
Abstract
Transitory starch metabolism is a nonlinear and highly regulated process. It originated very early in the evolution of chloroplast-containing cells and is largely based on a mosaic of genes derived from either the eukaryotic host cell or the prokaryotic endosymbiont. Initially located in the cytoplasm, starch metabolism was rewired into plastids in Chloroplastida. Relocation was accompanied by gene duplications that occurred in most starch-related gene families and resulted in subfunctionalization of the respective gene products. Starch-related isozymes were then evolutionary conserved by constraints such as internal starch structure, posttranslational protein import into plastids and interactions with other starch-related proteins. 25 starch-related genes in 26 accessions of Arabidopsis thaliana were sequenced to assess intraspecific diversity, phylogenetic relationships, and modes of selection. Furthermore, sequences derived from additional 80 accessions that are publicly available were analyzed. Diversity varies significantly among the starch-related genes. Starch synthases and phosphorylases exhibit highest nucleotide diversities, while pyrophosphatases and debranching enzymes are most conserved. The gene trees are most compatible with a scenario of extensive recombination, perhaps in a Pleistocene refugium. Most genes are under purifying selection, but disruptive selection was inferred for a few genes/substitutiones. To study transcript levels, leaves were harvested throughout the light period. By quantifying the transcript levels and by analyzing the sequence of the respective accessions, we were able to estimate whether transcript levels are mainly determined by genetic (i.e., accession dependent) or physiological (i.e., time dependent) parameters. We also identified polymorphic sites that putatively affect pattern or the level of transcripts.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Photosynthetic CO2 fixation enables photoautotrophic organisms to gain reduced carbon which allows growth and biomass production. This fundamental process is, however, restricted to the light period. During darkness the central carbon metabolism of plants strictly relies on carbon compounds deposited in a preceding light phase. Therefore, under natural conditions intra- and intercellular carbon fluxes of plants are largely altered twice per 24 h.
In the light period, plants preferentially store reduced carbon compounds as large size polymers which do not significantly affect cellular water potentials even when massively formed. Starch is the almost ubiquitous storage polysaccharide, in some plant species supplemented by other reduced compounds, such as fructans (Vijn and Smeekens 1999; Lattanzi et al. 2012). In those plant species that form leaf starch as the predominant carbon store, mutants lacking a single functional starch-related protein are often severely retarded in growth except when grown under prolonged light periods (for review see Zeeman et al. 2010; Graf and Smith 2011) and can exhibit significant changes in the hormonal status (Paparelli et al. 2013). Furthermore, altered leaf starch metabolism of the mother plant strongly affects fruit development and seed composition (Andriotis et al. 2012). Thus, functional transitory starch turnover is also crucial for propagation of the respective plant population.
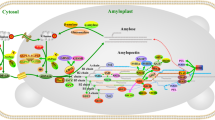
Transitory starch turnover originated very early in the evolution of chloroplast-containing cells and is based on a mosaic of polysaccharide-related genes that appear to be derived from the eukaryotic host cell, the prokaryotic endosymbiont, and Chlamydia (Ball et al. 2013; Cenci et al. 2013, 2014). Presumably, the ancestral state of starch metabolism was cytosolic and this subcellular distribution is still retained in Glaucophyta and Rhodophyceae, while in Chloroplastida it was rewired to plastids. Before and/or during translocation of starch metabolism to chloroplasts, several starch-related genes were duplicated (Fig. 1; for detailed informations see result section). Duplications are assumed to be essential to generate novel gene functions and to alter patterns of expression (Lynch and Conery 2000). Initially, many duplicated genes may have experienced relaxed selection, but—if duplicates acquired different functions—they were potentially exposed to gradually increasing selective constraints (Lynch and Conery 2000). Subsequently, starch-related isozymes underlied constraints related to the internal structure of native starch, the posttranslational import into the plastids, and to the interaction with other starch-related proteins. Both the internal structure of starch granules and the plastidial starch metabolism are highly conserved in Chloroplastida (Zeeman et al. 2010).

Schematic overview of starch metabolism and the involved genes in A. thaliana. A section of a chloroplast (including the two plastidial envelope membranes) and the cytosol is shown in light green and white, respectively. An intermediate of the Calvin cycle, fructose-6-phosphate (F6P; green) is converted by the plastidial phosphoglucoisomerase (pPGI; dark red) to glucose-6-phosphate (G6P) and, subsequently, to glucose-1-phosphate (G1P) by the plastidial phosphoglucomutase (pPGM). Following the reaction ATP + G1P ↔ ADPglucose (ADP-G) + pyrophosphate (PPi), the heterotetrameric ADPglucose pyrophosphorylase (AGPase) forms the principle glucosyl donor, ADP-G. In A. thaliana, four and two genes encode large (AtAPL1 to AtAPL4) and small (AtAPS1 and AtAPS2) subunits of AGPase, respectively. Unidirectional carbon flux from G1P to ADP-G requires pyrophosphate to be continuously removed which is achieved by conversion of pyrophosphate to orthophosphate as catalyzed by the plastidial inorganic pyrophosphatase (pPPA, AtPPA6). There is one further cytosolic (cPPA, AtPPA1) and four putatively cytosolic pyrophosphatases (AtPPA2 to AtPPA5) whose localization is not yet clearly verified (indicated with asteristics). ADP-G is used by all soluble starch synthases (SSs, AtSSI to AtSSIV) and the granule-bound starch synthase (GBSS, AtGBSS). Further modifications on glucan chains are performed by branching (BEs, AtBE1 to AtBE3) and debranching enzymes (ISAs, AtISA1 to AtISA3) as well as glucan phosphorylases (plastidial: Pho1/PHS1, AtPHS1; cytosolic: Pho2/PHS2, AtPHS2)
Plastidial starch turnover is based on close interactions of the products of more than 30 highly conserved gene products (Deschamps et al. 2008; Ball et al. 2011). In many higher plant species, such as maize and rice, additional duplications may permit a preferential expression of distinct starch synthase or branching isozymes in photoautotrophic and heterotrophic cells (Keeling and Myers 2010; Zeeman et al. 2010; Cheng et al. 2012).
In Arabidopsis thaliana, genes related to the central carbon metabolism exhibit similar levels of intraspecific sequence variability as genes involved in the secondary metabolism of plants (Schwarte and Tiedemann 2011; Schwarte et al. 2013). Given the fundamental importance of transitory starch for growth and development of A. thaliana (Zeeman et al. 2010; Andriotis et al. 2012; Pyl et al. 2012; Scialdone et al. 2013; Sulpice et al. 2014) and known differences in starch content among accessions (Sulpice et al. 2009), 26 accessions of A. thaliana were selected for sequence analyses of 25 starch-related genes that are members of six gene families (AGPase, PPA, SS, BE, ISA, PHS). Accessions were selected on the basis of physically separated natural populations exposed to different climates.
A. thaliana is a diploid plant species whose genome originates from a dicotyledon hexaploid ancestor and underwent massive alterations (Freeling 2009). Due to its reproduction predominantly by selfing, A. thaliana is supposed to be almost homozygous throughout the entire genome with an outbreeding rate of approximately 1–3 % (Abbott and Gomes 1989). The A. thaliana genome consists of five chromosomes which, despite their small size, contain numerous large duplicated segments and the genes belonging to six starch-related gene families are almost equally distributed over the five chromosomes (Fig. S1). These chromosomal rearrangements took place in the recent history of A. thaliana (Stein 2001; Blanc et al. 2003). Most of them took place after the split of monocots and eudicots which occurred about 150–200 million years ago (Mya; Wolfe et al. 1989; Blanc et al. 2003; Chaw et al. 2004). More precisely, there was a burst of duplications within the Rosids at the time of divergence between Rosid I (represented by Medicago truncatula) and Rosid II groups (represented by A. thaliana) (Blanc et al. 2003). Furthermore, more recently chromosomal duplications occurred with the order Brassicales between the separation of A. thaliana and Brassica rapa and the split of Brassicales (represented by A. thaliana) and Malvales (represented by cotton) approximately 24–40 Mya (Blanc et al. 2003). The signatures of these two rather recent chromosomal rearrangements are still visible in the genome of A. thaliana (Stein 2001; Blanc et al. 2003; Fig. S1). By contrast, the birth of starch-related genes can be traced back to the origin of Chloroplastida approximately 1,500 Mya (Blanc et al. 2003).
By sequence analyses of 25 starch-related genes in 26 accessions, we aimed to reveal (1) whether there is any correlation in the pattern of nucleotide diversity and/or the phylogenetic relationship among different starch-related genes/gene families whose products are all involved in the turnover of transitory starch, (2) whether there is an indication for positive selection and disruptive evolution in starch-related genes among accessions, and (3) whether there are putative associations between genetic divergence and the pattern or the level of transcripts. To answer the latter question, for selected genetically distinct A. thaliana accessions sequences of the putative promoter regions (about 1,000 bp upstream the translation start) were analyzed and transcript levels were quantified by Realtime PCR throughout the light period. Putative associations were evaluated on the basis of our data set consisting of 26 accessions as well as further 80 accessions retrieved from the 1001 Genome Project (Cao et al. 2011). Only one accession is contained in both data sets, such that our combined data set consists of 105 accessions in total.
Results
All starch-related genes analyzed in this study are encoded in the nuclear genome, but gene products are functional in either the plastid or the cytosol (Fig. 1). Genes whose products participate in both starch-related reactions and in other processes (such as pyrophosphatases) are also included. These genes are expected to face multiple evolutionary constraints. For each gene family, informations regarding the number of gene copies encoding small or large subunits of a given functional enzyme and the structure of the holoenzyme were collected. The KEGG database was checked for catalytic domains (Kanehisa and Goto 2000; Kanehisa et al. 2012) and the literature was screened for catalytical or regulatory motifs. For all nonsynonymous substitutions among accessions of A. thaliana (Table S1), known variation at the respective sites among other plant species (interspecies comparison; Table 1) and the inferred potential impact on functionality were discussed. Intraspecific genetic variation including nucleotide diversity, number of indels as well as synonymous and nonsynonymous substitutions was estimated for each gene (Table 2). The frequency of variable sites ranges from low (only very few accessions possessing the respective substitution) to high (numerous accessions exhibiting the same substitution). Multiple substitutions at the same site are rare. To increase the robustness of the genetic variation assessment, we extended our data set (26 accessions) by 80 additional accessions (Cao et al. 2011; Tables S1, S2).
Different tests to determine gene-wise selection modes were performed (Fig. 2). Any new variant of a gene can (1) drift randomly (neutral evolution), (2) be selected for (positive selection) or (3) be selected against (purifying selection). Positive selection refers to a number of selective processes by which multiple alleles (different gene variants/haplotypes) are maintained in the gene pool (Delph and Kelly 2014). Among them, disruptive selection (indicated by at least two divergent gene clusters/haplogroups in the respective genealogy) favors gene variants with distinct characteristics in different lineages. Positively selected genes or even positions can be indicative of adaptive processes which confer an advantage of fitness to the owner of the gene variant. By contrast, genes under purifying selection do not contribute to adaptive divergence due to marginal genetic variation. There are numerous established methods to identify modes of selection from the pattern of sequence variation, by comparing the relative occurrence of low frequency versus intermediate frequency substitutions, i.e., Tajima’s D (1989) as well as Fu and Li’s D*, F*, D, and F (Fu and Li 1993). An excess of rare polymorphisms (negative values) is indicative of purifying selection, a recent selective sweep or population expansion after a recent bottleneck, while an accumulation of intermediate-frequency polymorphisms (positive values) is indicative of ancestral or recent balancing selection or sudden population contraction. Among these tests, Tajima’s D is particularly influenced by demography and therefore less powerful to detect selection, if effective population sizes are not constant over time. Fu and Li’s statistics can be either performed with (D, F) or without an outgroup (D*, F*; Fu and Li 1993). The interspecific test (D, F) compares the number of single mutations (singletons) in internal and external branches by which ancestral and recent selective processes can be distinguished. Fu and Li’s D and F are most suitable to detect background/purifying selection. Another selection test that includes an outgroup is Fay and Wu’s H (2000). Here, intermediate- and high-frequency polymorphisms are compared. An excess of moderate- and high-frequency derived polymorphisms (negative values) is indicative of recent variation, while a deficit (positive values) indicates ancestral variation. As demographical changes can cause similar patterns of sequence variation as natural selection (see above), we also performed a powerful test to detect population expansion, i.e., R2 statistics (Ramos-Onsins and Rozas 2002). In combination of these tests, we (1) evaluate our data for deviation from the null hypothesis of neutral evolution in populations of constant size and (2) attempt to discern whether such deviations are rather due to demography or natural selection. If selection was inferred to occur in a particular gene, the putative mode of selection, i.e., purifying or disruptive selection, was inferred from the combined results of these tests (Fig. 2; Akey et al. 2004). Genes that possess an excess of low-frequency polymorphisms and a star-like genealogy (Figs. 3, 4, 5, 6, 7, 8, left panels) may be impacted by demography (i.e., population expansion, indicated by significant R2 values) or purifying selection. By contrast, genes with an excess of intermediate-frequency polymorphisms that often coincides with the occurrence of two or more distinct clusters in the genealogy (Figs. 3, 4, 5, 6, 7, 8, left panels) may be considered to be under positive selection, more specifically under disruptive selection among clusters (Fig. 2). For some genes, most accessions are assigned to a single main cluster from which only one or a few accessions deviate. In this case, the selection tests generally indicate strong purifying selection which may be considered the correct inference for the main cluster. The underlying pattern for single deviant accessions can be random noise or an altered selection regime; however, these two scenarios cannot be discerned with sufficient reliability, such that we refrain from presenting an evolutionary hypothesis for these deviations. Likewise, some genes did not yield a consistent pattern across selection tests. Here, we consider our data inconclusive with regard to the mode of selection.
Note that even if purifying selection was inferred for the entire gene, individual sites might be under positive selection and, therefore, a site-specific selection test was performed as well (Table 1).
