular context. Affinity purification coupled to mass spectrometry has proven to yield a wealth of information about protein complex constitutions for a broad range of organisms. For Oryza sativa, the technique has been initiated in callus and shoots, but has not been optimized ever since. We translated an optimized tandem affinity purification (TAP) approach from Arabidopsis thaliana toward Oryza sativa, and demonstrate its applicability in a variety of rice tissues. A list of non-specific and false positive interactors is presented, based on re-occurrence over more than 170 independent experiments, to filter bona fide interactors. We demonstrate the sensitivity of our approach by isolating the complexes for the rice ANAPHASE PROMOTING COMPLEX SUBUNIT 10 (APC10) and CYCLIN-DEPENDENT KINASE D (CDKD) proteins from the proliferation zone of the emerging fourth leaf. Next to APC10 and CDKD, we tested several additional baits in the different rice tissues and reproducibly retrieved at least one interactor for 81.4 % of the baits screened for in callus tissue and T1 seedlings. By transferring an optimized TAP tag combined with state-of-the-art mass spectrometry, our TAP protocol enables the discovery of interactors for low abundance proteins in rice and opens the possibility to capture complex dynamics by comparing tissues at different stages of a developing rice organ.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Proteins are the main “workhorse-entities” of cells. They exert their function by participating in or affecting macromolecular assemblies, resulting in complex dynamic networks. Plants have a sessile lifestyle and need to cope with different types of environmental changes. Plant cells therefore exploit the properties of these complex dynamic networks to pertain homeostasis, which is translated in a huge variety of cellular processes. Understanding these processes thus requires a deep understanding of the network topology behind it. One way to gather this type of information is through the identification of protein–protein interactions (PPIs). Three methods are the main drivers for the elucidation of PPIs in plants. The yeast two-hybrid (Y2H) method identifies binary protein interactions through screening of the interaction partners in yeast. The method enables both comprehensive screening of open reading frames (ORFs), as was done for Arabidopsis (Braun et al. 2011), and a more targeted approach focusing on specific pathways or tissues (Seo et al. 2011; Lumba et al. 2014). Alternatively, one-to-one interactions can be screened within plant cells, through protein complementation analysis (PCA) (Boruc et al. 2010). Affinity purification coupled to mass spectrometry (AP–MS) identifies all proteins that co-purify with the pull-down of a tagged “bait” protein under near-physiological conditions and thus also captures indirect interactions. There are some caveats however. As protein interactions depend on ionic and hydrophobic interactions, different interaction partners can be revealed depending on the buffer conditions used. In contrast to Y2H and PCA, AP–MS has been traditionally unable to detect transient interactions, which are often lost during the washing steps necessary to remove nonspecific binding. The technique already proved its merits though, in various plant species (Arabidopsis, rice, petunia, tomato, tobacco) (Dedecker et al. 2015) for different cellular processes, including the cell cycle (Van Leene et al. 2010), flowering (Smaczniak et al. 2012), leaf development (Vercruyssen et al. 2014) and endocytosis (Gadeyne et al. 2014).
Owing to its rather small (389 Mb), fully annotated genome, Oryza sativa is in addition to being the most important food crop in the world also an excellent model for biological research on cereals. In that frame, five AP–MS approaches have so far been presented using rice for screening PPIs. Three used cultured cells, and their performance has been proven with the isolation of interaction partners of the TATA-BINDING PROTEIN (Zhong et al. 2003), GIGANTEA (Abe et al. 2008) or the FERTILILZATION-INSENSITIVE ENDOSPERM 2-polycomb protein complex (Nallamilli et al. 2013). The fourth approach reported the purification of VIRESCENT YELLOW LEAF associated proteins from shoots of 6–8-week-old seedlings (Dong et al. 2013). A last, more high-throughput effort was presented by Rohila and co-workers. They identified interaction partners for 23 % of the 129 rice kinases screened, starting from the shoots of 6–8-week-old seedlings (Rohila et al. 2006, 2009). The rather low success rate in this study emphasizes that creating a comprehensive picture of possible interactions for a given complex in plants is a daunting task and that there is room for improvement of TAP protocols for plants. Indeed, plants contain a tremendous variety of cell types and cellular states, each of these shaped by specific PPI networks.
To help overcome this hurdle, we developed a more improved protocol utilizing the GSrhino TAP tag. It consists of a tandem repeat of the IgG-binding Z domain of protein G and a streptavidin-binding peptide (SBP), separated by a tandem repeat of the specific human rhinovirus 3C (HR3C) cleavage site for gentle elution. This TAP tag is a variant of the GS tag that has shown a higher efficiency in terms of purification specificity and yield in higher eukaryotic cells like mammalian cells (Bürckstümmer et al. 2006) or Arabidopsis cells (Van Leene et al. 2008), as compared to the classical TAP tag developed for yeast. In Arabidopsis, combination of the GSrhino tag and LC–MS/MS has allowed further optimization of the TAP procedure. In cell cultures, the approach has enabled the identification of on average 5.6 specific interactors in common per bait protein used in a duplicate TAP experiment, with a success rate of 65 % for identifying at least one interactor per bait protein, confirmed in both duplicate TAP experiments. Moreover, integration of sensitive LC–MS/MS has allowed extrapolation of the TAP procedure to Arabidopsis seedlings to study protein complexes in a developmental context (Van Leene et al. 2015).
We assessed the performance of the GS tag in rice callus, by comparing the interaction datasets resulting from purifications of the rice CYCLIN-DEPENDENT KINASES REGULATORY SUBUNIT 1 (CKS1) with the previously used ‘improved TAP tag’ or ‘TAPi’ tag (Rohila et al. 2006). Then we applied the GS tag-based procedure to screen interaction partners for the rice ANAPHASE PROMOTING COMPLEX SUBUNIT 10 (APC10) and CYCLIN-DEPENDENT KINASE D (CDKD) proteins in a variety of rice tissues, including dissected organ parts.
CKS1 has a conserved role in cell cycle regulation as scaffold protein. It functions as a docking factor for positive and negative regulators of CYCLIN-DEPENDENT KINASE (CDK) activity (Jacqmard et al. 1999; Boudolf et al. 2001; De Veylder et al. 2001).
The APC plays a regulatory role in the eukaryotic cell cycle controlling the specificity of sister-chromatid separation and exit from mitosis by ubiquitin-mediated proteolysis of cell cycle regulators, such as CYCLIN B and SECURIN. In addition to cell cycle regulation, the APC has a function in developmental processes in plants, as was demonstrated in Arabidopsis and rice (Vanstraelen et al. 2009; Lin et al. 2012; Xu et al. 2012). Apart from two subunits required for ubiquitin ligase activity, i.e. the CULLIN-related protein APC2 and the REALLY INTERESTING NEW GENE (RING) finger protein APC11, the APC contains at least nine additional subunits (Lima et al. 2010). The complex is guided toward its targets by either CELL DIVISION CONTROL 20 or CELL CYCLE SWITCH 52A (CCS52A) activators. These contain a WD40 protein-binding domain, which recognizes D-box, KEN-box or A-box destruction motifs (Eloy et al. 2006).
CDKD was previously shown to form a heterotrimeric CDK-activating kinase (CAK) complex with a regulatory CYCLIN H (CYCH) subunit and the assembly factor “MENAGE A TROIS 1” (MAT1) both in rice (Rohila et al. 2006) and Arabidopsis (Van Leene et al. 2010). In the same studies, the other sub-complex of the general transcription factor II H (TFIIH), i.e. the five-subunit core consisting of XERODERMA PIGMENTOSUM B (XPB), p34, p52, p62 and p44, was co-purified, together with another helicase subunit XERODERMA PIGMENTOSUM D (XPD), which links both complexes of the TFIIH. As part of the CAK complex, CDKD not only phosphorylates the C-terminal domain of RNA polymerase II, but also the T-loop of CDKs (Yamaguchi et al. 1998; Fabian-Marwedel et al. 2002). CDKD itself is thought to be activated by a CAK-activating kinase, CDKF (Ding et al. 2009).
For both APC10 and CDKD, we retrieved their core complexes throughout different rice tissues. We therefore believe that the proposed analytical procedure will set a benchmark for assaying protein complex constitutions, from cu ltured cells to different developmental contexts in crop plants. The proposed workflow provides a success rate of identifying at least one confirmed interactor per bait protein used of 81.4 %.
Results
Developing a portfolio of TAP–MS workflows to study rice protein complexes
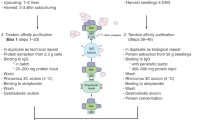
Targeted screening of protein complexes through AP–MS is based on four main steps: cloning, generation of sufficient biomass producing the tagged bait protein, protein complex purification and identification of the co-purified proteins through mass spectrometry. We developed a rice AP–MS workflow by implementing each step into a streamlined process. On top, we examined a portfolio of plant tissues for their efficiency to express the bait, enabling to screen complexes in the most suitable cellular environment, depending on the biological question and on prior knowledge of the bait protein. Our platform is built from the following consecutive steps: (1) flexible and Gateway-compatible cloning, (2) versatile generation of plant material producing the bait protein, (3) performant affinity purification to increase complex recovery and protocol sensitivity, (4) liquid chromatography coupled to tandem mass spectrometry, and (5) data analysis for identification of purified complex components. In the coming sections, we will describe all individual steps of the workflow and document its performance for systematic protein complex analysis covering different plant tissues.
Construction of TAP-fusion cassettes for use in rice
We constructed a rice-specific destination vector—named pTAP—compatible with MultiSite Gateway recombination-based cloning for both N- and C-terminal tagging (Fig . 1a). This destination vector contains in between the left and right T-DNA border sequences a kanamycin resistance gene (KmR) for selection and the Gateway cassette followed by the termination sequence from zein. Traditionally, a TAP construct consists of a desired promoter driving the expression of a translational fusion of the affinity tag and the protein of interest. The pTAP destination vector allows recombination of these elements once they are cloned into the appropriate entry vectors as shown in Fig. 1a.

Overview of the TAP procedure. a Schematic representation of the strategy for cloning TAP constructs using MultiSite Gateway. Both for N- and C-terminal cloning, a three fragment recombination strategy is performed, requiring only one type of destination vector that is suitable for any of the fusions. TT, zein-terminator; 35S, Cauliflower Mozaic Virus 35S promoter; Km R, kanamycin resistance gene for selection of transformed calli; LB and RB, resp. left and right border for T-DNA insertion; CcdB, toxic killer gene for negative selection; Cm R, chloramphenicol resistance gene. b Schematic representation of the time aspect of the different steps of the TAP–MS protocol starting from different plant resources
Since the affinity tag can interfere with the function of the bait protein, both N- and C-terminal fusions are tested for unknown bait proteins. This increases the chance to obtain a protein fusion that is still capable of binding its interaction partners, improving the success rate of the purifications. We already knew in advance which fusions worked best in Arabidopsis thaliana for the baits we chose (Van Leene et al. 2010) and therefore constructed only an N-terminal fusion for APC10 (LOC_Os05g50360, 81 % identity, 92 % similarity with Arabidopsis APC10) and a C-terminal fusion for CKS1 (LOC_Os03g05300, 97 % identity, 100 % similarity with Arabidopsis CKS1) and CDKD (LOC_Os05g32600, 70 % identity, 78 % similarity with Arabidopsis CDKD1;3).
We opted for an overexpression strategy as this is simple, maximizes the chance of having sufficient levels of bait expressed and favours competition with the endogenous counterpart for incorporation in the complex. We used the Cauliflower Mozaic Virus 35S promoter to drive expression of the fusion constructs. This in contrast to previous studies in rice, which used the ubiquitin promoter derived from maize (Zhong et al. 2003; Rohila et al. 2006, 2009). We argued that the lower activity of the 35S promoter in monocot tissues [10× less than the maize ubiquitin promoter in maize Christensen et al. (1992)] would prevent over-accumulation of non-complexed bait, ensuring a higher sensitivity of the protocol. Still, overexpressed proteins may exhibit protein misfolding, mislocalisation and/or misregulation on cellular level, and more false positive interactors (Gibson et al. 2013).
Assessment of the transferability of the GS tag to rice
We first estimated the compatibility of a GS tag-based workflow (Van Leene et al. 2010) in rice by comparing its performance to the current state-of-the-art in rice—a TAPi tag-based system (Rohila et al. 2009). We purified complexes from GS and TAPi tag-fusions with the cell cycle protein CKS1 using rice callus cells (Table S1; Fig. S1a). Our results show that the GS tag delivered a similar amount of interactors as the TAPi tag and confirmed the applicability of the procedure in rice.
Protein complex purification from a portfolio of rice tissues
With the transferability of the GS tag-based workflow confirmed, we further assayed the interaction partners for rice CDKD and APC10. These baits were tested as TAP constructs comprising the 35S promoter and a slightly optimised version of the GS tag, the GSrhino tag (Van Leene et al. 2015), which has a tandem repeat of the specific human rhinovirus 3C (HR3C) cleavage site replacing the TEV protease recognition sequence. We screened the two baits in callus cells and three additional types of biomass (“T0 shoots”, “T1 seedlings” and the proliferation zone of the emerging 4th leaf), varying throughout rice plant development (Fig. S1). In all cases, we used a series of different transformation events to level out positional effects from the T-DNA insertion site on bait expression, as previously suggested (Van Leene et al. 2007). Prior to purification, we confirmed bait accumulation by Western blotting in all tissues tested using an antibody against the Z domain (Fig. S2b, c). Sufficient protein extract was generated to perform two independent purifications in parallel, except for T0 shoots where we only performed a single purification. Protein input per experiment ranged from 7.5 mg protein extract from the proliferation zone over 50 mg from callus tissues to 150 mg from seedlings. The protein input from shoots varied depending on regeneration efficiency (130 mg protein extract for APC10 and 50 mg for CDKD). Because interactors found in two repeat experiments are more reliable as compared with interactors found only once, the technical repeats help minimizing false positive identifications caused by sample handling.
Protein complexes were isolated through a recently improved protocol (Van Leene et al. 2015). Bait and associated prey proteins are first retained by IgG-Sepharose beads through binding with the ZZ domain of the tagged protein. For callus and proliferation zone samples, this first purification step was performed “in-batch”, meaning that the affinity beads were simply added to the extract for binding. For shoots and seedlings, the relatively larger extract (around 25 mL on average) was brought consecutive times onto a column containing the affinity resin of the first affinity binding step, in order to optimize binding with the bait protein in the large extraction volume. The first affinity binding was followed by release from the immuno-precipitated ZZ domain through addition of human rhinovirus 3C protease (HR3C) and enrichment for a second time using streptavidin-Sepharose beads. Finally, bait and associated proteins are eluted by addition of desthiobiotin due to competitive binding. We added desthiobiotin immediately to the sample buffer. Consequently, the eluate can be applied directly on gel for further sample preparation MS. We performed in-gel trypsin digestion (Fig. S1e) prior to LC–MSMS analysis. The obtained mass spectra from the Q-Exactive mass spectrometer were applied for searches using the Mascot search engine against both the rice annotation project (RAP) database (Sakai et al. 2013) and against the Michigan State University (MSU) rice database (Kawahara et al. 2013), containing 82,162 and 66,338 entries, respectively. Only proteins identified with at least two rank-1 peptides with a peptide-spectrum matches confidence of >99 %, of which at least one is unique, were retained.
Establishment of a subtraction list of non-specific and false positive binders: separating the wheat from the chaff for co-purified interactors in rice
To filter out contaminant proteins which bind non-specifically to the beads or the tag and that complicate interpretation of the results, we built a dataset of re-occurring proteins generated from a large set of various baits analyzed with the same purification protocol. Background contaminants that bind to the tag and/or beads or non-specific interactors of bait proteins such as household proteins (e.g. chaperones, ribosomal proteins, cytoskeletal proteins, protein translation factors, etc.) are consistent across purifications with a particular TAP tag. We exploited this characteristic by compiling all interaction data from in total 174 TAP experiments in rice including those from different additional baits.