In promoter regions, substitutions can affect cis-regulatory elements and, thereby, influence transcript levels directly. For six accessions representing different haplogroups and/or individual deviant haplotypes, transcript levels were quantified by Realtime PCR at the beginning (BOL), in the middle (MOL), and at the end of the light period (EOL). Transcript levels were calculated relative to the transcript level of the housekeeping gene ubiquitin. Deviations are given on a log2 scale (=∆Ct values), i.e., ∆Ct values of +1 or −1 correspond to transcript levels twofold elevated (=21) or 50 % decreased (=2−1), respectively (Figs. 3, 4, 5, 6, 7, 8, right panels). The respective influences of genetics (transcript levels differing between accessions at a given time point) or physiology (transcript levels differing between time points in a given accession) as well as the interaction between genetics and physiology were disentangled in a two-way analysis of variance (ANOVA; Table 3). Finally, site-specific correlation analyses were performed to identify positions putatively associated with either the pattern or the level of transcription (hereinafter referred to as putative associations; Table S2).
ADPglucose pyrophosphorylase
Background
In higher plants, ADPglucose pyrophosphorylase (AGPase; EC 2.7.7.27) exists often as a heterotetrameric holoenzyme (α2β2) composed of two so-called large (often designated as L or β) and two small subunits (S or α). The size of the two subunit types is approximately 51 and 50 kDa, respectively (Georgelis et al. 2007; Ventriglia et al. 2008). Genes encoding α or β subunits originate from the same ancient gene (Georgelis et al. 2007). Leaves from A. thaliana mutants that lack either subunit type (α or β) of the AGPase holoenzyme accumulate very little transitory starch (Lin et al. 1988). Both subunits carry a single catalytic domain (pf:NTP_transferase; residues see below; KEGG) containing highly conserved amino acid residues. In A. thaliana, six AGPase genes exist, four of which encode large subunits (AtAPL1 to AtAPL4) and two genes small subunits (AtAPS1 and AtAPS2; Crevillén et al. 2003, 2005; Georgelis et al. 2007; Ventriglia et al. 2008). The heteromeric AGPase holoenzyme from leaves is assumed to mainly consist of AtAPS1 and AtAPL1 (Crevillén et al. 2003, 2005; Hädrich et al. 2012).
Sequence diversity
At the amino acid (aa) level, identity among the four large subunits from A. thaliana ranges from 57 to 88 %. AtAPL3 and AtAPL4 exhibit the highest degree of aa identity. The small subunits, AtAPS1 and AtAPS2, share 41 % of their aa residues. With respect to intraspecific variability, both AtAPL1 and AtAPL2 exhibit only 3 inferred aa substitutions and a highly conserved catalytic domain (pf:NTP_transferase; AtAPL1: residues 92-368; AtAPL2: 87-364; KEGG; Table 1), lacking any nonsynonymous substitutions. By contrast, in the four other AGPase subunits more aa substitutions were inferred, some of which occurring at positions otherwise highly conserved among plant species (pf:NTP_transferase; AtAPL3: residues 91-365; AtAPL4: 93-368; AtAPS1: 92-365; AtAPS2: 57-320; KEGG; Table 1). However, none of these nonsynonymous substitutions affect sites with known catalytic or regulatory functions (i.e., in AtAPL1: R102, K112, K271; in AtAPL2: R97, K107, K267; in AtAPL3: K101, T111; in AtAPL4: K103, T113; in AtAPS1: C81, R102, K112, D214, K267; Ballicora et al. 2004; Hädrich et al. 2012). Crevillén et al. (2003) describe functional motifs of the small subunits, i.e., ATP binding site (residues 185-193 in AtAPS1; 148-156 in AtAPS2), catalytic site (211-221; 174-184), G1P binding site (261-272 in AtAPS1; 224-233 in AtAPS2), and activator site (503-520 in AtAPS1; 459-476 in AtAPS2). In seven accessions, two simultaneously occurring nonsynonymous substitutions were observed within the G1P binding site of AtAPS1 (I262V, H272Q; Table 1; Suppl. Table 1). These sites are also found in numerous other accessions (Gan et al. 2011; Suppl. Table 1). In AtAPS2, none of these functional motifs is affected by any amino acid substitution.
All six AGPase genes exhibit significant intraspecific nucleotide diversities (Table 2). In both promoter and gene, highest diversity was observed for AtAPL3 (Table 2). Nucleotide diversity within AtAPL3 is five times higher than in AtAPL1 and AtAPL2. Comparing the two small subunit genes, AtAPS1 is more variable than AtAPS2 (Table 2).
Inferred selection and phylogenetic clustering
As it is the case for all starch-related genes, each gene possesses its own evolutionary history. AtAPL1 yields negative test statistics of Tajima and Fu and Li, but none of them are significant (Fig. 2). In the genealogy, there is a single cluster from which several accessions deviate (Fig. 3a, left panel). AtAPL2 shows an excess of low-frequency polymorphisms as determined by the test statistics of Tajima and Fu and Li, albeit again not significant (Fig. 2). The distribution of substitutions across numerous accessions leads to a star-like genealogy (Fig. 3b, left panel). The selective and/or demographic processes shaping the genealogies of AtAPL1 and AtAPL2 could not be inferred with sufficient certainty. Most polymorphisms of AtAPL3 show intermediate frequencies as indicated by positive Tajima and Fu and Li test statistics (Fig. 2). The respective genealogy is structured into small but separated clusters (Fig. 3c, left panel). Jointly, selection test statistics and phylogeny are indicative of disruptive selection among clusters in AtAPL3. AtAPL4 shows an excess of low-frequency polymorphisms as determined by the test statistics of Tajima and Fu and Li (Fig. 2). Substitutions mainly occur in a single deviating accession (Can; Fig. 3d, left panel) and the test statistics are highly significantly negative. The extension of the data set by 80 additional accessions does not yield further deviating accessions (Cao et al. 2011; Tables S1, S2). Therefore, purifying selection seems to be the most plausible scenario for the evolution of AtALP4. AtAPS1 exhibits several intermediate-frequency polymorphisms (Fig. 2). There are four deviating accessions apart from the main cluster (Fig. 3e, left panel). However, the mode of selection could not be unequivocally inferred. AtAPS2 possesses an excess of low-frequency polymorphisms as indicated by significant Tajima and Fu and Li values (Fig. 2) leading to a star-like genealogy (Fig. 3f, left panel). However, R2 statistics for population expansion are significant (Fig. 2). For this reason, a particular mode of either selection and/or demography shaping this genealogy cannot be inferred.

Gene-wise selection tests on starch-related genes among accessions of A. thaliana. Intraspecific and interspecific (including the outgroup A. lyrata) selection tests were performed. Tajima’s D as well as Fu and Li’s D*, F*, D and F compare low- and intermediate-frequency variants. Fay and Wu’s H compares medium- and high-frequency variants. Fu and Li’s D and F as well as Fay and Wu’ H are interspecific selection tests that additionally compare ancestral and derived polymorphisms. R2 statistics is a powerful tool to detect population expansion. Genes are ordered from left to right based on the results of Tajima and Fu and Li test statistics, started with the lowest ones. Statistical significance for all tests was calculated by using coalescent simulations with 10,000 simulations. The probability is indicated with asteristics (*p < 0.05, **p < 0.02, ***p < 0.01)

Phylogenetic trees and transcript levels of AGPase genes in A. thaliana. a AtAPL1; b AtAPL2; c AtAPL3; d AtAPL4; e AtAPS1; f: AtAPS2. Left panels unrooted maximum likelihood trees among accessions based on promoter and gene sequences. Clusters are highlighted with circles. Right panels differences in transcript levels among selected accessions are displayed by ∆Ct values (normalized with a housekeeping gene; see text). As measure of the reliability, confidence intervals are integrated as shadings. The selected six accessions are members of different phylogenetic clusters of respective genes and are marked with the same color in both the left and the right panel
In all AGPase genes except AtAPS2, most polymorphisms arose in the recent history of A. thaliana (Fay and Wu’s H, Fig. 2).
The mode of selection of an entire gene can deviate from that of distinct codons. In both small subunits of the AGPase, positively selected positions were identified (Table 1, marked with asteristics). In AtAPS1, five out of 10 nonsynonymous substitutions were inferred to occur at positively selected sites (PSS). All these PSS co-occur in seven accessions of our data set and numerous other accessions (Table S1; Cao et al. 2011). However, these accessions are not directly related in the respective gene tree (Fig. 3e, left panel). AtAPS2 exhibits a single inferred PSS. All PSS both in AtAPS1 and AtAPS2 are found at positions that are known to be variable among a wide range of plant species (Table 1).
Transcript levels relative to accession and time-of-the-day
Of all large subunit genes, AtAPL1 has highest relative transcript levels (Fig. 3a, right panel) followed by AtAPL2, AtAPL3, and AtAPL4 (Fig. 3b–d, right panels). These data are consistent with previous studies according to which AtAPL1 is the dominant isoform in leaves (Crevillén et al. 2005; Gan et al. 2011). Differences in transcript levels of the four large AGPase subunits can largely be attributed to variation among accessions (Table 3) and may be related to the genetic variation observed. Among the AGPase small subunits, AtAPS1 is the dominant isoform (Fig. 3e, right panel) which again is consistent with previous studies (Crevillén et al. 2005; Gan et al. 2011). Transcript levels of this gene are even higher than those of the dominant large subunit, AtAPL1. Differences in transcript levels can be attributed to both accession and time-of-the-day in AtAPS1, while the interaction between both factors accounts for about one-third of the variation (Table 3). Throughout the entire light period, transcript levels of AtAPS2 are lowest of all AGPase subunits (Fig. 3f, right panel). More than 50 % of the variation in AtAPS2 transcript levels occurred within biological and/or technical replicates within accessions (Table 3).
For AtAPL1, we could not identify any genetic variation (putative associations) that correlates with the two transcript patterns which could be observed throughout the light period, i.e., either highest (Can, El) or lowest levels (Ws, Gre, Mt, Er) at MOL (Fig. 3a, right panel). We further compared accessions with overall high vesus low transcript levels (Can, Ws vs. El, Er). Two sites in the putative promoter correlate with high transcript levels that co-occur in 23 and 25 accessions, respectively (Table S2; Cao et al. 2011).
In AtAPL2, two accessions (Er, Nok) exhibit deviating transcript patterns, but no sites were detected whose variation correlates to the transcript patterns observed (Fig. 3b, right panel). After arranging the accessions with regard to transcript levels from high to low, two sites could be identified that are correlated with the level of transcripts (one position in the promoter; one in an exon; Table S2). Interestingly, the substitution in the exon occurs in 78 out of 105 accessions (Table S2; Cao et al. 2011).
In AtAPL3, accessions exhibit two different transcript patterns, i.e., either an increase or a decrease throughout the light period (Fig. 3c, right panel), but polymorphic sites correlating with this pattern were not found. Comparative analyses of all accessions, however, revealed sites that are correlated with the overall level of transcripts. Accessions with low transcript levels (El, Ler, Sap) possess specific sites that differ from those accessions with high transcript level (one site in an exon; three in introns; Table S2). All of these sites co-occur in more than half of the analyzed 105 accessions (Table S2; Cao et al. 2011).
Different transcript patterns were also observed for AtAPL4 as accessions show increasing, decreasing transcript levels as well as lowest levels at MOL (Fig. 3d, right panel), but comparative sequence analyses did not reveal any coincidence with particular substitutions or indels, neither for the temporal pattern of transcription nor for the overall transcript levels.
In AtAPS1, transcript levels follow two different patterns throughout the light period (Fig. 3e, right panel), i.e., either peaking at MOL (Bur, Oy) or steadily decreasing from BOL towards EOL (Ct, Nok, Stw, Van). No polymorphisms were associated with this pattern or the level of transcripts.
The prevalent transcript pattern of AtAPS2 is the peak at MOL (Fig. 3f, right panel). Cvi shows a deviating pattern and exhibit six individual sites that differ from all other accessions analyzed in this study, including the 80 accessions subset of the 1001 Project (Table S2; Cao et al. 2011).
Inorganic pyrophosphatases
Background
Unidirectional carbon flux from glucose-1-phosphate (G1P) to ADPglucose requires pyrophosphate to be continuously removed which is achieved by hydrolyzing pyrophosphate to orthophosphate as mediated by the plastidial inorganic pyrophosphatase (PPA; EC 3.6.1.1; Schulze et al. 2004; Meyer et al. 2012). In a functional state, PPA is a monomeric protein that occurs in several compartments. Increased activity of one of the cytosolic PPA isozymes is associated with elevated levels of ascorbate, sucrose, and glucose, but decreased starch contents (Osorio et al. 2013). In A. thaliana, six PPA encoding genes exist (Schulze et al. 2004; Navarro-De la Sancha et al. 2007; Meyer et al. 2012). The PPA isozymes share a common catalytic domain comprising approximately 150 amino acid residues which covers almost the entire sequence of the polypeptide (pf:pyrophosphatase; AtPPA1: residues 50-203; AtPPA2: 56-209; AtPPA3: 54-207; AtPPA4: 54-207; AtPPA5: 54-207; AtPPA6: 106-285; KEGG). Products of AtPPA1 to AtPPA5 are non-plastidal proteins with a high degree of amino acid (aa) identity (68–89 %). AtPPA6 encodes the plastidal PPA including a transit peptide of approximately 66 aa residues (ChloroP; Emanuelsson et al. 1999). The mature plastidial AtPPA6 protein deviates from all other isozymes in having only approximately 24 % aa identity to them (24.8 % identity to AtPPA1; 23.2 % to AtPPA2; 23.6 % to AtPPA3; 23.6 % to AtPPA4; 23.6 % to AtPPA5).