Some caution regarding the approach is advised, since this way of filtering does not take into account whether the baits are expected to function in the same pathway or biological process and thus might share some common interactors. We therefore assigned the baits to ‘bait classes’, based on shared membership in the same protein complex, the same gene family (Van Bel et al. 2012) or the same biological process according to their gene ontology annotation (Carbon et al. 2009). We set an arbitrary threshold as such that proteins present in two or more different bait groups were considered nonspecific. A similar approach was recently successfully applied in Arabidopsis (Van Leene et al. 2015).
The majority of the purifications considered were performed on callus tissue (115 purifications using 27 different baits), while purifications from plant tissue varied for shoots, seedlings and proliferation zone with respectively 25, 26 and 8 experiments on 7,7 and 2 baits. Because the large dataset contains a smaller number of experiments derived from plants than from callus tissue, we assigned a separate cut-off for background and non-specific proteins for experiments performed with plant tissues. Here, the cut-off was set for proteins present in more than one bait class. Importantly, this way of filtering out non-specific interactors might remove some bona fide interactors, for example interacting proteins that are genuinely in common between seemingly unrelated processes. Also, this method only works for proteins that are not common background proteins, which of course also will have interaction partners.
The resulting list of non-specific proteins in rice holds up to 951 potential contaminants (Table S2), allowing more efficient filtering (Fig. S3) as compared to a previous background list of 152 proteins (Rohila et al. 2009) that resulted from screening solely kinases in 129 experiments.
Investigation of complex composition throughout different tissues
To benchmark our method, we followed the constitution of two already established complexes throughout plant development using the four proposed types of plant tissues. The CAK complex of the TFIIH containing CDKD was unravelled both in rice shoots and in Arabidopsis cell suspensions by TAP (Rohila et al. 2006; Van Leene et al. 2010). The composition of the conserved APC in plants has been identified by TAP experiments with Arabidopsis cell suspension cultures and seedlings (Van Leene et al. 2010; Eloy et al. 2012). In the latter experiment, the plant-specific APC regulator SAMBA was used as bait protein instead of APC10. We performed a total of seven purifications each for APC10 and CDKD, covering two technical TAP repeats on extracts derived from calli, seedlings and tissues from the proliferation zone, and a single experiment on extracts from T0 shoots. Non-specific and background proteins were filtered out by subtracting non-specific proteins as described above. The remaining identified proteins were considered specific and could be divided in a group of experimentally confirmed interactions (Table 1) and a group of proteins identified in only one purification (Table S3). For both APC10 and CDKD, we were able to retrieve the interaction partners identified from previous experiments (Rohila et al. 2006; Van Leene et al. 2010), with on top novel candidate interaction partners, indicating the efficacy of our protocol.
CDKD
In first instance, we confirmed the participation of rice CDKD into the general TFIIH complex. TFIIH is responsible for two separate functions in eukaryotes: it melts the DNA around a lesion during nucleotide excision repair and helps to open the DNA template during the process of gene transcription. Yeast and human TFIIH consist of two sub-complexes: a trimeric sub-complex containing CDKD, CYCH and MAT1, and a core complex built from XPB, p34, p52, p44 and p62; both sub-complexes are linked by the helicase XPD (Chang and Kornberg 2000; Schultz et al. 2000; Gibbons et al. 2012).
Similar to a previous report that used rice CDKD for TAP purifications using rice leaves (Rohila et al. 2006), we retrieved only eight of the nine expected subunits of TFIIH. The subunit we could not retrieve is the XPB subunit. Low resolution models for yeast and human TFIIH show that XPB is the most distal to the CAK trimer, and only linked to the complex through binding with p52 (Chang and Kornberg 2000; Schultz et al. 2000; Gibbons et al. 2012). Probably this association is too weak to withstand the lengthy TAP protocol.
The complex composition was clearly very stable throughout plant development, since we were able to retrieve all components in all assayed tissues, including the leaf proliferation zone. In addition, we identified an alternative MAT1 assembly factor as potential interactor and purified the CDKF1 activating subunit. The interaction with the latter represents a kinase-substrate interaction (Shimotohno et al. 2004), which is a typical transient short-living interaction, explaining why we found it only once in a callus purification experiment. In rice, the direct interaction of CDKD with CDKF1 has been detected earlier using a Y2H assay (Ding et al. 2009).
APC
The APC is a conserved multi-subunit E3 ligase complex required for sister chromatin separation during anaphase and establishment of the G1 phase in the cell cycle (Eloy et al. 2006). A study using TAP–MS in Arabidopsis cell suspension cultured cells identified at least 11 APC subunits (Capron et al. 2003; Van Leene et al. 2010). Three dimensional reconstruction of human APC has shown that the complex adopts a triangular shape, a bit similar-looking as an open shell (Chang et al. 2014). The backbone contains a lobe of tetratricopeptide (TPR) domain-containing subunits APC3, APC6, APC7 and APC8, and a platform built out of APC1, APC4 and APC5. The APC10 subunit functions in recognizing and recruiting D-box containing proteins for ubiquitination (Heyman and De Veylder 2012). APC10 docks on the APC through interaction with the tetratricopeptide domain of APC3. In Arabidopsis thaliana, APC3 is encoded by two isoforms, namely APC3a/CDC27a and APC3b/HOBBIT (HBT), whereas rice contains only one APC3 paralog. The catalytic core of APC is built from APC2, a CULLIN domain subunit, and APC11, a RING domain subunit.
Our experiments using rice APC10 largely confirmed this complex constitution (Table 1). In callus, containing a population of mainly dividing cells, we purified all complex constituents found in Arabidopsis, together with the recently identified APC regulator called SAMBA (Eloy et al. 2012). We detected the APC11 subunit, previously reported to be very notorious to identify from TAP purifications because of its small size (Eloy et al. 2012). Also the known APC activator CCS52A was identified in callus tissue and seedlings, however only once in callus.
The retrieval of the APC from complicated plant tissues (shoots and seedlings) was more challenging even with the use of more than double the amount of protein extract (130 and 150 mg, respectively) as compared to callus tissue (50 mg), since sampling of whole plants results in a mixed population of different cell types, hampering the detection sensitivity of interactors. We failed for example to detect the APC11 subunit from both shoots and seedlings, APC6 from shoots, and APC4 from seedlings (Table 1). Also none of the CCS52A activators were retrieved from shoots, but CC52A1 was identified and experimentally validated from seedlings.
By using specifically the proliferation zone of the leaf instead of whole T0 seedlings or T1 shoots, the ratio of relevant proliferating tissue is favoured, leading to the purification of the whole core of the APC (Fig. 2, red highlighted subunits), even with a six times lower protein input as compared to callus tissues.

Visualization of the detected APC10 interactors over different rice tissues. Nodes are colored according to the tissues in which they were detected. The APC holo-complex as discovered in this study is highlighted and colored according to their function: subunits highlighted in red are APC/C backbone subunits, catalytic subunits are marked orange, activator subunits in green and negative regulators in blue. TPR-lobe tetratricopeptide-lobe
Discussion
Rice is, next to Arabidopsis, a very successful model species in plant biology. Quite large sets of genetic, molecular and genomic resources are already available, and being a monocot and a crop species itself, it also provides an excellent model for cereal biology. One of the key aspects in understanding biological processes is identifying the interactions between proteins to form complexes. Different technologies have been developed to screen for these types of interactions, such as Y2H, PCA and AP–MS. The former two certainly have shown their merits in the identification of binary interactions; AP–MS is complementary to Y2H and PCA, since it isolates and identifies protein complexes rather than binary interactions.
Several studies reported the development of an AP–MS approach in rice, two based on purification from cultured cells (Zhong et al. 2003; Abe et al. 2008), and three based on experiments on 6–8-week-old seedlings (Rohila et al. 2006, 2009; Dong et al. 2013). However, the majority only purified one complex, which makes it difficult to evaluate these individual platforms. Only the two studies on rice kinases (Rohila et al. 2006, 2009) demonstrated the screening of a significant amount of baits, with 23 % of these baits yielding actual interaction partners. Their relatively low success rate can however partly be attributed to the screening of kinases, which might be a tricky class of proteins to screen interactors from, and the fact that they only tested N-terminal fusions. Both parameters might have compromised the true potential of their platform. Rohila et al. used the traditional TAP tag developed in yeast to design their bait proteins, whereas more optimal tags are currently available (Bürckstümmer et al. 2006; Van Leene et al. 2008, 2015). We implemented these recent technical advances and combined them with the latest and most sensitive MS technology to an AP–MS protocol in rice that significantly improves the state-of-the-art in rice.
To generate a reliable dataset of potential interactors, a clear estimation of the false-positive interactions is required. We built a contaminant dataset based on occurrence over independent purifications of unrelated bait groups to efficiently remove the remainder of the contaminants. Importantly, there are drawbacks to this type of heuristic management of the interaction data. First, frequency filters are readily applicable to larger scale studies, but in lesser extent when smaller numbers of baits were used. This is illustrated in our dataset of plant purifications. The limited set of experiments forced us to also consider callus experiments and required the filter to be set very stringently. Second, the frequency filters are often chosen in a context or data-dependent manner, and are hard to apply if baits are functionally related and co-purify with similar sets of proteins. To address this issue, we grouped our baits according to co-membership in protein complexes, gene families of biological processes and considered the resulting bait groups for applying the frequency filters. In addition, the filtering method is only based on binary data (presence or absence of the interactor in each bait dataset), and ignore quantitative features of the mass spectrometry results. Genuine abundant interactors of any given bait are often missed since they are occasionally identified as low abundance interactors with several other baits. Quantitative measures of protein abundance in interaction data sets are therefore important to resolve whether the interaction bait-prey is specific (Keilhauer et al. 2015; Van Leene et al. 2015). Another quality parameter in TAP–MS experiments is reproducibility. To offer the best quality interaction dataset, we only considered interactions confirmed in at least two replicate experiments as bona fide.
Next to rice CKS1, APC10 and CDKD, several additional baits were tested in the different rice tissues, showing a success rate of 81.4 % of the baits screened for in callus tissue and T1 seedlings (Fig. S4). Five of the baits we screened are annotated as protein kinase. From this subset, we retrieved all bait proteins and we could find at least one interactor for all of them. These baits were all assayed from callus cells and suggest that our protocol in callus cells provides higher sensitivity compared to the former study on rice kinases.
These improvements are required to satisfy needs not only for screening complexes in tissues that provide sufficient protein extract, such as cultured cells, but also for screening in more technically demanding tissues, such as whole plants, or even specifically isolated organs or tissues. To our opinion, this versatility is key for elucidating biological processes, since protein complexes are known to be dynamic rather than static entities. The APC, for example, is constitutively present over the plant’s life cycle, but has been shown to exist in different constitutions during development in Arabidopsis (Eloy et al. 2006). Dependent on the developmental context, different interaction partners will result in a different biological signalling and outcome. As a proof of concept, we implemented our AP–MS protocol in rice callus tissues, shoots immediately regenerated from the callus, 2-week-old seedlings and the proliferation zone of the emerging fourth leaf, and show the constitution of the APC and the CDKD-associated complex in these different tissues. We postulate that all tissues tested have their merit. Cultured cells in the form of callus tissue provide fast means for an unlimited supply of protein extract. Since they represent only one (undifferentiated) cell type, their protein extract will be equally relevant for obtaining complexes. This reasonably low sample complexity, together with the absence of functional chloroplasts (and in particular RubisCo), results in not too much sensitivity requirements from the mass spectrometer, which is reflected in the identification of the smaller SAMBA and APC11 proteins as interaction partners of APC10 and of the more transient interactor CDKF for the CDKD-associated complex.
Since undifferentiated cells mainly support active division and basal pathways, interaction partners that are expressed more discretely in developmental context might be missed. To circumvent this, interaction data from callus tissue can be complemented with interaction data from plant samples. In first instance, we tested two types of ‘brute force’ biomass sources, containing young areal plant tissues. Shoots immediately regenerated from callus (T0 shoots) provide a way to have this more complex tissue type in a comparable timeframe as callus material. The downside is that we suffered from a high dependency on the regeneration efficiency for the generation of sufficient shoot biomass. Nevertheless, we were able to retrieve all core interactors with CDKD and the majority of the core subunits from the APC. To circumvent this dependency, we generated seeds from 60 individual transgenic events and selected lines having only one transgene copy for growing of plants. TAP experiments with 2-week-old seedlings revealed the core CDKD complex and, apart from APC11 and APC4, also the APC’s backbone and catalytic sub-complexes.
A combination of lowering the sample complexity and isolating interaction partners from the relevant developmental and anatomical context is provided when the specific tissue of interest is isolated and used for purification. We provided a proof of concept by isolating the proliferation zone of the emerging fourth leaf for performing our TAP experiments. Both the complexes containing CDKD and APC10 were isolated with a rather small input of 7.5 mg of protein, extracted from the proliferation zone of the fourth leaf of 550 seedlings. Especially for the APC10, we detected interesting potential interaction partners, like SEUSS and two YT521-B domain containing proteins (Table 1), subject to further validation.
Conclusion
In conclusion, we transferred an improved AP–MS procedure to rice and exploited these advances to provide a portfolio of possible biological tissues to screen interaction partners for proteins of interest. These range from tissues that provide a large amount of protein extract such as cultured cells, to more technically demanding tissues, such as whole plants and even specifically isolated parts of the plant. The latter should be envisioned as a proof of concept for assaying protein complexes from any plant organ. Indeed, our optimized protocol in combination with the state-of-the-art MS now allows to identify complexes from minute samples. This opens possibilities for elucidating biological processes by comparing protein complexes assayed from different organs or from organs at different developmental stages. This could be key in gaining a comprehensive view on the biology behind the interactors, since protein complexes are known to be dynamic rather than static entities. For this, rice is a more suitable model compared to Arabidopsis, since assaying plant organs in Arabidopsis is more difficult, given that some of the plant’s organs are too small at the stages when e.g. proliferation is occurring.
Methods
Construction of TAP expression vectors
The destination vector for creating TAP expression constructs was derived from p05050, a destination vector used for Oryza sativa transformation (WO2011114279A1) by replacing the attR1–attR2, the GFP cassette and the GOS2 promoter region by the attR3–attR4 Gateway cassette. The final destination vector pTAP was verified by sequence analysis.
Sequences for the N-terminal and C-terminal TAP tags with the necessary recombination sites (att sites) for Gateway® cloning were created by gene synthesis. The gateway cassettes containing the N-terminal and C-terminal TAP tags were cloned in compatible pDONR221 and pDONR R2–L3 vectors (Life Technologies, Carlsbad, CA) respectively according to the manufacturer’s instructions.
ORFs from genes of interest were isolated with Phusion Hot Start II High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA) on complement DNA (cDNA) of O. sativa (ecotype japonica). Primers used to isolate the different ORFs were designed using vector NTI software (Life Technologies, Carlsbad, CA) and sequences of the necessary recombination sites (att sites) for Gateway® cloning were added to the primers. To verify amplification, PCR products were loaded on a 1 % agarose gel and run for 25 min at 100 V. Subsequently, fragments of the corresponding gene size were excised from gel and then extracted using the QIAquick gel extraction kit (Qiagen, Venlo, The Netherlands). Next, ORFs were cloned in compatible pDONR221 (for C-terminal tagging) or pDONRR2–L3 (for N-terminal tagging) vectors (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Further cloning was performed using the standard MultiSite Gateway™ cloning technology to generate the TAP destination vectors. Entry vectors were verified by sequence analysis. TAP expression vectors were then transferred to Agrobacterium tumefaciens lba4404/pal4404 by electroporation. Transformed bacteria were selected on yeast extract broth medium with the appropriate antibiotics and verified by colony PCR with Takara Taq polymerase (Takara Bio Inc, Shiga, Japan) according to the manufacturer’s instructions.