Sequence diversity
The active site of PPAs contains a motif consisting of seven amino acid residues, DNDPIDV, located between position 100 and 110 of the mature PPA proteins (Schulze et al. 2004). In AtPPA5, position five (DNDPIDV, highlighted in bold) is affected by a synonymous substitution (position 316-318 in the coding sequence, ATA to ATT) that is found only in Cha. AtPPA3 and AtPPA6 are the only PPAs possessing nonsynonymous substitutions (Table 1; Table S1). In AtPPA3, the substitution N54H occurs in the catalytic domain. As revealed by interspecies comparison, these positions are rather variable. Overall, the PAA gene family is much more conserved than any other starch-related gene analyzed here (Tables 1, 2). Presumably, this is due to the fact that most of the sequence represents the highly conserved catalytic domain. AtPPA4 is the only exception, as its exhibits both a considerable nucleotide diversity and numerous indels (Table 2).
Inferred selection and phylogenetic clustering
Significant negative values of Tajima and Fu and Li test statistics in AtPPA1, AtPPA3, and AtPPA5 are indicative of an excess of low-frequency polymorphisms (Fig. 2). These substitutions are mainly found in single accessions, i.e., Sha, Cvi, and Cha, which appear separate from the main phylogenetic cluster (Fig. 4a, c, e, left panels). The extension of the data set by 80 additional accessions does not yield additional deviating accessions (Cao et al. 2011, Tables S1, S2). Therefore, we assume purifying selection to act on these genes. AtPPA2 possesses an excess of low-frequency polymorphisms, which is supported by negative Tajima and partially significant Fu and Li values (Fig. 2). These substitutions are spread over several accessions leading to a star-like genealogy (Fig. 4b, left panel). In addition, R2 statistics for population expansion are significant (Fig. 2). Our data are hence inconclusive as to whether the pattern of polymorphisms is indicative of selective or demographic processes. AtPPA4 exhibits an excess of intermediate-frequency polymorphisms (Fig. 2). The substitutions are distinct among groups of accessions in the genealogy (Fig. 4d, left panel). This evidence is compatible with the scenario of disruptive selection among phylogenetic clusters acting on AtPPA4. In AtPPA6, there is an excess of low-frequency polymorphisms (Fig. 2), which are distributed over several accessions that form again three individual small clusters (Fig. 4f, left panel). The underlying selective or demographic modes, however, could not be determined with sufficient confidence.

Phylogenetic trees and transcript levels of pyrophosphatase genes in A. thaliana. a AtPPA1; b AtPPA2; c AtPPA3; d AtPPA4; e AtPPA5; f AtPPA6. Left panels unrooted maximum likelihood trees among accessions based on promoter and gene sequences. Clusters are highlighted with circles. Right panels differences in transcript levels among selected accessions are displayed by ∆Ct values (normalized with a housekeeping gene; see text). As measure of the reliability, confidence intervals are integrated as shadings. The selected six accessions belong to different clusters of the respective genes and are marked with the same color in the left and the right panel
Site-specific selection analysis revealed that none of the few nonsynonymous substitutions are inferred to be positively selected. Most polymorphisms found in AtPPA1, AtPPA3, AtPPA5, and AtPPA6 originated recently in the lineage of A. thaliana, while those of AtPPA2, and AtPPA4 are mainly older and occurred in A. lyrata as well (Fay and Wu’s H, Fig. 2).
Transcript levels relative to accession and time-of-the-day
Based on transcript levels, AtPPA1 and AtPPA6 are likely to be the main isoforms, followed by AtPPA2, AtPPA5, AtPPA4, and AtPPA3 (Fig. 4a–f, right panels). These data are consistent with a previous study (Gan et al. 2011). The two last ones exhibit highest intraspecific variation in transcript levels. Throughout the light period, transcript levels of AtPPA3 are very low. Differences in transcript levels of AtPPA1, AtPPA3, and AtPPA6 are almost equally due to accession and time-of-the-day (Table 3). AtPPA2 transcripts are mostly influenced by time-of-the-day, while AtPPA4 and AtPPA5 are more affected by differences among accessions (Table 3).
Generally, AtPPA1 exhibits lowest transcript levels at MOL but transcript profiles among accessions differ in details (Fig. 4a, right panel). Can is the only accession whose AtPPA1 transcript levels do not increase during the light period. This accession exhibits four positions which putatively are associated with differences in transcript levels. Two of them also occur in one and three further accessions, respectively (Table S2; Cao et al. 2011). Putative associations that are correlated with the level of transcripts among all analyzed accessions were not identified.
AtPPA2 shows lowest transcript levels at MOL and a similar transcript pattern across all accessions (Fig. 4b, right panel) but levels differ among accessions. Two putative associations in the putative promoter correlate with the level of transcripts and are altogether present in 40 and 18 accessions, respectively, out of the total dataset of 105 accessions (Table S2; Cao et al. 2011).
In AtPPA3, two transcript patterns were observed. Both Cvi and Sap exhibit decreasing transcript levels during the light period, while the remaining accessions show an increase of transcripts towards EOL (Fig. 4c, right panel). No correlations were found between polymorphic sites and patterns or levels of transcripts.
In AtPPA4, Sha, Stw, and Te possess highest transcript levels at MOL, while Cha, Rsch, and Van have low levels at MOL (Fig. 4d, right panel). Sha and Stw exhibit a similar pattern throughout the light period, while in Te transcript levels particularly decline towards EOL. A distinct position was identified in the 3′-UTR that differs in Te as compared to Sha and Stw. Sha exhibits much higher transcript levels as compared to Stw and Te. Sha possesses three unique sites in the putative promoter (Table S2). Among the accessions with low transcript levels at MOL, Rsch is the only one with lowest transcript levels at BOL while the expression at other times follows the pattern of Cha and Van. Site-specific sequence analyses revealed four positions in introns and the 3′-UTR that are altered in Rsch as compared to Cha and Van (Table S2). Of all other PPAs, transcript levels of AtPPA4 are most variable among the accessions studied and putative associations between substitutions and transcript pattern were found in 2–85 accessions (Table S2; Cao et al. 2011).
In AtPPA5, most analyzed accessions show largely constant transcript levels throughout the light period, with few exceptions (Fig. 4e, left panel): Cha exhibits decreasing transcript levels and has also a unique position in the phylogenetic tree. In this accession, three unique sites were identified in the putative promoter (Table S2). Additionally, there are 22 variable sites in the gene and the 3′-UTR, both substitutions and indels (Table S2). Screening all the 105 accessions, only one of these sites was found to be variable in eight further accessions (Table S2; Cao et al. 2011). Er and El exhibit deviating transcript patterns, but putative associations between substitutions and transcript pattern were not found.
Transcript patterns of AtPPA6 are similar among accessions with lowest levels at MOL (Fig. 4f, left panel) but both Er and Rsch deviate. Er exhibits much lower transcript levels, while in Rsch transcript levels are almost constant throughout the light period. Er shows a single unique site in the putative promoter region, while in Rsch carries several sites that have unique nucleotides (one position in the promoter; four in introns; Table S2). All putative associations are also present in 10–52 other accessions (Table S2; Cao et al. 2011).
Starch synthases Footnote 1
Background
The quantitatively predominant process in starch biosynthesis is the successive elongation of α-glucan chains by several starch synthase isozymes (SSs; EC 2.4.1.21; ADPglucose: [1 → 4] α-d-glucan 4-α-d-glucosyl transferase; Tenorio et al. 2003; Delvallé et al. 2005; Zhang et al. 2005, 2008; Roldán et al. 2007) that repetitively transfer the glucosyl moiety from ADPglucose to non-reducing chain ends. Massively and unidirectionally elongated α-glucan chains form a helical structure. Higher plants possess at least five classes of SSs comprising four classes of soluble synthases (SSI to SSIV) and a single class of granule bound synthase (GBSS). In A. thaliana, each class is represented by a single gene (AtSSI to AtSSIV and AtGBSS; Tenorio et al. 2003; Delvallé et al. 2005; Zhang et al. 2005, 2008; Roldán et al. 2007). It is widely accepted that GBSS synthesizes amylose whereas SSI to III catalyze distinct, but partly overlapping steps in the amylopectin biosynthesis (Fujita et al. 2011; Szydlowski et al. 2011; Bertoft 2013). SSIV is important for the initiation of starch granule biosynthesis, but can be functionally replaced by SSIII to some extent. Double knock-out A. thaliana mutants lacking both AtSSIII and AtSSIV possess largely diminished starch content, elevated ADPglucose levels, and are severely compromised in growth even under long-day conditions (Ragel et al. 2013). AtSSIV appears to be essential for the coordination of the initiation of starch granule formation and chloroplast division in rapidly expanding leaf cells (Crumpton-Taylor et al. 2013). SSs possess two catalytic domains, designated as GT5 (pf:Glyco_transf_5; AtSSI: residues 144-401; AtSSII: 302-545; AtSSIII: 647-837; AtSSIV: 544-783; AtGBSS: 86-345; KEGG) and GT1 (pf:Glycos_transf_1; AtSSI: 455-607; AtSSII: 605-743; AtSSIII: 896-1019; AtSSIV: 839-995; AtGBSS: 399-527; KEGG). AtSSIII is unique in possessing three copies of a carbohydrate binding module (CBM25) in its N-terminal region, each comprising 85–95 amino acid residues (KEGG). Therefore, AtSSIII appears to be capable of multiply binding to carbohydrate targets.
Sequence diversity
For all SSs, the GT5 domain is more variable than GT1 (Table 1). In AtSSIII, all three CBM binding modules exhibit nonsynonymous substitutions, but the second CBM is most variable (N-terminal CBM25: T279I; second CBM25: F392V, L393M, G398S, Q408R, L410I, N421D, R425 K, D430E, R431 K, M438T, E451K; C-terminal CBM25: I571F, V616I, I623L; Suppl. Table 1). Generally, all SSs exhibit high numbers of nonsynonymous substitutions, some of them at positions highly conserved in other plant species (Table 1). AtSSI and AtSSIV have the highest levels of nucleotide diversity, while variability is lowest in AtSSII (Table 2). AtSSI possesses, however, the lowest number of nonsynonymous substitutions (Table 1). In AtSSII, nonsynonymous substitutions are twofold more abundant than synonymous ones. For AtSSIII, three accessions (Can, Ct, El) deviate from all other accessions (Table S1). The genetic alteration includes one indel of 21 nucleotides (7 amino acids) in exon 1. The majority of these substitutions and indels, including that in exon 1, are shared with A. lyrata, the sister species of A. thaliana (Grigoriev et al. 2012). AtSSIV and AtGBSS possess intermediate intraspecific nucleotide diversities (Table 2).
Inferred selection and phylogenetic clustering
The Tajima as well as the Fu and Li test statistics reveal an excess of intermediate-frequency polymorphisms in AtSSI (Fig. 2). The substitutions are restricted to a group of accessions, which results in a bifurcation in the genealogy indicative of disruptive selection (Fig. 5a, left panel). AtSSII shows an excess of low-frequency polymorphisms (Fig. 2), which occurs occasionally across accessions leading to a genealogy with several deviating accessions (Fig. 5b, left panel). Furthermore, R2 statistics for population expansion are significant (Fig. 2). For this reason, the mode of evolution underlying the AtSSII diversity pattern, i.e., selection and/or demography is not clear. In AtSSIII and AtGBSS, slightly positive Tajima and Fu and Li test statistics indicate an excess of intermediate-frequency polymorphisms (Fig. 2). For both genes, a group of accessions clearly deviates from the main phylogenetic cluster (Fig. 5c, e, left panel). The extension of the data set revealed additional accessions possessing these substitutions (Cao et al. 2011; Tables S1, S2). However, a particular mode of selection or demography could not be inferred. In AtSSIV, there is an excess of both low- and intermediate frequency polymorphisms (Fig. 2). The substitutions mainly occur in four accessions (Fig. 5d, left panel). In addition, the test for population expansion is significant (Fig. 2). Therefore, we cannot infer the respective impact of selection and demography on the genetic variation of AtSSIV.

Phylogenetic trees and transcript levels of starch synthase genes in A. thaliana. a AtSSI; b AtSSII; c AtSSIII; d AtSSIV; e AtGBSS. Left panels unrooted maximum likelihood trees among accessions based on promoter and gene sequences. Clusters are highlighted with circles. Right panels differences in transcript levels among selected accessions are displayed by ∆Ct values (normalized with a housekeeping gene; see text). As measure of the reliability, confidence intervals are integrated as shadings. The selected six accessions representing different clusters of the respective genes are marked with the same color in the left and the right panel
Overall, starch synthases constitute one of the most variable starch-related gene families. All genes show numerous nonsynonymous substitutions, but none of these codons are inferred to be positively selected (Table 1). Most polymorphisms of starch synthases originated recently in the lineage of A. thaliana (Fig. 2).
Transcript levels relative to accession and time-of-the-day
Transcript levels of AtSSI are primarily affected by the time-of-the-day, while transcript levels of AtSSII, AtSSIII, and AtSSIV mainly vary among accessions (accounting for 40–47 % of the variation; Table 3). Differences in transcript levels of AtGBSS are equally affected by accession and time-of-the-day (38 and 42 %, respectively; Table 3).
In AtSSI, the transcription pattern in Bl, Can, and Edi is similar as highest transcript levels were obtained at MOL, while Gre, Sha, and Wil exhibit individual patterns (Fig. 5a, left panel). Genetic variation specific to individual accessions is found in Gre (two positions in the promoter; four in introns; Table S2), Wil (one position in the promoter; four in introns; Table S2), and Sha (eight positions in the promoter; one in an exon; two in introns; Table S2). For most of these sites, 2–55 additional accessions were found to possess the same genetic variant (Table S2; Cao et al. 2011).
In AtSSII, the transcript pattern is similar among accessions as lowest transcript levels were obtained at MOL (Fig. 5b, left panel). Col, however, shows a deviating transcript pattern. Polymorphisms associated with either patterns or levels of transcripts were not found.