Generation of plant tissues expressing the TAP constructs
A. tumefaciens-mediated transformation of O. sativa seeds was done according to patent WO2001006844 A1 with minor modifications. After mechanical dehusking using rice husker Kett US TR120, 150–200 seeds were surface sterilized with 6 % sodium hypochlorite solution for 45 min and washed with sterile water. Afterwards, seeds were transferred to induction medium (pH 5.8, 4 g/L MS salts, 1 mL/L MS vitamins, 2878 mg/L l-Proline, 300 mg/L Casamino Acids, 30 g/L sucrose, 4 g/L gelrite, 2 mg/L 2,4-D) and allowed to germinate at 32 °C under continuous light of 3000 lux. Six days after germination, the seeds were briefly submerged in liquid infection medium (pH 5.2, 4 g/L MS salts, 1 mL/L MS vitamins, 300 mg/L Casamino Acids, 68.5 g/L sucrose, 36 g/L D+ glucose-monohydrate, filter sterilised) containing 100 µM acetosyringone and transgenic A. tumefaciens lba4404/pal4404 containing the TAP destination vector (OD600 0.05–0.1) and transferred to co-cultivation medium (pH 5.2, 4 g/L MS salts, 1 mL/L MS vitamins, 300 mg/L Casamino Acids, 30 g/L sucrose, 10 g/L D+ glucose-monohydrate, 4 g/L gelrite, 2 mg/L 2,4-dichlorophenoxyacetic acid, 100 µM acetosyringone). Co-cultivation was allowed for 3 days at 25 °C in darkness. Thereafter, the explants were removed from the seeds, washed with 250 mg/L cefotaxime and transferred to selection medium (pH 5.8, 4 g/L MS salts, 1 mL/L MS vitamins, 2878 mg/L l-Proline, 300 mg/L Casamino Acids, 30 g/L sucrose, 7 g/L agarose type 1, 2 mg/L 2,4-dichlorophenoxyacetic acid, 100 mg/L cefotaxime, 100 mg/L vancomycin, 35 mg/L G418 disulfate) for incubation under continuous light (3000 lux) at 32 °C. Twelve days later, microcalli were isolated and transferred onto fresh selection medium, refreshed every 10 days, and grown until 30 g of callus was obtained. The callus material was then harvested in liquid nitrogen and stored at −80 °C for subsequent analysis.
For the generation of T0 shoot material, the same transformation protocol was utilized as described above. After isolation, the microcalli were transferred onto pre-regeneration medium (pH 5.8, 4 g/L MS salts, 1 mL/L MS vitamins, 500 mg/L l-Proline, 300 mg/L CasaminoAcids, 30 g/L sucrose, 7 g/L agarose type 1, 2 mg/L kinetin, 1 mg/L α-naphthaleneacetic acid, 5 mg/L abscisic acid, 100 mg/L cefotaxime, 100 mg/L vancomycin, 20 mg/L G418 disulfate) and incubated for 1 week at 32 °C under continuous light (3000 lux). Resistant calli were first brought to regeneration medium I (pH 5.8, 4.3 g/L MS salts, 1 mL/L MS vitamins, 2 g/L CasaminoAcids, 30 g/L sucrose, 30 g/L sorbitol, 10 g/L agarose type 1, 2 mg/L kinetin, 0.02 mg/L α-naphthaleneacetic acid, 100 mg/L cefotaxime, 100 mg/L vancomycin, 20 mg/L G418 disulfate) for 7 days and then transferred to regeneration medium II (pH 5.8, 4.3 g/L MS salts, 1 mL/L MS vitamins, 2 g/L Casamino Acids, 30 g/L sucrose, 30 g/L sorbitol, 7 g/L agarose type 1, 2 mg/L kinetin, 0.02 mg/L α-naphthaleneacetic acid, 100 mg/L cefotaxime, 100 mg/L vancomycin, 20 mg/L G418 disulfate) for two additional weeks at 32 °C under continuous light (lux 3000). Plants whose shoot and root grew more than 1 cm in length were ultimately transferred to growth medium (pH 5.8, 2.15 g/L MS medium micro and macro, 0.5 mL (0.5 x) B5 vitamins, 10 g/L sucrose, 0.05 mg/L α-naphthaleneacetic acid, 0.75 g/L MgCl2·6H2O, 2.5 g/L gelrite) and incubated for 2 weeks at 32 °C under continuous light (3000 lux) before being harvested in liquid nitrogen and stored at −80 °C for subsequent analysis.
Transgenic seeds containing the TAP expression vector were derived from the transformation protocol generating shoots described above. Instead of harvesting, 60 plants, each derived from an individual transformation event, were transferred to the greenhouse and further grown until seeds could be harvested. For growing seedlings, seeds were first dehusked and sterilized as previously described before sowing them on growth medium containing the selective agent (pH 5.8, 2.15 g/L MS medium micro and macro, 0.5 mL (0.5×) B5 vitamins, 10 g/L sucrose, 0.05 mg/L α-naphthaleneacetic acid, 0.75 g/L MgCl2·6H2O, 7 g/L agarose, 20 mg/L G418 disulfate). T1 seedlings were grown in a growth chamber under short day conditions at 32 °C, and harvested 2 weeks after sowing in liquid nitrogen and stored at −80 °C for subsequent analysis. Plants for isolation of the proliferation zone were grown in jiffies in the greenhouse under short day light conditions. Once the 4th leaf started emerging (approximately after 2 weeks), leafs were carefully separated and the 4th leaf was collected. The first cm from the base was then collected in liquid nitrogen.
Expression analysis of the bait proteins
Plant material was ground to homogeneity in liquid nitrogen with mortar and pestle. About 200 μL of extraction buffer (25 mM Tris–HCl pH 7.6, 15 mM MgCl2, 150 mM NaCl, 15 mM p-Nitrophenyl phosphate, 60 mM β-glycerophosphate, 0.1 % NP-40, 0.1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 1 μM E64, EDTAfree Ultra Complete tablet (1/10 mL) (Roche Diagnostics, Brussels, Belgium), 5 % ethylene glycol) was added and homogenized with a 1.5-mL pellet mixer. Homogenized samples were flash frozen in liquid nitrogen, thawed on ice and centrifuged twice for 15 min at 4 °C at 20,800 g. Protein concentrations were determined by Bradford assay (Bio-rad, Hercules, CA). Fifty μg of total protein extract was loaded for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 0.75 mm 12 % Mini-PROTEAN® TGX™ precast gels (Bio-Rad, Hercules, CA) for 20 min at 300 V in TGX running buffer (25 mM Tris–HCl, pH 8.3, 1.92 M glycine, 35 mM SDS). Resolved proteins were transferred to PVDF membranes using Trans-Blot® Turbo™ Mini PVDF transfer packs and the Trans-Blot® Turbo™ Transfer system (Bio-rad, Hercules, CA) according to instructions of the manufacturer. Blotted PVDF membranes were then incubated in blocking buffer [3 % Difco™ skimmed milk (w/v) in TBS-T buffer (50 mM Tris, 150 mM NaCl pH8.0, 0.1 % Triton X-100)] overnight at 4 °C or 1 h at room temperature (RT) on an orbital shaker. After this blocking step, membranes were incubated for 1 h at RT with peroxidase anti-peroxidase antibody (Sigma-Aldrich, Saint-Louis, MO) or anti-CKS1 antibody in blocking buffer on an orbital shaker. Membranes were washed 1 × 15 min and 4 × 5 min with TBS-T buffer. Bound antibody was detected by mixing equal amounts of the two chemiluminescent reagents from the ECL-kit (Perkinelmer, Waltham, MA) and incubating for 1 min. Membranes were placed in a film cassette and exposed to an Amersham hyperfilm™ ECL film (GE Healthcare, Wauwatosa, WI) in a dark room, where autoradiograms were also developed.
Tandem affinity purification of protein complexes
Callus material was ground to homogeneity in liquid nitrogen with mortar and pestle. Plant material (shoots, seedlings and tissues from the proliferation zone) was ground to homogeneity in liquid nitrogen with a hand blender (Braun GmbH, Kronberg, Germany). Crude protein extracts were prepared in two volumes of extraction buffer (25 mM Tris–HCl pH 7.6, 15 mM MgCl2, 150 mM NaCl, 15 mM p-nitrophenyl phosphate, 60 mM β-glycerophosphate, 0,1 % NP-40, 0.1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 1 µM E64, EDTA-free Ultra Complete tablet Easypack (1/10 mL) (Roche Diagnostics, Brussels, Belgium), 5 % Ethylene glycol) at 4 °C using an Ultra-Turrax T25 mixer (IKA Works, Wilmington, NC). Soluble fraction was obtained from isolating the supernatants after double centrifugation at 36,900g for 20 min at 4 °C. The extract was passed through four layers of miracloth (Merck KGaA, Darmstadt, Germany) and kept on ice.
Purifications on TAPi tagged CKS1 bait were performed as previously described (Van Leene et al. 2007). The GS tagged CKS1 bait was purified as described below, with the only exception that AcTEV protease (Life Technologies, Carlsbad, MA) was used instead of rhinovirus 3C protease.
Purifications were performed as described by Van Leene et al. (Van Leene et al. 2015) with some modifications. For material from callus material and tissues from the proliferation zone, all purifications were performed in batch. The protein extract was added to 25 µL of effective immunoglobulin G (IgG) Sepharose 6 Fast Flow beads (GE Healthcare, Wauwatosa, WI), pre-equilibrated with 3 × 250 µL extraction buffer. After incubation for 1 h at 4 °C under gentle rotation, the beads were transferred to a Poly-Prep column (Bio-Rad, Hercules, CA) mounted to a two-way valve in a vacuum manifold system (Grace, Columbia, MD) and washed with 375 µL or 150 column volumes wash buffer (10 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.1 % NP-40, 0.5 mM EDTA, 1 μM E64, 1 mM PMSF, 5 % Ethylene glycol). Bound complexes were eluted by digestion in a mobicol column (MoBiTec GmbH, Göttingen, Germany) using 100 µL wash buffer and 2 × 10 U (2 × 1 µL, second boost after 30 min) rhinovirus 3C protease (GE Healthcare, Wauwatosa, WI) for 1 h at 4 °C on a shaker. Eluate was collected by two consecutive spinning steps of the mobicol column (MoBiTec GmbH, Göttingen, Germany) in a 2–mL Eppendorf tube for 30 s at 1500 rpm at 4 °C. In between, 100 µL wash buffer was added to the beads to collect residual eluate. The resulting eluate was incubated for 1 h at 4 °C under gentle rotation with 25 µL effective Streptavidin-Sepharose High Performance beads (GE Healthcare, Wauwatosa, WI), pre-equilibrated with 3 × 250 µL wash buffer. Streptavidin beads were transferred to a mobicol column and washed with 100 column volumes or 2,5 mL wash buffer. Complexes were eluted in 40 µL NuPAGE sample buffer containing 20 mM desthiobiotin (Sigma-Aldrich, Saint-Louis, MO) by 5 min incubation on ice, followed by centrifugation at 1500 rpm at 4 °C.
For seedling and shoot material, the first affinity step was performed on column. Briefly, protein extract was applied on a Poly-Prep column (Bio-Rad, Hercules, CA) containing 100 µL of effective IgG Sepharose 6 Fast Flow beads (GE Healthcare, Wauwatosa, WI), pre-equilibrated with 3 × 1 mL extraction buffer, with a peristaltic pump (GE Healthcare, Wauwatosa, WI), at flow rate 1 mL/min. The rest of the protocol proceeded similarly, reckoning with adjusted column volumes, as described above. Purified protein samples were loaded and separated with a short 7-min run on a precast 4–12 % gradient NuPAGE Bis–Tris gel (Life Technologies, Carlsbad, CA), fixed in 50 % EtOH/2 % H3PO4 and visualized with colloidal Coomassie Brilliant Blue G-250 (Sigma-Aldrich, Saint-Louis, MO) staining.
Proteolysis and peptide isolation
NuPAGE gel containing purified protein samples was destained twice in HPLC-grade water (Thermo Fisher Scientific, Waltham, MA) for 1 h and incubated in 25 mL of reducing buffer (6.66 mM DTT plus 50 mM NH4HCO3 in HPLC-grade water) for 40 min to reduce the polypeptide disulphide bridges. Subsequently, thiol groups were alkylated by incubating the gel for 30 min in 25 mL of alkylating buffer (55 mM iodoacetamide, 50 mM NH4HCO3 in HPLC-grade water) in the dark before washing with HPLC-grade water. The zone containing the protein sample was sliced from the gel and sectioned into different gel plugs. These were washed twice with 600 µL of HPLC-grade water and dehydrated in 600 µL 95 % acetonitrile twice for 10 min. The dehydrated gel plugs were submerged and rehydrated in 90 µL trypsin digest buffer (12.5 µg/mL trypsin (MS gold; Promega, Madison, WI) in 50 mM NH4HCO3 and 10 % (v/v) acetonitrile in HPLC-grade water) for 30 min at 4 °C. Afterwards, trypsin digestion was allowed for 3.5 h at 37 °C. Resulting peptide samples were sonicated for 5 min in a sonication bath and the solution covering the gel plugs (containing trypsinized peptides) were kept aside. Remaining gel plugs were completely dehydrated in 95 % acetonitrile for 10 min and the remaining acetonitrile solution was added to the first fraction of trypsin digests. The resulting trypsin-digested sample was completely dried in a SpeedVac for 2–3 h at 4 °C.
LC–MS/MS analysis
A nano LC system (Ultimate 3000 RSLC nano, Dionex, Amsterdam, The Netherlands) was set up connected in-line to the Q-Exative (Thermo Fisher Scientific, Waltham, MA), with a trapping column (PepMap 100) of 100–µm internal diameter (I.D.) × 20 mm (length) with 5–µm C18 Reprosil-HD beads (Dionex, Amsterdam, The Netherlands), a flow rate 6 µL/min and, 5 min after injection, switched in-line with a reverse phase analytical separating column (Acclaim, PepMap 100) of 75 µm I.D. × 150 mm with 3 µm C18 Reprosil-HD beads (Dionex, Amsterdam, The Netherlands).
Peptides were solubilized in 15 µL of loading Solvent A (0.1 % (v/v) trifluoroacetic acid, 2 % (v/v) acetonitrile in HPLC grade water), and 5 µL of the peptide sample was loaded on the trapping column. Peptide samples were separated with a 30–min gradient from 2 % mobile phase solvent A’ (0.1 % (v/v) formic acid in HPLC-grade water) to 50 % mobile phase solvent B’ (0.1 % (v/v) formic acid and 80 % acetonitrile in HPLC-grade water) at a flow rate of 300 nL/min. The Q-Exactive was operated in data-dependent mode to automatically switch between MS and MS/MS acquisition for the ten most abundant peaks in a given MS spectrum. Full-scan MS spectra were acquired in the Orbitrap™ at a target value of 1E6 with a resolution of 60,000. The ten most intense ions were isolated for fragmentation in the quadrupole, with a dynamic exclusion of 20 s. Target value for filling the quadrupole was set to 1E4 ion counts.
Analysis of the protein interaction data
Mascot generic files were created with the Mascot Distiller software. Grouping of spectra with a maximum intermediate retention time of 30 s and 0.005-Da precursor tolerance was allowed together with a maximum intermediate scan count of five if possible. No de-isotoping was used and the relative signal-to-noise limit was set to 2. A peak list was generated only when the MS/MS spectrum contained more than ten peaks. The Mascot Daemon interface was used to search peak lists with the Mascot search engine against the RAP database (Sakai et al. 2013) and against the Michigan State University rice database (MSU) (Kawahara et al. 2013). Search parameters in Mascot Daemon were as follows: variable modifications set to methionine oxidation and methylation of aspartic acid and glutamic acid, fixed modifications to carbamidomethylation of cysteines, mass tolerance on MS 10 ppm, MS/MS tolerance 20 mmu, ESI-QUAD as instrument, 2+ and 3+ as peptide charge and protease trypsin/P, allowing for 1 missed cleavage. In the Mascot result URL, a Mascot select summary was created with following settings: significance threshold p > 0.01, maximum number of hits AUTO, Mudpit scoring, Ion score or expectancy cut-off >0.01, require bold red. Identifications were retained when at least two peptides matched a high confidence rank, with at least one peptide unique to the protein. The resulting protein identification list was cross-checked against the list of non-specific proteins.