In AtSSIII, accessions show different patterns throughout the light period, having either lowest (Ct, Ler, Yo) or highest levels (Can, El) at MOL, or constantly decreasing transcript levels from BOL towards EOL (Stw; Fig. 5c, left panel). Specific sites that correlate with either lowest or highest transcript levels at MOL were not found, but Stw exhibits substitutions that are found in further accessions as well (two positions in the promoter; two in exons; one in the 3′-UTR; Table S2; Cao et al. 2011). For all three time points, Yo exhibits highest transcript levels as compared to all other analyzed accessions. This accession has several specific substitutions (four positions in exons; one in the 3′-UTR; Table S2). The deviant substitution pattern (putative associations) was found in 17–21 further accessions (Table S2; Cao et al. 2011).
In AtSSIV, three different transcript patterns exist. Highest levels at MOL are found in Can and Cvi, lowest levels at MOL in Gre and Ws or constantly decreasing throughout the light period in Bur and Er (Fig. 5d, left panel). There are numerous sites that differ among accessions with increasing (Can, Cvi) and decreasing transcript levels (Bur, Er, Gre, Ws) towards MOL, i.e., 13 in exons, 22 in introns, and 6 in the 3′-UTR. There are a few additional accessions that share most of the putative associations (Table S2; Cao et al. 2011). Putative associations between sequence and transcript pattern are not observed in the putative promoter, as the promoter sequence of Bur is similar to those of Can and Cvi. For this gene, the position of accessions in the phylogenetic tree correlates with transcript levels, especially at MOL. Er, Gre, and Ws show low transcript levels and belong to cluster I. Can and Cvi exhibit highest transcript levels and are clearly separated from cluster I, while Bur is intermediate, both in phylogenetic position and transcript levels.
In AtGBSS, transcripts tend to decrease towards EOL (most variation due to time-of-the-day; Table 3), but the level of transcripts also differs among accessions (Fig. 5e, left panel). Sequence variation and level of transcripts appear to be uncorrelated.
Branching enzymes
Background
Branching enzymes (BEs; EC 2.4.1.18; α-1,4-glucan: α-1,4-glucan 6-glucosyl transferase) are monomeric proteins possessing two distinct enzymatic activities that are attributed to two domains of the polypeptide (Dumez et al. 2006). BEs from higher plants are often grouped into two classes, designated as I and II. Class I BEs are thought to transfer longer chains as compared to class II isozymes (Tomlinson and Denyer 2003), but not all BEs follow this classification. In A. thaliana, three genes (AtBE1 to AtBE3) encode branching enzymes. The sequence of AtBE1 is related to the starch branching enzyme family but it cannot be assigned to either class I or II and, therefore, does not code for a true branching enzyme (Dumez et al. 2006). Sequence similarity to the two other gene products is low (compared to AtBE2 and AtBE3, aa identity and similarity is approximately 26 and 35 %, respectively). Both AtBE2 and AtBE3 belong to BE class II, sharing 73 % aa identity (Dumez et al. 2006). Like α-amylases, they cleave internal α-1,4-interglucose bonds using the N-terminal domain (pf:Alpha-amylase; AtBE1: residues 425-498; AtBE2: 318-474; AtBE3: 353-425; KEGG). In addition, BEs transfer the oligoglucanyl residue liberated to the same or a vicinal α-glucan chain forming an α-1,6-glucosidic bond. This catalytic activity is attributed to the C-terminal beta domain (pf:Alpha-amylase_C; AtBE1: 800-893; AtBE2: 707-801; AtBE3: 742-835; KEGG). Furthermore, BEs often possess a carbohydrate binding module (pf:CBM_48; AtBE1: unknown; AtBE2: 169-252; AtBE3: 205-287; KEGG). In addition to the classical domains designated by KEGG, the (β/α)8-barrel is another structural feature typical for branching enzymes (Jespersen et al. 1993; Svensson 1994). It consists of eight β-strands and eight α-helices which alternate along the polypeptide chain and fold the inner cylindrical β-strands surrounded by α-helices. Here, the (β/α)8-barrel structure described for the BE from E. coli was used to determine the approximate position of the respective motifs in the branching enzymes of A. thaliana.
Sequence diversity
Like in most starch-related gene families, several nonsynonymous substitutions were observed in the three BE genes (Table 1). In AtBE1 and AtBE3, both catalytic domains (according to KEGG) lack mutations. In AtBE2, however, nonsynonymous substitutions were identified in both catalytic domains (Table 1; Table S1). Based on interspecific comparison, these positions are generally highly conserved (Table 1). The amino acid sequence of the CBM48 that features AtBE2 and AtBE3 (but not AtBE1) exhibits only synonymous substitutions. Within the (β/α)8-barrel, nonsynonymous substitutions were identified in each BE (AtBE1: V405I, N649S, A671S, N754S; AtBE2: P273L, S354T; AtBE3: K479L; Table S1). Promoter regions of all BEs exhibit similar nucleotide diversities, but regarding the gene sequence, AtBE1 is by far more variable than AtBE2 and AtBE3 (Table 2).
Inferred selection and phylogenetic clustering
For AtBE1, the positive test statistics of Tajima and Fu and Li are indicative of an excess of intermediate-frequency polymorphisms (Fig. 2), which affect about half of the accessions visible as two clusters in the genealogy (Fig. 6a, left panel). For this reason, we assume disruptive selection for AtBE1. Both AtBE2 and AtBE3 possess an excess of low-frequency polymorphisms as indicated by significant negative Tajima and Fu and Li values (Fig. 2). The substitutions are distributed over numerous accessions, which lead to a star-like genealogy (Fig. 6b, c, left panel). In addition, there are significant signs of population expansion (Fig. 2). Whether the star-like shape of the AtBE2 and AtBE3 genealogy is caused by purifying selection and/or population expansion is not clear.

Phylogenetic trees and transcript levels of branching enzyme genes in A. thaliana. a AtBE1; b AtBE2; c AtBE3. Left panels unrooted maximum likelihood trees among accessions based on promoter and gene sequences. Clusters are highlighted with circles. Right panels differences in transcript levels among selected accessions are displayed by ∆Ct values (normalized with a housekeeping gene; see text). As measure of the reliability, confidence intervals are integrated as shadings. The selected six accessions are members of different clusters of the respective genes and are marked with the same color in the left and the right panel
The comparison with A. lyrata revealed an excess of derived polymorphisms (Fay and Wu’s H, Fig. 2).
Transcript levels relative to accession and time-of-the-day
Transcript levels of AtBE2 are highest, followed by AtBE3 (Fig. 6b, c, right panels). AtBE1 whose metabolic function is not yet clear shows lowest transcript levels (Fig. 6a, right panel). This general tendency is consistent with a previous study (Gan et al. 2011). In all BE genes, transcript levels strongly differ among accessions throughout the entire light period. The influence of time-of-the-day on AtBE2 transcription is relatively small, while AtBE1 and AtBE3 transcript levels are highly affected by time-of-the-day (explaining 33 and 58 % of the variation, respectively; Table 3).
In most accessions, transcript levels of AtBE1 increase during the light period (Fig. 6a, right panel). With regard to the pattern of transcripts throughout the light period, Cvi and Nok deviate from most accessions as they show either highest or lowest transcript levels at MOL. Cvi differs from all other analyzed accessions by several unique sites (one in the promoter; five in introns; one in the 3′-UTR; Table S2). Nok shows individual substitutions as well (one position in an exon; one in an intron; Table S2). Some substitutions of both Cvi and Nok were found in 2–65 further accessions in the 105 accessions data set (Table S2; Cao et al. 2011).
In AtBE2, most accessions follow the same transcript pattern throughout the light period, i.e., slightly lower transcript levels at MOL and an increase towards EOL, except for Bl that exhibits highest transcript levels at MOL (Fig. 6b, right panel). Unique sites were found in Bl (one position each in the promoter, exon, and intron; Table S2). These putative associations are also found in 4–12 further accessions (Table S2; Cao et al. 2011). No positions were found to correlate with transcript levels.
The pattern of transcription in AtBE3 generally coincides among accessions as lowest transcript levels are found at MOL (Fig. 6c, right panel). Three positions that correlate with high transcript levels (Oy, Wil, Yo) were found in 31 and 63 further accessions, respectively (one position in the promoter; two in exons; Table S2; Cao et al. 2011).
Debranching enzymes
Background
All plant debranching enzymes (DBEs) selectively hydrolyze α-1,6-interglucose linkages (direct debranching). Based on sequence comparison and substrate specificities, two types of DBEs are distinguished, isoamylases (ISAs; EC 3.2.1.68) and pullulanases, also named limit-dextrinases (PUs/LDAs; E3.2.1.142; Streb et al. 2012). The ISA type covers three classes designated as ISA1, ISA2, and ISA3. In A. thaliana, each class is represented by a single gene (AtISA1 to AtISA3; Delatte et al. 2005; Wattebled et al. 2005, 2008). In several plants species, both ISA1 and ISA2 have been shown to mainly or exclusively participate in the biosynthesis of amylopectin by forming homomeric and/or heteromeric functional complexes (Kubo et al. 2005, 2010; Utsumi and Nakamura 2006; Utsumi et al. 2011; Hennen-Bierwagen et al. 2012; Facon et al. 2013; Sundberg et al. 2013; Sim et al. 2014) and selectively hydrolyze excess branchings (i.e., α-1,6 interglucose bonds) during starch granule biosynthesis. Thereby, they facilitate the formation of the branching clusters of amylopectin and, presumably, mediate the low degree of branching in amylose. The latter designates a complex mixture of linear and poorly branched α-glucans widely differing in size. Starch formed by A. thaliana mutants lacking ISA1 and/or ISA2 are structurally altered (Delatte et al. 2005; Wattebled et al. 2005; Streb et al. 2012). By contrast, ISA3 and PU1 act during starch degradation hydrolyzing α-1,6 glucosidic bonds and liberating full length side chains (Delatte et al. 2005; Li et al. 2007; Wattebled et al. 2008; Streb et al. 2012). All ISAs have the carbohydrate binding module CBM48 in common (pf:CBM_48; AtISA1: residues 76-170; AtISA2: 234-328; AtISA3: 92-190; KEGG) and a catalytic domain which resembles that of α-amylase (pf:Alpha-amylase; AtISA1: 240-417; AtISA2: 388-476; AtISA3: 260-616; KEGG).
Sequence diversity
The CBM48 domain of AtISA1 is affected by nonsynonymous substitutions, while the CBMs of AtISA2 and AtISA3 are highly conserved (Table 1; Table S1). In both AtISA2 and AtISA3 the catalytic domains exhibit amino acid substitutions (Table 1; Table S1). Surprisingly, almost all substitutions in the catalytic domain are found in regions that, based on the interspecies comparison, are considered to be highly conserved (Table 1). In promoter regions, the ISA genes possess similar intraspecific nucleotide diversity (Table 2). The genes of the three ISA isoforms differ in their variability with AtISA2 exhibiting the lowest degree of variation.
Inferred selection and phylogenetic clustering
Both AtISA1 and AtISA3 exhibit an excess of low-frequency polymorphisms as determined by negative test statistics of Tajima and Fu and Li (Fig. 2). However, these substitutions are restricted to individual accessions, which lead to at least two separated clusters in the genealogy of these genes (Fig. 7a, c, left panel). Due to the lack of significant results we could not clearly determine the selective or demographic mode underlying the diversity pattern of these genes. In AtISA2, there is an excess of low-frequency polymorphisms as well, but the Tajima and Fu and Li values are significant (Fig. 2). Due to an even distribution of polymorphisms over all accessions, the genealogy is star-like (Fig. 7b, left panel). Additionally, the R2 statistic for population expansion is significant (Fig. 2). For this reason, the genealogy could be shaped by both purifying selection and/or population expansion.

Phylogenetic trees and transcript levels of debranching enzyme genes in A. thaliana. a AtISA1; b AtISA2; c AtISA3. Left panels unrooted maximum likelihood trees among accessions based on promoter and gene sequences. Clusters are highlighted with circles. Right panels differences in transcript levels among selected accessions are displayed by ∆Ct values (normalized with a housekeeping gene; see text). As measure of the reliability, confidence intervals are integrated as shadings. The selected six accessions representing different clusters of the respective genes are marked with the same color in the left and the right panel
Most substitutions occurring in AtISA1 and AtISA3 are of recent history, while the majority of substitutions in AtISA2 are shared with A. lyrata (Fay and Wu’s H, Fig. 2).
ISAs exhibit several presumably PSS (highlighted with asteristics in Table 1; Table S1). In AtISA1, four out of 10 substitutions are inferred to be positively selected. N22Y is almost exclusively present in cluster I (14 accessions; not present in Bl, Col, Er, and Ler) and also in Rsch which is an intermediate haplotype (Fig. 7a, left panel). Likewise, E154D is mainly found in cluster II (not present in Stw) and the intermediate Rsch. In AtISA3 two out of 11 substitutions are inferred to be positively selected, but—unlike in AtISA1—they do not coincide with the inferred clusters. F209L occurs in only four accessions (Cvi, Edi, Gre, and Van) that represent both cluster I and II (Fig. 7c, left panel). S683T is mainly present in cluster II (not present in Mt, and Sap) but also in Ct and Stw (phylogenetically intermediate accessions) and Gre which belongs to cluster I.
Transcript levels relative to accession and time-of-the-day
When comparing the two ISAs that certainly are involved in starch synthesis, i.e., AtISA1 and AtISA2, both genes exhibit similar transcript levels (Figs. 6b, 7a, right panels), which is consistent with a previous study (Gan et al. 2011). Transcript levels of AtISA3 differ between accessions, especially at the middle of light phase (Fig. 7c, right panel). Differences in transcript levels of AtISA1 are strongly influenced by time-of-the-day (Table 3). By contrast, for AtISA2, genetic variations across accessions are the most prominent factor, accounting for about one-third of the variation (Table 3). Transcript levels of AtISA3 are rather equally affected by differences among accessions and time-of-the-day. During the light period, AtISA3, which is involved in starch breakdown, shows similar transcript levels as AtISA1 and AtISA2 (Fig. 7, right panels). This is surprising, as starch synthesis and breakdown are antagonistically regulated.
In AtISA1, three different transcript patterns were found as there are accessions with increasing (Edi, Rsch, Te), relatively constant (Stw) or lowest levels at MOL (Bur, Ler; Fig. 7a, right panel) but differences between the three groups are minor. Edi, Rsch, and Te share a specific substitution in an exon that occurs in 59 further accessions (Table S2; Cao et al. 2011). Further correlations between substitutions and transcript pattern or level were not found.
In AtISA2, transcripts follow two contrary patterns as accessions exhibit either highest (Sap, Te) or lowest transcript levels at MOL (Bl, Bur, Oy, Rsch; Fig. 7b, right panel). Specific sites that correlate with either the pattern or the level of transcripts were not identified.
In AtISA3, transcript levels generally increase throughout the light period (Fig. 7c, right panel). The pattern slightly differs among accessions. Oy exhibit lowest transcript levels as compared to all analyzed accessions. Here, five accession-specific substitutions were identified, all of which are located in introns (Table S2; Cao et al. 2011).
Phosphorylases
Background
The α-glucan phosphorylases (PHS/Pho; EC 2.4.1.1; α-1,4-d-glucan: orthophosphate α-d-glucosyl transferase) catalyze the reversible transfer of terminal glucosyl residues at the non-reducing end(s) of α-1,4-glucan chain and orthophosphate (Albrecht et al. 2001; Rathore et al. 2009). In higher plants, two distinct phosphorylase types exist, the plastidal (PHS1/Pho1) and the cytosolic (PHS2/Pho2) transferases. In A. thaliana each isoform is represented by a single nuclear gene (plastidal: AtPHS1; cytosolic: AtPHS2; Zeeman et al. 2004; Lu et al. 2006; Rathore et al. 2009). According to KEGG, both phosphorylases share the same catalytic domain (pf:Phosphorylase; AtPHS1: residues 180-504 and 559-957; AtPHS2: 114-836). For posttranslational import into the plastid, AtPHS1 possesses an approximately 62 aa comprising N-terminal transit peptide (ChloroP; Emanuelsson et al. 1999). Additionally, plastidal glucan phosphorylases from higher plants (Pho1/PHS1) contain an about 80 aa insertion that is located between the N- and the C-terminal domains, first described for potato (Nakano and Fukui 1986). Consistent with high levels of interspecific variation, the insertion might originate from an intron ancestor (Rathore et al. 2009). It is enriched in charged residues (Rathore et al. 2009). Within the insertion there is a region called PEST site that has been identified in potato, maize, and rice (Rathore et al. 2009). This region which is enriched in proline, glutamic acid, serine, and threonine is considered as primary target of proteolytic degradation (Rathore et al. 2009). AtPHS2 is restricted to the cytosol and is involved in maltose metabolism preferring high molecular weight glycans as glucosyl acceptors (Lu et al. 2006).
Sequence diversity
The transit peptide of AtPHS1 (ChloroP; Emanuelsson et al. 1999) exhibits 8 variable positions (Table 1). Most nonsynonymous substitutions in AtPHS1 and AtPHS2 occur in the catalytic domain (Table 1; Table S1). Another functionally important motif is the PLP binding site (E-A-[S or C]-G-X-[G or S]-X-M–K-X(2)-[L or M]-N; Rathore et al. 2009). This motif is invariable in AtPHS1, while in AtPHS2 a substitution occurred in one of these positions (E-A-[S or C]-G-X-[G or S]-X-M–K-X(2)-[L or M]-N; in bold) in the accession Cvi. At this position, different aa are known to occur among plant species (indicated by X). The substitution in Cvi might be nonetheless interesting as threonine is substituted by proline which is known to specifically affect the secondary structure of proteins by introducing kinks.
In AtPHS1, the insertion has a putative length of 51 aa (residues 511-561) as revealed by comparison with potato sequences. In total, almost 40 % of the insertion consists of charged residues (20 out of 51 residues; acid residues: 3 aspartic acid residues, 11 glutamic acid residues; basic residues: 6 lysine residues). In the intraspecific comparison, the insertion of AtPHS1 exhibits a considerably high number of substitutions. Two synonymous (position 531, 559) and 5 nonsynonymous substitutions are observed (Table 1). The prominent PEST region is essential for proteolytic degradation (Rathore et al. 2009). However, in AtPHS1 the PEST region appears to be absent. Among starch-related gene families analyzed in this study, the PHS genes exhibit the highest intraspecific nucleotide diversity in the promoter region (Table 2). Nucleotide diversity of the two AtPHS genes is similar to other high diversity gene families, such as starch synthases.
Inferred selection and phylogenetic clustering
AtPHS1 exhibits an excess of low- and intermediate-frequency polymorphisms (Fig. 2). Substitutions are mostly found in three accessions that deviate from the main cluster (Bl, Cha, Wil; Fig. 8a, left panel). Furthermore, the R2 test for population expansion is significant. Therefore, it is unclear whether selective and/or demographic influences affect the genealogy of AtPHS1. Significant negative values for Tajima and Fu and Li test statistics in AtPHS2 are indicative of an excess of low-frequency polymorphisms (Fig. 2). These substitutions occur almost exclusively in a single accession (Cvi, Fig. 8b, left panel). Therefore, we assume purifying selection as major actor shaping the genealogy of AtPHS2.Besides SSs, PHSs are the most variable starch-related gene family harboring a large number of nonsynonymous substitutions (Table 1; Table S1). However, none of these substitutions are inferred to be positively selected. As determined by a significant negative Fay and Wu value, there is an excess of derived substitutions that originated in the lineage of A. thaliana (Fig. 2).

Phylogenetic trees and transcript levels of phosphorylase genes in A. thaliana. a AtPHS1; b AtPHS2. Left panels unrooted maximum likelihood trees among accessions based on promoter and gene sequences. Clusters are highlighted with circles. Right panels differences in transcript levels among selected accessions are displayed by ∆Ct values (normalized with a housekeeping gene; see text). As measure of the reliability, confidence intervals are integrated as shadings. The selected six accessions are members of different clusters of the respective genes and are marked with the same color in the left and the right panel
Transcript levels relative to accession and time-of-the-day
Transcript levels of the two phosphorylases are quantitatively similar and exhibit essentially the same time dependency (Fig. 8, right panels). Time-of-the-day is the major determinant of transcript levels, accounting for more than 60 % of the variation. Differences among accessions account for 15–20 % of transcript level variation (Table 3).
In AtPHS1, the transcript pattern generally coincides among accessions as it increases throughout the entire light period, except for Est that exhibits highest levels at MOL (Fig. 8a, right panel). Several sites are unique to Est (five positions in the promoter; three in introns; Table S2). Cha and Wil follow the general transcript pattern, but exhibit highest transcript levels. In the phylogenetic analysis, these two accessions are separated from the main cluster. They share numerous sites that differ from cluster I accessions (32 positions in the promoter; 36 in exons; 81 in introns; six in the 3′-UTR; Table S2). These putative associations are found in one to 29 further accessions within the 105 accessions data set (Table S2; Cao et al. 2011).
AtPHS2 shows increasing transcript levels throughout the entire light period (Fig. 8b, right panel). In Cvi, however, AtPHS2 is differently transcribed. This accession also stands apart in the phylogenetic analysis due to numerous specific substitutions and/or indels (20 positions in the promoter; 37 in exons; 71 in introns, two in the 3′-UTR; Table S2). Most of these sites are found in other accessions, but they are rare (occurring in up to 7 accessions; Table S2; Cao et al. 2011).
Discussion
Given the fundamental importance of transitory starch metabolism in A. thaliana and the variations present in starch content among accessions (Sulpice et al. 2009), intraspecific variability has been studied for 25 starch-related genes whose products mediate essential reactions in storage carbohydrate metabolism. This combined genetic/transcription study aimed to elucidate the evolutionary history with regard to genetic diversity within gene families and to identify putative associations between genetic variation and patterns as well as levels of transcripts to finally correlate them with starch content. In order to report more comprehensively the distribution of putative associations, i.e., substitutions that are associated with transcript patterns, among A. thaliana accessions, we extended our sequence data set of 26 accessions by additional 80 accessions (Cao et al. 2011)—an easily accessible subset of the 1001 Genomes project (Weigel and Mott 2009), yielding a data set of 105 accessions in total (as one accession was covered in both our and the Cao data set; Table S1, S2). The Cao data set does not provide sequence information for all sites (i.e., it contains numerous N’s for some genes) and the length of indels/repetitive sequences was not determined, such that any statements about indels are based on our 26 accessions data set only. We also looked for comparable transcript data on further A. thaliana accessions in the public domain. We identified one data set based on sufficiently similar environmental conditions and tissue harvest regime, i.e., the transcriptomes of the 19 founders of the MAGIC lines (Gan et al. 2011). As 12 accessions are included in both data sets, this data set yields transcription information on additional 7 accessions.
Our study on the variability of starch-related genes in altogether 105 accessions and the transcript levels in a relevant subset of them may serve as a starting point for functional investigations on naturally occurring variation in the starch metabolism of higher plants.
Evolutionary history of starch-related genes
Among the genes of the gene families analyzed here, the level of amino acid similarity greatly varies. Presumably, these differences are attributed to different phylogenetic gene ages, i.e., duplication events (Deschamps et al. 2007; Ball 2012) which apparently preceded those duplications in the more recent history of A. thaliana as the positions of the genes did not concur with these chromosomal duplications, not even for the more closely related genes (Fig. S1).
For SSs, ISAs, and PHSs, the common ancestor of all Chloroplastida likely already possessed the full set of genes found in A. thaliana (Deschamps et al. 2007; Ball 2012; Schwarte et al. 2013; Nougué et al. 2014). Due to the high phylogenetic age of the respective gene family members, the amino acid (aa) identity (i.e., percentage of shared aa residues) is relatively low among members (11–36 % for SSs; 23–42 % for ISAs; 55 % for PHSs). In other gene families, some copies originated later, as the common ancestor of Chloroplastida only possessed two AGPase genes (encoding for the large and the small subunit, respectively; Deschamps et al. 2007; Ball 2012), but higher plants possess at least three APL genes and one APS gene (Georgelis et al. 2007). This is again reflected in our data set as the four AtAPLs share between 57 and 84 % of their aa residues, while aa identity between AtAPLs and AtAPSs is only 33–49 %. Likewise, the common ancestor of Chloroplastida only had two BE genes (Deschamps et al. 2007). In A. thaliana, AtBE2 and AtBE3 have 73 % of their aa residues in common, but they share only about 26 % with AtBE1. This rather low sequence similarity is not surprising due to the fact that AtBE1 is related to the branching enzymes family, but cannot be assigned to one of the two classes. For PPAs, informations about the onset of genes in the common ancestor are missing to our knowledge. By using KEGG (Kanehisa and Goto 2000; Kanehisa et al. 2012), we searched for gene copy numbers in lower plant species (Ostreococcus tauri, Chlamydomonas reinhardtii, and Volvox cateri) as well as higher plants (A. thaliana, A. lyrata, Populus trichocarpa, Zea mays, Oryza sativa). In lower plant species, the PPA gene copy number varies between two and four (Ostreococcus tauri: 4, Chlamydomonas reinhardtii: 3, and Volvox cateri: 2), while it increases in higher plants. There are fewer gene copies in eudicots (A. thaliana: 6, A. lyrata: 7, Populus trichocarpa: 9) as compared to monocots with up to 14 gene copies (Zea mays: 11, Oryza sativa: 14) which is related to the differential expression of Group I and Group II genes which are preferentially expressed in endosperm and in the vegetative tissues, respectively (Yan et al. 2009).
Notwithstanding all functional constraints during evolution of starch-related genes, our analysis with regard to gene copy number and similarity at protein level demonstrate that the divergence between protein isoforms within one family in one accession of a single species (here, A. thaliana Col-0) still contains a phylogenetic signal that provides informations with regard to the course of gene duplications by which they originated.
Genetic diversity in starch-related genes among A. thaliana accessions
At the gene level, the degree of nucleotide diversity varies depending on the respective gene family as well as on the gene itself. Diversity shows interdependency at gene family level rather than below. The two phosphorylases (average nucleotide diversity of promoter and gene sequence [πtotal] = 0.01622) and the five starch synthases (πtotal = 0.00671) are highly diverse across accessions. By contrast, the six genes encoding pyrophosphatase (πtotal = 0.00274) and the family of the three DBEs (πtotal = 0.00224) are more conserved. Within gene families, variability differs among genes (Tables 1, 2) confirming previous studies (Schwarte and Tiedemann 2011; Schwarte et al. 2013). Genes related to central carbon metabolism are equally variable as genes involved in secondary metabolism, even at the protein level. Sequence analysis with regard to functional implications on specific sites, however, revealed that sites which are involved in either function or regulation are highly conserved (Jespersen et al. 1993; Svensson 1994; Crevillén et al. 2003; Tenorio et al. 2003; Ballicora et al. 2004; Schulze et al. 2004; Delatte et al. 2005; Delvallé et al. 2005; Wattebled et al. 2005, 2008; Zhang et al. 2005, 2008; Dumez et al. 2006; Roldán et al. 2007; Rathore et al. 2009; Hädrich et al. 2012; Sonnewald and Kossmann 2013; Nougué et al. 2014). Detailed selection tests on starch-related gene families revealed a phase of subfunctionalization directly after gene duplication that is followed by a further and independent occurring improvement (Nougué et al. 2014). For this reason, PSS do not match with known functional domains but probably with regulatory domains and/or sites that are involved in enzyme conformation or activity (Nougué et al. 2014).