Abbreviations
- AP–MS:
-
Affinity purification coupled to mass spectrometry
- TAP:
-
Tandem affinity purification
- APC10:
-
ANAPHASE PROMOTING COMPLEX SUBUNIT 10
- CDKD:
-
CYCLIN-DEPENDENT KINASE D
- PPI:
-
Protein–protein interaction
- Y2H:
-
Yeast two-hybrid
- ORF:
-
Open reading frame
- PCA:
-
Protein complementation analysis
- RING:
-
REALLY INTERESTING NEW GENE
- CCS52A:
-
CELL CYCLE SWITCH PROTEIN 52 A
- CAK:
-
CDK-activating kinase
- CYCH:
-
CYCLIN H
- MAT1:
-
MENAGE A TROIS
- TFIIH:
-
General transcription factor II H
- XPB:
-
XERODERMA PIGMENTOSUM B
- XPD:
-
XERODERMA PIGMENTOSUM D
- SBP:
-
Streptavidin-binding peptide
- MS:
-
Mass spectrometry
- TEV:
-
Tobacco etch virus
- IgG:
-
Immunoglobulin G
- RAP:
-
Rice annotation project
- MSU:
-
Michigan State University
- CBP:
-
Calmodulin-binding peptide
- EGTA:
-
Ethylene glycol tetra-acetic acid
References
Abe M, Fujiwara M, Kurotani K-I, Yokoi S, Shimamoto K (2008) Identification of dynamin as an interactor of rice GIGANTEA by tandem affinity purification (TAP). Plant Cell Physiol 49:420–432. doi:10.1093/pcp/pcn019
Boruc J, Van den Daele H, Hollunder J, Rombauts S, Mylle E, Hilson P, Inzé D, De Veylder L, Russinova E (2010) Functional modules in the Arabidopsis core cell cycle binary protein-protein interaction network. Plant Cell 22:1264–1280. doi:10.1105/tpc.109.073635
Boudolf V, Rombauts S, Naudts M, Inzé D, De Veylder L (2001) Identification of novel cyclin-dependent kinases interacting with the CKS1 protein of Arabidopsis. J Exp Bot 52:1381–1382
Braun P, Carvunis AR, Charloteaux B, Dreze M, Ecker JR, Hill DE, Roth FP, Vidal M, Galli M, Balumuri P, Bautista V, Chesnut JD, Kim RC, de los Reyes C, Gilles P, Kim CJ, Matrubutham U, Mirchandani J, Olivares E, Patnaik S, Quan R, Ramaswamy G, Shinn P, Sw VM (2011) Evidence for network evolution in an Arabidopsis interactome map. Science 333:601–607. doi:10.1126/science.1203877
Bürckstümmer T, Bennett KL, Preradovic A, Schütze G, Hantschel O, Superti-Furga G, Bauch A (2006) An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods 3:1013–1019. doi:10.1038/nmeth968
Capron A, Serralbo O, Fülöp K, Frugier F, Parmentier Y, Dong A, Lecureuil A, Guerche P, Kondorosi E, Scheres B, Genschik P (2003) The Arabidopsis anaphase-promoting complex or cyclosome: molecular and genetic characterization of the APC2 subunit. Plant Cell 15:2370–2382. doi:10.1105/tpc.013847
Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25:288–289. doi:10.1093/bioinformatics/btn615
Chang WH, Kornberg RD (2000) Electron crystal structure of the transcription factor and DNA repair complex, core TFIIH. Cell 102:609–613
Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D (2014) Molecular architecture and mechanism of the anaphase-promoting complex. Nature 513:388–393. doi:10.1038/nature13543
Christensen AH, Sharrock RA, Quail PH (1992) Maize polyubiquitin genes: structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol Biol 18:675–689
De Veylder L, Beeckman T, Beemster GT, Krols L, Terras F, Landrieu I, van der Schueren E, Maes S, Naudts M, Inzé D (2001) Functional analysis of cyclin-dependent kinase inhibitors of Arabidopsis. Plant Cell 13:1653–1668
Dedecker M, Van Leene J, De Jaeger G (2015) Unravelling plant molecular machineries through affinity purification coupled to mass spectrometry. Curr Opin Plant Biol 24:1–9. doi:10.1016/j.pbi.2015.01.001
Ding X, Richter T, Chen M, Fujii H, Seo YS, Xie M, Zheng X, Kanrar S, Stevenson RA, Dardick C, Li Y, Jiang H, Zhang Y, Yu F, Bartley LE, Chern M, Bart R, Chen X, Zhu L, Farmerie WG, Gribskov M, Zhu J-K, Fromm ME, Ronald PC, Song W-Y (2009) A rice kinase-protein interaction map. Plant Physiol 149:1478–1492. doi:10.1104/pp.108.128298
Dong H, Fei G-L, Wu C-Y, Wu F-Q, Sun Y-Y, Chen M-J, Ren Y-L, Zhou K-N, Cheng Z-J, Wang J-L, Jiang L, Zhang X, Guo X-P, Lei C-L, Su N, Wang H, Wan J-M (2013) A rice virescent-yellow leaf mutant reveals new insights into the role and assembly of plastid caseinolytic protease in higher plants. Plant Physiol 162:1867–1880. doi:10.1104/pp.113.217604
Eloy NB, Coppens F, Beemster GTS, Hemerly AS, Ferreira PCG (2006) The Arabidopsis anaphase promoting complex (APC): regulation through subunit availability in plant tissues. Cell Cycle 5:1957–1965
Eloy NB, Gonzalez N, Van Leene J, Maleux K, Vanhaeren H, De Milde L, Dhondt S, Vercruysse L, Witters E, Mercier R, Cromer L, Beemster GTS, Remaut H, Van Montagu MCE, De Jaeger G, Ferreira PCG, Inzé D (2012) SAMBA, a plant-specific anaphase-promoting complex/cyclosome regulator is involved in early development and A-type cyclin stabilization. Proc Natl Acad Sci USA 109:13853–13858. doi:10.1073/pnas.1211418109
Fabian-Marwedel T, Umeda M, Sauter M (2002) The rice cyclin-dependent kinase-activating kinase R2 regulates S-phase progression. Plant Cell 14:197–210. doi:10.1105/tpc.010386
Gadeyne A, Sánchez-Rodríguez C, Vanneste S, Di Rubbo S, Zauber H, Vanneste K, Van Leene J, De Winne N, Eeckhout D, Persiau G, Van De Slijke E, Cannoot B, Vercruysse L, Mayers JR, Adamowski M, Kania U, Ehrlich M, Schweighofer A, Ketelaar T, Maere S, Bednarek SY, Friml J, Gevaert K, Witters E, Russinova E, Persson S, De Jaeger G, Van Damme D (2014) The TPLATE adaptor complex drives clathrin-mediated endocytosis in plants. Cell 156:691–704. doi:10.1016/j.cell.2014.01.039
Gibbons BJ, Brignole EJ, Azubel M, Murakami K, Voss NR, Bushnell DA, Asturias FJ, Kornberg RD (2012) Subunit architecture of general transcription factor TFIIH. Proc Natl Acad Sci USA 109:1949–1954. doi:10.1073/pnas.1105266109
Gibson TJ, Seiler M, Veitia RA (2013) The transience of transient overexpression. Nat Methods 10:715–721. doi:10.1038/nmeth.2534
Heyman J, De Veylder L (2012) The anaphase-promoting complex/cyclosome in control of plant development. Mol Plant 5:1182–1194. doi:10.1093/mp/sss094
Jacqmard A, De Veylder L, Segers G, de Almeida EJ, Bernier G, Van Montagu M, Inze D (1999) Expression of CKS1At in Arabidopsis thaliana indicates a role for the protein in both the mitotic and the endoreduplication cycle. Planta 207:496–504
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, Childs KL, Davidson RM, Lin H, Quesada-Ocampo L, Vaillancourt B, Sakai H, Lee SS, Kim J, Numa H, Itoh T, Buell CR, Matsumoto T (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (NY) 6:4. doi:10.1186/1939-8433-6-4
Keilhauer EC, Hein MY, Mann M (2015) Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP–MS). Mol Cell Proteomics 14:120–135. doi:10.1074/mcp.M114.041012