At genomic level, A. thaliana exhibits levels of polymorphisms that are lower than for Drosophila melanogaster, but higher than for humans (Nordborg et al. 2005). Indeed, considerable mutation rates (7 × 10−9 bp × generation−1; Ossowski et al. 2010) and recombination rates (3.6 cM × Mb−1; Salomé et al. 2012) have been reported which explain both the generation of genetic diversity and its spread across populations/accessions. Thus, each starch-related gene family and even each individual gene seems to follow its own evolutionary trajectory driven by mutation, random drift, selective constraints, and extensive recombination.
The phylogenetic relationship of each starch-related gene is independent from each other as each gene possesses its own gene tree that does not necessarily follow the species tree. If species assignments in Arabidopsis follow the phylogenetic species concept, we would expect that (1) A. thaliana and A. lyrata represent two reciprocally monophyletic terminal taxa and that (2) the majority of gene trees for individual genes mirrors the history of the species (Kizirian and Donnelly 2004); exceptions of this pattern—if occurring—could be caused by ancestral lineage sorting or introgression through occasional hybridization. In sexually reproducing species, this pattern of reciprocal monophyly typically breaks down among evolutionary lineages below the species level (e.g., geographical populations). For this reason, it is likely that most of the variation found in A. thaliana originated after the divergence of the two sister species 3.8–10 million years ago (Kuittinen and Aguadé 2000; Kuittinen et al. 2004; Clauss and Koch 2006; Hu et al. 2011). Because A. thaliana became a selfer, accessions are typically homozygous for most loci. Under obligate selfing, genetic exchange among lineages (or accessions) should be negligible and the phylogenetic history of all genes within an accession would be synchronized. This should result in a high congruence among gene trees. According to our data, however, there is a different lineage (=accession)-specific evolutionary trajectory for any gene. Although genomic analyses performed with A. thaliana have revealed a global geographic population structure according to isolation-by-distance (Sharbel et al. 2000), we did not find any congruence among any pair of gene trees, neither for genes of the same family nor for different gene families (cf. Figs. 3, 4, 5, 6, 7, 8, left panels). Hence, any hypothesis of long-term divergent evolution among accessions can be clearly rejected. In fact, the pattern of gene diversity among accessions appears similar to that in a sexual reproductive population in which extensive recombination (3.6 cM × Mb−1; Salomé et al. 2012) generates mosaic patterns of allelic combinations among loci and individuals as it has been previously observed both for individual genes (Schwarte and Tiedemann 2011) and large proportions of the genome (Nordborg et al. 2005). It has been reported that, due to postglacial population expansion by human disturbance, gene-wise analyses do not reveal any correlation between genotype and geography (Nordborg et al. 2005).
Gene-wise selection patterns of starch-related gene families
Given the high phylogenetic age and the fundamental importance of starch-related genes, one would generally expect highly adapted genes under stringent evolutionary constraints. Generally, the genealogies exhibit no signs of phylogeographical pattern and, hence, clearly differ from the genomic background. The combination of different selection tests revealed that each gene possesses its individual evolutionary history and even members of the same gene family behave differently (Fig. 2). Our combination of selection tests, demographic inference, and phylogenetic analyses assigned any starch-related gene to one of the four groups: (1) genes under purifying selection, (2) genes that are influenced by selective and/or demographic processes, (3) genes under disruptive selection, and (4) genes with inconclusive results with regard to the evolutionary pattern. All genes inferred to be under purifying selection (indicated by significant negative Tajima and Fu and Li test statistics) exhibit genealogies with one main cluster from which single accessions deviate (AtAPL4, AtPPA1, AtPPA3, AtPPA5, AtPHS2; Figs. 2, 3, 4, 5, 6, 7, 8). In several other genes with a significant excess of low-frequency polymorphisms, significant R2 statistics are indicative of population expansion as the underlying evolutionary pattern (AtAPS2, AtPPA2, AtSSII, AtSSIV, AtBE2, AtBE3, AtISA2, AtPHS1; Fig. 2). In this case, the major actor shaping genealogy is demography, potentially (but not necessarily) together with purifying selection. By contrast, some starch-related genes (AtAPL3, AtPPA4, AtSSI, AtBE1; Fig. 2) showed an excess of intermediate-frequency polymorphisms leading to two clearly separated clusters in the genealogy. Here, selection tests yield positive values. By combining phylogeny and selection tests we assume that disruptive selection might contribute to the divergence among the inferred phylogenetic lineages. Post-hoc selection tests within these lineages revealed an excess of low-frequency polymorphisms. Hence, purifying selection is likely to act within clusters, while disruptive selection is acting among them. Finally, there is a group of genes where our analyses remained inconclusive with regard to the evolutionary pattern underlying the observed variation (AtAPL1, AtAPL2, AtAPS1, AtPPA6, AtSSIII, AtGBSS, AtISA1, AtISA3; Fig. 2).
The classification of different modes of selection should be treated with cautions. However, by combining different test statistics, we could identify at least two selective modes, i.e., purifying selection with star-like genealogies accompanied by single deviating accessions and disruptive selection with bifurcated genealogies (Figs. 2, 3, 4, 5, 6, 7, 8).
Site-specific selection analyses revealed that two gene families (AGPase: AtAPS1, and AtAPS2; ISAs: AtISA1, and AtISA3) exhibit positively selected sites (PSS; Table 1; Table S1). In AtAPS1, all PPSs occurred in the same seven accessions and by expanding the sequence data set to 105 accessions, 20 were found carrying these mutations (Table S1). The PSS in AtAPS2 was found in 6 accessions in total (Table S1). Nonsynonymous substitutions in AGPase genes occur randomly across the respective gene trees and do not correlate with distinct clusters (cf. Fig. 3; left panels), such that selection on these sites would fail to explain the clustering in the phylogenetic analysis. PSSs in both AtISA1 and AtISA3 are shared by 2–66 accessions and are hence not evenly distributed across accessions (Table S1). The substitution pattern is generally congruent with the respective clustering of the gene trees. Possibly, the genetic variants at these sites are positively selected for in the respective evolutionary lineages in which they occur, such that these substitutions may constitutes further examples for disruptive selection among lineages/accessions (cf. Fig. 7; left panels).
In summary, a common evolutionary pattern is apparently a combination of purifying selection and demographic changes. This is not unexpected, as (1) starch-related genes are functionally important and deleterious mutations are likely to be counterselected and (2) A. thaliana is known to have experienced population expansion (Sharbel et al. 2000; Nordborg et al. 2005). Nonetheless, we identified a few candidate regions in which divergence may be driven by positive selection, i.e., different substitutions may be selected for in different evolutionary lineages. None of these deviating gene variants among accessions, neither entire alleles nor individual sites, correlate completely with high (Cvi, Nok, Stw, Te, Van, Yo; Sulpice et al. 2009) or low starch content (Est, Mt; Sulpice et al. 2009). Therefore, functional implications of the found genetic variation among A. thaliana accessions remain to be elucidated.
Putative associations between genetic and transcriptional variation
Differences in transcript levels can be affected by both genetic factors divergent among accessions and physiological conditions, such as the time point of harvesting the leaves used for transcription analysis. Therefore, transcript levels were measured at three different time points during the light period for six accessions that are genetically most divergent. Among the genetic factors, cis-regulatory elements affecting transcript levels are likely to be located within the putative promoter, but regions in introns are known as transcriptional regulators as well (Hir et al. 2003; Casas-Mollano et al. 2006; Spasovski et al. 2013). For this reason, the entire sequence of a gene was screened for putative associations that correlate with either the pattern or the level of transcripts.
In some starch-related genes, such as AtAPL2, AtAPL3, AtPPA1, AtSSII, AtSSIII, AtSSIV, and AtBE2, variation in transcript levels is mainly due to genetic factors (>40 % of the variation explained by differences among accessions; Table 3). Putative associations between transcript pattern and genetic variation could be found in AtAPL1, AtAPL2, AtAPL3, AtPPA2, AtPPA4, AtPPA6, AtSSIV, AtBE3, and AtPHS1. Moreover, sites that putatively correlate with transcript levels were identified in AtPPA1, AtPPA5, AtPPA6, AtSSI, AtSSIII, AtSSIV, AtBE1, AtBE2, AtISA1, and AtPHS2. Putative associations are considered as being verified if they were found in at least two accessions across the extended data set of 105 accessions (Suppl. Table 2). For two additional genes, either the pattern (AtISA3) or the level of transcripts (AtAPS2) is putatively associated with sites exclusively found in single accessions. These sites still need to be confirmed. To what extent the putative associations are truly involved in transcript regulation needs to be evaluated. With regard to gene regulatory elements, most of these sites did not affect known cis-regulatory elements (Yilmaz et al. 2011). Only in AtPHS1, one substitution in the promoter is located in the binding site of the LEAFY (LFY) transcription factor (consensus sequence: CCANTG; variable position in bold; Moyroud et al. 2011; Yilmaz et al. 2011), but downstream effects are unlikely, as this substitution occurs at the only position in the consensus sequence known to be variable (Moyroud et al. 2011). Finally, we screened all putative associations regarding co-occurrences across starch-related genes to possibly identify global transcript regulating sites that affect the formation of protein complexes. However, such substitutions could not be found in the investigated starch-related genes. For several genes, i.e., AtAPL4, AtAPS1, AtPPA3, AtSSII, AtGBSS, and AtISA2, there was no correlation between any substitution and and transcript pattern.
Given that (1) genetics explain a large part of transcription variation in many starch-related genes, (2) several differences in transcript patterns correlate with specific sites in the respective gene, but (3) none of these sites affect cis-regulatory elements known to influence transcription, there are two not mutually exclusive explanations: (1) starch-related genes are generally cis-regulated, but the description of respective binding sites is incomplete so far and/or (2) starch-related genes are trans-regulated by global transcription factors that regulate the entire or parts of this complex metabolic pathway. A combination of these two mechanisms is likely.
Transcript patterns or levels of individual genes that correlate with high (Cvi, Nok, Stw, Te, Van, Yo; Sulpice et al. 2009) or low starch content (Est, Mt; Sulpice et al. 2009) could not be identified. The lack of any correlation of starch content with the expression pattern of single genes might be due to the complex metabolic pathway of starch synthesis and degradation. Furthermore, a possible incongruence between transcript and protein levels may also be relevant in this context. To some extent our transcript results could be influenced by the experimental design as in a long day treatment as implemented here, starch metabolism can be relaxed as plants may not consume all their starch during the night (Sulpice et al. 2014). It is conceivable that this effect may influence transcript patterns of starch-related genes. It would be hence valuable to verify the transcript patterns revealed in our study in a study with shorter light period.
Detailed analyses of AGPase and starch synthase genes
A detailed analysis of genetic and transcript variation can provide further clues to the evolution of individual genes or gene families. For example, the AGPase holoenzyme in eukaryotes is typically a heterotetramer (α2β2), consisting of small (α) and large (β) subunits. All genes encoding the small and large subunits originated from a single ancestral gene during an ancient duplication in angiosperms (Georgelis et al. 2007; Ventriglia et al. 2008). The number of genes encoding small (1–2) and large (3–6) subunits differ depending on plant species (Georgelis et al. 2007). In A. thaliana, four genes encode the large subunits (AtAPL1-4) and two genes the small subunits (AtAPS1-2). Genes of the large subunits underwent more duplication events as compared to those of the small subunits, permitting distinct yet closely related and tissue-specific functions (Georgelis et al. 2007). AtAPL1 is highly expressed in source tissues while AtAPL2 has a broader expression level but is mainly functional in sink tissues (Georgelis et al. 2007; Ventriglia et al. 2008). AtAPL3 and AtAPL4 are minor forms only expressed in sink tissues (Georgelis et al. 2007; Ventriglia et al. 2008). For all accessions analyzed here, transcript levels of the various large subunits of the AGPase concur with these results, as AtAPL1 appears to be most strongly expressed throughout the light period (Fig. 3a–d, right panels). Transcript levels did, however, fluctuate depending on the accession analyzed. For all large subunits, differences in transcript levels are mainly determined by variations among accessions rather than by time-of-the-day (Table 3). More specifically, transcript levels of AtAPL3, especially to the time point EOL, correlate with the clustering of accessions in the phylogenetic tree. This observation points to cis-regulatory mechanisms that affect transcript levels of AtAPL3. The two small subunits of the AGPase seem to undergo a divergent evolution. In fact, it has been proposed that AtAPS1 encodes the only functional small subunit whereas AtAPS2 is thought to be a pseudogene (Crevillén et al. 2003, 2005; Ventriglia et al. 2008). For identifying nuclear pseudogenes, the pattern and/or frequency of nucleotide substitutions are generally believed to be a prime criterion (Bailey et al. 2003), which can be supported by transcription analysis. Specifically, pseudogenes can be expected to exhibit low transcript levels and to harbor excess nonsynonymous substitutions. Indeed, AtAPS2 has the lowest transcript levels, the highest (random) transcript level variation among biological replicates (accounting for 57 % of the variation; Table 3), and the highest percentage of nonsynonymous mutations (piN/piS 1:1) among all AGPase genes, while AtAPS1 reveals a higher overall genetic diversity (Tables 1, 2). Altogether, identity among the two AGPase small subunits is rather low for both DNA (52 %) and inferred aa (41 %) sequences. Thus, our data tentatively confirm the pseudogenic nature of AtAPS2. Given the only moderate overall diversity level of AtAPS2 and the lack of frame shift mutations in exons, this putative pseudogenization may be considered evolutionary recent.