Lima MDF, Eloy NB, Pegoraro C, Sagit R, Rojas C, Bretz T, Vargas L, Elofsson A, de Oliveira AC, Hemerly AS, Ferreira PC (2010) Genomic evolution and complexity of the anaphase-promoting complex (APC) in land plants. BMC Plant Biol 10:254. doi:10.1186/1471-2229-10-254
Lin Q, Wang D, Dong H, Gu S, Cheng Z, Gong J, Qin R, Jiang L, Li G, Wang JL, Wu F, Guo X, Zhang X, Lei C, Wang H, Wan J (2012) Rice APC/C(TE) controls tillering by mediating the degradation of MONOCULM 1. Nat Commun 3:752. doi:10.1038/ncomms1716
Lumba S, Toh S, Handfield L-F, Swan M, Liu R, Youn J-Y, Cutler SR, Subramaniam R, Provart N, Moses A, Desveaux D, McCourt P (2014) A mesoscale abscisic acid hormone interactome reveals a dynamic signaling landscape in Arabidopsis. Dev Cell 29:360–372. doi:10.1016/j.devcel.2014.04.004
Nallamilli BRR, Zhang J, Mujahid H, Malone BM, Bridges SM, Peng Z (2013) Polycomb group gene OsFIE2 regulates rice (Oryza sativa) seed development and grain filling via a mechanism distinct from Arabidopsis. PLoS Genet 9:e1003322. doi:10.1371/journal.pgen.1003322
Rohila JS, Chen M, Chen S, Chen J, Cerny R, Dardick C, Canlas P, Xu X, Gribskov M, Kanrar S, Zhu J-K, Ronald P, Fromm ME (2006) Protein–protein interactions of tandem affinity purification-tagged protein kinases in rice. Plant J 46:1–13. doi:10.1111/j.1365-313X.2006.02671.x
Rohila JS, Chen M, Chen S, Chen J, Cerny RL, Dardick C, Canlas P, Fujii H, Gribskov M, Kanrar S, Knoflicek L, Stevenson B, Xie M, Xu X, Zheng X, Zhu J-K, Ronald P, Fromm ME (2009) Protein–protein interactions of tandem affinity purified protein kinases from rice. PLoS ONE 4:e6685. doi:10.1371/journal.pone.0006685
Sakai H, Lee SS, Tanaka T, Numa H, Kim J, Kawahara Y, Wakimoto H, Yang C, Iwamoto M, Abe T, Yamada Y, Muto A, Inokuchi H, Ikemura T, Matsumoto T, Sasaki T, Itoh T (2013) Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol 54:e6. doi:10.1093/pcp/pcs183
Schultz P, Fribourg S, Poterszman A, Mallouh V, Moras D, Egly JM (2000) Molecular structure of human TFIIH. Cell 102:599–607
Seo Y-S, Chern M, Bartley LE, Han M, Jung K-H, Lee I, Walia H, Richter T, Xu X, Cao P, Bai W, Ramanan R, Amonpant F, Arul L, Canlas PE, Ruan R, Park C-J, Chen X, Hwang S, Jeon J-S, Ronald PC (2011) Towards establishment of a rice stress response interactome. PLoS Genet 7:e1002020. doi:10.1371/journal.pgen.1002020
Shimotohno A, Umeda-Hara C, Bisova K, Uchimiya H, Umeda M (2004) The plant-specific kinase CDKF;1 is involved in activating phosphorylation of cyclin-dependent kinase-activating kinases in Arabidopsis. Plant Cell 16:2954–2966. doi:10.1105/tpc.104.025601
Smaczniak C, Immink RGH, Muiño JM, Blanvillain R, Busscher M, Busscher-Lange J, Dinh QDP, Liu S, Westphal AH, Boeren S, Parcy F, Xu L, Carles CC, Angenent GC, Kaufmann K (2012) Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc Natl Acad Sci USA 109:1560–1565. doi:10.1073/pnas.1112871109
Van Bel M, Proost S, Wischnitzki E, Movahedi S, Scheerlinck C, Van de Peer Y, Vandepoele K (2012) Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol 158:590–600. doi:10.1104/pp.111.189514
Van Leene J, Stals H, Eeckhout D, Persiau G, Van De Slijke E, Van Isterdael G, De Clercq A, Bonnet E, Laukens K, Remmerie N, Henderickx K, De Vijlder T, Abdelkrim A, Pharazyn A, Van Onckelen H, Inzé D, Witters E, De Jaeger G (2007) A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Mol Cell Proteomics 6:1226–1238. doi:10.1074/mcp.M700078-MCP200
Van Leene J, Witters E, Inzé D, De Jaeger G (2008) Boosting tandem affinity purification of plant protein complexes. Trends Plant Sci 13:517–520. doi:10.1016/j.tplants.2008.08.002
Van Leene J, Hollunder J, Eeckhout D, Persiau G, Van De Slijke E, Stals H, Van Isterdael G, Verkest A, Neirynck S, Buffel Y, De Bodt S, Maere S, Laukens K, Pharazyn A, Ferreira PCG, Eloy N, Renne C, Meyer C, Faure J-D, Steinbrenner J, Beynon J, Larkin JC, Van de Peer Y, Hilson P, Kuiper M, De Veylder L, Van Onckelen H, Inzé D, Witters E, De Jaeger G (2010) Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol Syst Biol 6:397. doi:10.1038/msb.2010.53
Van Leene J, Eeckhout D, Cannoot B, De Winne N, Persiau G, Van De Slijke E, Vercruysse L, Dedecker M, Verkest A, Vandepoele K, Martens L, Witters E, Gevaert K, De Jaeger G (2015) An improved toolbox to unravel the plant cellular machinery by tandem affinity purification of Arabidopsis protein complexes. Nat Protoc 10:169–187. doi:10.1038/nprot.2014.199
Vanstraelen M, Baloban M, Da O, Cultrone A, Lammens T, Brown SC, De VL, Mergaert P, Kondorosi E (2009) APC/C CCS52A complexes control meristem maintenance in the Arabidopsis root. Proc Natl Acad Sci USA 106(28):11806–11811. doi:10.1073/pnas.0901193106
Vercruyssen L, Verkest A, Gonzalez N, Heyndrickx KS, Eeckhout D, Han S-K, Jégu T, Archacki R, Van Leene J, Andriankaja M, De Bodt S, Abeel T, Coppens F, Dhondt S, De Milde L, Vermeersch M, Maleux K, Gevaert K, Jerzmanowski A, Benhamed M, Wagner D, Vandepoele K, De Jaeger G, Inzé D (2014) ANGUSTIFOLIA3 binds to SWI/SNF chromatin remodeling complexes to regulate transcription during Arabidopsis leaf development. Plant Cell 26:210–229. doi:10.1105/tpc.113.115907
Xu C, Wang Y, Yu Y, Duan J, Liao Z, Xiong G, Meng X, Liu G, Qian Q, Li J (2012) Degradation of MONOCULM 1 by APC/C(TAD1) regulates rice tillering. Nat Commun 3:750. doi:10.1038/ncomms1743
Yamaguchi M, Umeda M, Uchimiya H (1998) A rice homolog of Cdk7/MO15 phosphorylates both cyclin-dependent protein kinases and the carboxy-terminal domain of RNA polymerase II. Plant J 16:613–619
Zhong J, Haynes PA, Zhang S, Yang X, Andon NL, Eckert D, Yates JR, Wang X, Budworth P (2003) Development of a system for the study of protein–protein interactions in planta: characterization of a TATA-box binding protein complex in Oryza sativa. J Proteome Res 2:514–522
Acknowledgments
Maarten Dedecker is supported by a predoctoral fellowship from the Agency for Innovation by Science and Technology and CropDesign N.V. (‘Baekeland’ funding, IWT 100222). The authors thank Annick Bleys for help in preparing the manuscript.
Author contributions
M.D., S.V. and G.D.J. designed the research. N.D.W., G.P., E.V.D.S, B.C., L.V., L.D. and N.W. performed experiments. M.D., D.E. and G.D.J. analyzed the data. K.G. provided protocols for LC–MS/MS analysis. J.V.L. and K.G. commented on the manuscript. M.D. and G.D.J. wrote the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dedecker, M., Van Leene, J., De Winne, N. et al. Transferring an optimized TAP-toolbox for the isolation of protein complexes to a portfolio of rice tissues. Plant Mol Biol 91, 341–354 (2016). https://doi.org/10.1007/s11103-016-0471-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-016-0471-x