As a second example of evolutionary implications of our combined genetic/transcription analysis, starch synthases (SSs) were identified as those starch-related genes exhibiting the highest levels of intraspecific diversity including both synonymous and nonsynonymous substitutions (Tables 1, 2). AtSSI has already been described as a high diversity gene (Cork and Purugganan 2005). Some substitutions even occur at positions that, based on interspecific comparison, are considered to be highly conserved (Table 1). For all SSs, gene-specific phylogenetic trees were obtained. They indicate that the intraspecific diversity does not follow a common pattern although three gene products are interacting within the biosynthesis of amylopectin. Likewise, transcript levels of the various SSs vary depending on both the target gene and the accession. For both AtSSIII and AtSSIV, individual sites could be identified that might be responsible for differences in transcript levels (see results). Further biochemical analyses are needed to analyze the functional implications of specific sites of the various starch synthases but are, for several reasons, difficult to be performed. Firstly, detailed structural analyses of the transitory starch require large quantities of leaf material and cannot be performed with a high number of accessions. Secondly, at protein level AtSSI and AtSSIII constitute the vast majority of the detectable enzyme activity whereas the activity of both AtSSII and AtSSIV is largely undetectable. Therefore, sequence-dependent modulations of the activity of the different isozymes are very challenging to follow. Thirdly, several starch synthases are known to reversibly interact with other starch-related enzymes (Tetlow et al. 2004, 2008; Zeeman et al. 2010; Emes and Tetlow 2012). Their actual in vivo function appears to be closely related to that of the herteromeric protein complexes rather than to the individual starch synthase isoforms. Detailed analyses of biochemical functions of the complexes are difficult to perform and largely lacking.
Conclusions
In 26 accession of A. thaliana, 25 starch-related genes representing six gene families were sequenced including the putative promoter regions. All starch-related genes have their individual evolutionary history that can be reconstructed by the analysis of gene copy number and amino acid similarity. The genes exhibit significant intraspecific variations and contain numerous synonymous and nonsynonymous substitutions. Sequence variability strongly depends of the gene family, with starch synthases and phosphorylases being the most variably genes. Despite the fact that all gene products act in assimilatory starch turnover, sequence variability does not follow any common pattern, but any gene follows its own evolutionary trajectory, resulting in gene-specific genealogies. This confirms earlier hypotheses of mixture and recombination among the ancestors of current accessions (perhaps in a pleistocene refugium; Sharbel et al. 2000), followed by a human-assisted worldwide expansion. Purifying selection can be assumed as general pattern, but some genes experienced divergent evolution among groups of accessions, potentially caused by positive (disruptive) selection. Likewise, putative promoter regions exhibit considerable sequence diversity. Transcript levels of the starch-related genes vary largely. Each starch-related gene family or rather each member experiences its individual evolutionary constraint that is influenced by internal structure of native starch, the posttranslational import into the plastids and the interaction with other starch-related proteins. In some occasions, putative associations between genetic variation and transcript patterns were identified. They may serve as candidates for future functional analyses to unravel the impact of genetic variation on the complex network of starch metabolism. However, the verification of functional implications for distinct sites is challenging due to the fact that during starch biosynthesis many enzymes closely interact and, therefore, any disturbance of this concerted action may result in complex alterations of the starch structure.
Methods and material
Plant materials and cultivation
Arabidopsis accessions were kindly provided by Prof. Altmann (IPK Gatersleben, Germany). Prior to germination, seeds were kept for at least 2 days at 4 °C. Seedlings and plants were grown in 1:1 (w/w) mixture of GS 90 soil and vermiculite. For sequencing plants were grown under controlled short day conditions (12 h light [120 µE m−2 s−1], 20 °C; 12 h dark, 18 °C). Leaves were harvested after 4 weeks. For Realtime PCR experiments, plants were grown for 4 weeks under controlled long day conditions (16 h light [120 µE m−2 s−1], 21 °C; 8 h dark, 18 °C). Within the vegetative stage (plants did not started bolting yet), leaves were harvested at three time points during the light phase, i.e., beginning of the light period (BOL; after 30 min light), middle of light (MOL; after 8 h light), and end of light (EOL; 30 min before the onset of dark).
DNA isolation, PCR amplification, and sequencing
Genomic DNA was extracted from a pool of leaves collected from three plants per accession using a modified CTAB procedure (Rogers and Bendich 1985). Primers were designed based on the Col-0 sequence. All analyses were performed with the following genes: ADPglucose pyrophosphorylase [AtAPL1 (At5g19220), AtAPL2 (At1g27680), AtAPL3 (At4g39210), AtAPL4 (At2g21590), AtAPS1 (At5g48300), AtAPS2 (At1g05610)], inorganic pyrophosphorylases [AtPPA1 (At1g01050), AtPPA2 (At2g18230), AtPPA3 (At2g46860), AtPPA4 (At4g53620), AtPPA5 (At4g01480), AtPPA6 (At5g09650)], soluble starch synthases [AtSSI (At5g24300), AtSSII (At3g01180), AtSSIII (At1g11720), AtSSIV (At4g18240)], granule bound starch synthase [AtGBSS (At1g32900)], branching enzymes [AtBE1 (At3g20440), AtBE2 (At5g03650), AtBE3 (At2g36390)], DBEs [AtISA1 (At2g39930), AtISA2 (At1g03310), AtISA3 (At4g09020)], glycan phosphorylases [AtPHS1 (At3g29320), AtPHS2 (At3g46970)]. For amplification and sequencing primers were placed about 1.0 kb upstream the translation start. The fragments of 26 worldwide distributed accessions (Bl-1, Bur-0, Can-0, Cha-0, Col-0, Ct-1, Cvi-0, Edi-0, El-0, Er-0, Est-1, Gre-0, Ler-1, Mt-0, Nok-2, Oy-0, Rsch-0, Sap-0, Sha(kdara), Stw-0, Te-0, Tsu-1, Van-0, Wil, Ws-3, Yo-0) were amplified with the proof-reading polymerase Phusion (Finnzymes) and purified enzymatically by using Exonuclease I and Antarctic Phosphatase (New England Biolabs). The templates were directly used for sequencing on an ABI 3130xl automated sequencer (Life Technologies), using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies).
RNA isolation, cDNA synthesis, and Realtime PCR
RNA was isolated with the Invitrap® Spin Plant RNA Mini Kit (STRATEC Molecular) using the DCT lysis solution. For each accession, three RNA preparations were independently isolated (three biological replicates). RNA (2 μg each) was reversely transcribed using the RevertAidTM First Strand cDNA Synthesis Kit (Fermentas). For each gene, 6 out of 26 accessions were selected for Realtime PCR experiments. Accessions were selected representing all haplotype clusters identified in the maximum likelihood analysis with combined promoter and gene sequences. cDNA (0.2 μl per Realtime PCR run; 10 μl reaction volume) was amplified using SensiMix™ SYBR Low-ROX (Bioline) and a LightCycler® 480 (Roche). For each biological replicate, three technical replicates were performed. Transcript levels of starch-related genes were normalized relative to transcript level of the housekeeping gene ubiquitin (UBC21, At5g25760). Primer pairs were exclusively designed in coding regions lacking any kind of genetic variation among accessions and flanking an intronic region. Primer sequences were as follows: 5′-TTTCCAAGGAGGAAGCTGAA-3′ and 5′-TGAACCAAAGTCGTTTGCTG-3′ for AtAPL1, 5′-TGCAGAAGCACATTGAGTCC-3′ and 5′-CCCTTTGGTTTTTCCGAGA-3′ for AtAPL2, 5′-TCGGTAATCTCACACGGATG-3′ and 5′-TTTGTATCTCGGCCAATGC-3′ for AtAPL3, 5′-TGGAGGTGGTTTCGTAGAGG-3′ and 5′-ATCCTCAAACACCCAGAGGA-3′ for AtAPL4, 5′-GTGCCACTTCCCTTGGACT-3′ and 5′-GCTGTGTGTAGATCGGAGCA-3′ for AtAPS1, 5′-TGATCGGTCGAGAACAGATG-3′ and 5′-AGCTCCAATGCTCCTCACAT-3′ for AtAPS2, 5′-GGTTTTGTTCCTCGCACATT-3′ and 5′-CACCCTGGTCAATCATAGGC-3′ for AtPPA1, 5′-GTGTACCCCCACAACTACGG-3′ and 5′-GAGGAATGATCCGGTTAGCA-3′ for AtPPA2, 5′-AACAACTCGCTCCTCATCGT-3′ and 5′-TGATGGCAAGAAATCGTTGA-3′ for AtPPA3, 5′-GGAAAAGGGAGCAAGGTGA-3′ and 5′-ACCGGTTCCTGCATAATGAC-3′ for AtPPA4, 5′-CTCCAGTGATTTTCAATGTGGTT-3′ and 5′-ACATAGTGTGCGTGGGACAA-3′ for AtPPA5, 5′-GGAAGATCCATCTCATGCAAA-3′ and 5′-CATCCAAAGAAATGGCAACA-3′ for AtPPA6, 5′-TTCACGTTACTTTGCCATGC-3′ and 5′-ACTTTGCGGCCAAAAGTATG-3′ for AtSSI, 5′-CCTGAATTTCGGCATCTGAG-3′ and 5′-AAGCCAAATTTCCATCACCA-3′ for AtSSII, 5′-CGGAATGGACAGGTTGTCTT-3′ and 5′-CCCCAGCATAAATCAAATGG-3′ for AtSSIII, 5′-CTGGCAAACAGCTTTTGTTG-3′ and 5′-TGATCCTGCATTCTGTCTGG-3′ for AtSSIV, 5′-CAAACGAGGAGTTGATCGTG-3′ and 5′-AACTGAACCGGAGTTGGTTG -3′ for AtGBSS, 5′-CTTCTTTGCTGCCAGTAGCC-3′ and 5′-GAGAAAGCCCAACCATCTGA-3′ for AtBE1, 5′-CGGAGATTTCAACAACTGGAA-3′ and 5′-TACACGTGAGCCATGAGGAA-3′ for AtBE2, 5′-TGGTTTGGAGGCATTTTCTC-3′ and 5′-CGATCAGTGATGCTGCCTTA-3′ for AtBE3, 5′-CAACTGCAATCATCCTGTGG-3′ and 5′-CCTTCTACATCAGCCCCGTA-3′ for AtISA1, 5′-TCCTGTTGTTCAGCAGTTGG-3′ and 5′-GGACGAGAGAGCTGTTCACC-3′ for AtISA2, 5′-GGAGCTTCAGAGGCGTTCTA-3′ and 5′-GGTCTCCCTCACCACTAGCA-3′ for AtISA3, 5′-AATTTTCGATGAACGGTTGC-3′ and 5′-CCCTCTGCTCTCTCCTTCCT-3′ for AtPHS1, 5′-CAGTCTCCGTCTCTGGGAAG-3′ and 5′-TTCCCATTCTCGGTAGCATC-3′ for AtPHS2, and 5′-CTGCGACTCAGGGAATCTTCTAA-3′ and 5′-TTGTGCCATTGAATTGAACCC-3′ for UBC21.
Data analysis
Alignment
Sequences were assembled with BioEdit version 7.0.5 (Hall 1999). For each accession, all variable sites were checked manually during the construction of a sequence contig. As reference, all sequences were aligned to those of Col-0.
Estimation of genetic variation
By using DnaSP version 5 (Librado and Rozas 2009), intraspecific genetic analyses were performed. Nucleotide diversity (π), number of insertions/deletions (indel), synonymous (syn) and nonsynonymous (nonsyn) substitutions were estimated.
Selection at particular codons
Within a codon for a single amino acid, the ratio of nonsynonymous to synonymous substitution rate (ω) can be used for assessing selection, as values for ω < 1, =1, and >1 are indicative of purifying selection, neutral evolution, and positive selection, respectively. PSS, suggested by ω > 1, were searched by using maximum-likelihood-based random-sites model analysis implemented in PAML 3.14 package (Yang 1997; Yang et al. 2000). For each gene, analyses were performed using run code “user tree” in codeml. The utilized maximum likelihood trees were constructed by RAxML 7.0.4 (Stamatakis 2006) under the GTR + G + I model with 1,000 bootstrap replicates. A Likelihood Ratio Test (LRT) for positive selection (M7-8) was performed. M7 (beta) assumes a beta distribution of ω over sites, whereas model M8 (beta & ω) adds an additional site class (free ω ratio) which is estimated from the data set (Yang et al. 2000). If the alternative model M8 (BEB) results in a better fitting (p < 0.05) as compared null model, codons were considered as being positive selected.
Evaluation of gene-wise selection
The numbers of polymorphisms of different frequency (low, intermediate, high) are indicative of selection and/or demographic changes. To test for natural selection, we applied a combination of test statistics, as suggested, e.g., Akey et al. (2004). Firstly, the number of low- and intermediate-frequency polymorphisms was compared. This comparison was determined by Tajima’s D (Tajima 1989) as well as Fu and Li’s D*, F*, D, and F (Fu and Li 1993). An excess of rare polymorphisms is indicative of negative (=purifying) selection, a recent selective sweep or population expansion after a recent bottleneck. By contrast, an accumulation of intermediate-frequency polymorphisms is indicative of ancestral or recent positive (balancing or disruptive) selection or sudden population contradiction. Fu and Li’s statistics can be either performed with an outgroup (D, F) or without an outgroup (D*, F*; Fu and Li 1993). The interspecific test compares the number of single mutations (singletons) in internal and external branches by which ancestral and recent selective processes can be distinguished. Another selection test that includes an outgroup is Fay and Wu’ H (2000), by which intermediate- and high-frequency polymorphisms are compared. An excess of moderate- and high-frequency derived polymorphisms is indicative of recent variation, while a deficit is indicative of ancestral variation. To assess the potentially confounding effect of demographic changes, R2 statistics were calculated as well (Ramos-Onsins and Rozas 2002). This test is very powerful to detect population expansion. The putative modes of selection for the respective gene, i.e., purifying or disruptive selection, was inferred by comparing the results of these seven tests. Statistical significance for all tests was calculated by using coalescent simulations with 10,000 replicates. All selection tests were performed with DnaSP v5 (Librado and Rozas 2009). When the different tests yielded incongruent results and/or the R2 statistic indicated significant demographic impacts, the results were considered inconclusive with regard to the mode of selection.
Maximum likelihood gene tree
By using RAxML 7.0.4 (Stamatakis 2006) maximum likelihood gene trees for the combined promoter and gene data set of each gene were constructed. The trees were generated under the GTR + G + I model of sequence evolution with 1,000 bootstrap replicates.
Estimation of differences in transcript levels among accessions
ΔCt values were estimated by subtracting the Ct values of the target gene from those of the housekeeping gene. Positive values are indicative of transcript levels that are higher than the housekeeping gene and negative values are lower transcript levels compared to the housekeeping gene. As within each cycle of Realtime PCR target fragments will be doubled, differences in ΔCt values of factor 1 means twofold, factor 2 means fourfold, factor 3 means eightfold etc. higher transcript levels (i.e., ΔCt is scaled by log2). As measure of the reliability of the data, we calculated confidence intervals and integrated them as shadings into the graphs (Figs. 3, 4, 5, 6, 7, 8). In addition, a two-way analysis of variance (ANOVA) of model I (fixed effects) was performed to determine the influence of genetics (transcript levels differing between accessions at a given time point), physiology (transcript levels differing between time points in a given accession) and the interaction between both of them. The ANOVA framework was used both to establish statistical significance of the influence of the two factors (and their interaction) on transcript level differences and to apportion the total variance in transcription to (1) genetics, (2) physiology, (3) interaction genetics x physiology, and (4) random differences among biological replicates (variation within groups).
Notes
Note that SS sequence data for 26 accessions and transcript data for middle of the light period (MOL) for 6 accessions are taken from a previous study (Schwarte et al. 2013) and are included here to provide a comprehensive coverage of genes and transcripts underlying starch metabolism.
References
Abbott RJ, Gomes MF (1989) Population genetic structure and the outcrossing rate of Arabidopsis thaliana. Heredity 62:411–418
Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L (2004) Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol 2:e286
Albrecht T, Koch A, Lode A, Greve B, Schneider-Mergener J, Steup M (2001) Plastidic (Pho1-type) phosphorylase isoforms in potato (Solanum tuberosum L.) plants: expression analysis and immunochemical characterization. Planta 213:602–613
Andriotis VME, Pike MJ, Schwarz SL, Rawsthorne S, Wang TL, Smith AM (2012) Altered starch turnover in the maternal plant has major effects on Arabidopsis fruit growth and seed composition. Plant Physiol 160:1175–1186
Bailey CD, Carr TG, Harris SA, Hughes CE (2003) Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Mol Phylogenet Evol 29:435–455
Ball SG (2012) Evolution of the starch pathway. In: Tetlow IJ (ed) Essential reviews in experimental biology 5. Starch: origins, structure and metabolism, 1st edn. Society for Experimental Biology, London, pp 29–54
Ball SG, Colleoni C, Cenci U, Raj JN, Tirtiaux C (2011) The evolution of glycogen and starch metabolism in eukaryotes gives molecular clues to understand the establishment of plastid endosymbiosis. J Exp Bot 62:1776–1801
Ball SG, Subtil A, Bhattacharya D, Moustafa A, Weber APM, Gehre L, Colleoni C, Arias M-C, Cenci U, Dauvillée D (2013) Metabolic effectors secreted by bacterial pathogens: essential facilitators of plastid endosymbiosis? Plant Cell 25:7–21
Ballicora MA, Iglesias AA, Preiss J (2004) ADP-Glucose pyrophosphorylase: a regulatory enzyme for plant starch synthesis. Photosynth Res 79:1–24
Bertoft E (2013) On the building block and backbone concepts of amylopectin structure. Cereal Chem 90:294–311
Blanc G, Hokamp K, Wolfe KH (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res 13:137–144
Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, Wang X, Ott F, Müller J, Alonso-Blanco C, Borgwardt K, Schmid KJ, Weigel D (2011) Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 43:956–963
Casas-Mollano JA, Lao NT, Kavanagh TA (2006) Intron-regulated expression of SUVH3, an Arabidopsis Su(var)3-9 homologue. J Exp Bot 57:3301–3311
Cenci U, Chabi M, Ducatez M, Tirtiaux C, Nirmal-Raj J, Utsumi Y, Kobayashi D, Sasaki S, Suzuki E, Nakamura Y, Putaux JL, Roussel X, Durand-Terrasson A, Bhattacharya D, Vercoutter-Edouart AS, Maes E, Cecilia Arias M, Palcic M, Sim L, Ball SG, Colleoni C (2013) Convergent evolution of polysaccharide debranching defines a common mechanism for starch accumulation in Cyanobacteria and plants. Plant Cell 25:3961–3975
Cenci U, Nitschke F, Steup M, Minassian BA, Colleoni C, Ball SG (2014) Transition from glycogen to starch metabolism in Archaeplastida. Trends Plant Sci 19:18–28
Chaw SM, Chang CC, Chen HL, Li WH (2004) Dating the monocot-dicot divergence and the origin of core eudicots using whole chloroplast genomes. J Mol Evol 58:424–441
Cheng J, Khan MA, Qiu W-M, Li J, Zhou H, Zhang Q, Guo W, Zhu T, Peng J, Sun F, Li S, Korban SS, Han Y (2012) Diversification of genes encoding granule-bound starch synthase in monocots and dicots is marked by multiple genome-wide duplication events. PLoS ONE 7:e30088
Clauss MJ, Koch MA (2006) Poorly known relatives of Arabidopsis thaliana. Trends Plant Sci 11:449–459
Cork JM, Purugganan MD (2005) High-diversity genes in the Arabidopsis genome. Genetics 170:1897–1911
Crevillén P, Ballicora MA, Mérida A, Preiss J, Romero JM (2003) The different large subunit isoforms of Arabidopsis thaliana ADP-glucose pyrophosphorylase confer distinct kinetic and regulatory properties to the heterotetrameric enzyme. J Biol Chem 278:28508–28515
Crevillén P, Ventriglia T, Pinto F, Orea A, Mérida A, Romero JM (2005) Differential pattern of expression and sugar regulation of Arabidopsis thaliana ADP-glucose pyrophosphorylase-encoding genes. J Biol Chem 280:8143–8149
Crumpton-Taylor M, Pike M, Lu K-J, Hylton CM, Feil R, Eicke S, Lunn JE, Zeeman SC, Smith AM (2013) Starch synthase 4 is essential for coordination of starch granule formation with chloroplast division during Arabidopsis leaf expansion. New Phytol 200:1064–1075
Delatte T, Trevisan M, Parker ML, Zeeman SC (2005) Arabidopsis mutants Atisa1 and Atisa2 have identical phenotypes and lack the same multimeric isoamylase, which influences the branch point distribution of amylopectin during starch synthesis. Plant J 41:815–830
Delph LF, Kelly JK (2014) On the importance of balancing selection in plants. New Phytol 201:45–56
Delvallé D, Dumez S, Wattebled F, Roldán I, Planchot V, Berbezy P, Colonna P, Vyas D, Chatterjee M, Ball S, Mérida A, D’Hulst C (2005) Soluble starch synthase I: a major determinant for the synthesis of amylopectin in Arabidopsis thaliana leaves. Plant J 43:398–412
Deschamps P, Colleoni C, Nakamura Y, Suzuki E, Putaux JL, Buléon A, Haebel S, Ritte G, Steup M, Falcón LI, Moreira D, Löffelhardt W, Raj JN, Planck C, d’Hulst C, Dauvillée D, Ball S (2007) Metabolic symbiosis and the birth of the plant kingdom. Mol Biol Evol 25:536–548
Deschamps P, Haferkamp I, d’Hulst C, Neuhaus HE, Ball SG (2008) The relocation of starch metabolism to chloroplasts: when, why and how. Trends Plant Sci 13:574–582
Dumez S, Wattebled F, Dauvillee D, Delvalle D, Planchot V, Ball SG, D’Hulst C (2006) Mutants of Arabidopsis lacking starch branching enzyme II substitute plastidial starch synthesis by cytoplasmic maltose accumulation. Plant Cell 18:2694–2709
Emanuelsson O, Nielsen H, von Heijne G (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8:978–984
Emes MJ and Tetlow IJ (2012) The role of heteromeric protein complexes in starch synthesis. In: Tetlow IJ (ed) Essential reviews in experimental biology 5. Starch: origins, structure and metabolism, 1st edn. Society for Experimental Biology, London, pp 255-278
Facon M, Lin Q, Azzaz AM, Hennen-Bierwagen TA, Myers AM, Putaux J-L, Roussel X, D’Hulst C, Wattebled F (2013) Distinct functional properties of isoamylase-type starch debranching enzymes in monocot and dicot leaves. Plant Physiol 163:1363–1375
Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155:1405–1413
Freeling M (2009) Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol 60:433–453
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Fujita N, Satoh R, Hayashi A, Kodama M, Itoh R, Aihara S, Nakamura Y (2011) Starch biosynthesis in rice endosperm requires the presence of either starch synthase I or IIIa. J Exp Bot 62:4819–4831
Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, Kahles A, Bohnert R, Jean G, Derwent P, Kersey P, Belfield EJ, Harberd NP, Kemen E, Toomajian C, Kover PX, Clark RM, Rätsch G, Mott R (2011) Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477:419–423
Georgelis N, Braun EL, Shaw JR, Hannah LC (2007) The two AGPase subunits evolve at different rates in angiosperms, yet they are equally sensitive to activity-altering amino acid changes when expressed in bacteria. Plant Cell 19:1458–1472
Graf A, Smith AM (2011) Starch and the clock: the dark side of plant productivity. Trends Plant Sci 16:169–175
Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, Otillar R, Poliakov A, Ratnere I, Riley R, Smirnova T, Rokhsar D, Dubchak I (2012) The genome portal of the department of energy joint genome institute. Nucleic Acids Res 40(Database issue):D26–D32
Hädrich N, Hendriks JH, Kötting O, Arrivault S, Feil R, Zeeman SC, Gibon Y, Schulze WX, Stitt M, Lunn JE (2012) Mutagenesis of cysteine 81 prevents dimerization of the APS1 subunit of ADP-glucose pyrophosphorylase and alters diurnal starch turnover in Arabidopsis thaliana leaves. Plant J 70:231–242
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Series 41:95–98
Hennen-Bierwagen TA, James MG, Myers AM (2012) Involvement of debranching enzymes in starch biosynthesis. In: Tetlow IJ (ed) Essential reviews in experimental biology 5. Starch: origins, structure and metabolism, 1st edn. Society for Experimental Biology, London, pp 179–215
Hir HL, Nott A, Moore MJ (2003) How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci 28:215–220
Hu TT, Pattyn P, Bakker EG, Cao J, Cheng JF, Clark RM, Fahlgren N, Fawcett JA, Grimwood J, Gundlach H, Haberer G, Hollister JD, Ossowski S, Ottilar RP, Salamov AA, Schneeberger K, Spannagl M, Wang X, Yang L, Nasrallah ME, Bergelson J, Carrington JC, Gaut BS, Schmutz J, Mayer KF, Van de Peer Y, Grigoriev IV, Nordborg M, Weigel D, Guo YL (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet 43:476–481
Jespersen HM, MacGregor EA, Henrissat B, Sierks MR, Svensson B (1993) Starch- and glycogen-debranching and branching enzymes: prediction of structural features of the catalytic (beta/alpha)8-barrel domain and evolutionary relationship to other amylolytic enzymes. J Protein Chem 12:791–805
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res 40:D109–D114
Keeling PL, Myers AM (2010) Biochemistry and genetics of starch synthesis. Annu Rev Food Sci Technol 1:271–303
Kizirian D, Donnelly MA (2004) The criterion of reciprocal monophyly and classification of nested diversity at the species level. Mol Phylogenet Evol 32:1072–1076
Kubo A, Rahman S, Utsumi Y, Li Z, Mukai Y, Yamamoto M, Ugaki M, Harada K, Satoh H, Konik-Rose C, Morell M, Nakamura Y (2005) Complementation of sugary-1 phenotype in rice endosperm with the wheat isoamylase1 gene supports a direct role for isoamylase1 in amylopectin biosynthesis. Plant Physiol 137:43–56
Kubo A, Colleoni C, Dinges JR, Lin Q, Lappe RR, Rivenbark JG, Meyer AJ, Ball SG, James MG, Hennen-Bierwagen TA, Myers AM (2010) Functions of heteromeric and homomeric isoamylase-type starch-debranching enzymes in developing maize endosperm. Plant Physiol 153:956–969
Kuittinen H, Aguadé M (2000) Nucleotide variation at the CHALCONE ISOMERASE locus in Arabidopsis thaliana. Genetics 155:863–872
Kuittinen H, de Haan AA, Vogl C, Oikarinen S, Leppälä J, Koch M, Mitchell-Olds T, Langley CH, Savolainen O (2004) Comparing the linkage maps of the close relatives Arabidopsis lyrata and A. thaliana. Genetics 168:1575–1584
Lattanzi FA, Ostler U, Wild M, Morvan-Bertrand A, Decau M-L, Lehmeier CA, Meuriot F, Prud’homme M-P, Schäufele R, Schnyder H (2012) Fluxes in central carbohydrate metabolism of source leaves in a fructan-storing C3 grass: rapid turnover and futile cycling of sucrose in continuous light under contrasting nitrogen nutrition status. J Exp Bot 63:2363–2375
Li L, Ilarslan H, James MG, Myers AM, Wurtele ES (2007) Genome wide co-expression among the starch debranching enzyme genes AtISA1, AtISA2, and AtISA3 in Arabidopsis thaliana. J Exp Bot 58:3323–3342
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lin TP, Caspar T, Somerville C, Preiss J (1988) Isolation and characterization of a starchless mutant of Arabidopsis thaliana (L.) Heynh lacking ADPglucose pyrophosphorylase activity. Plant Physiol 86:1131–1135
Lu Y, Steichen JM, Yao J, Sharkey TD (2006) The role of cytosolic alpha-glucan phosphorylase in maltose metabolism and the comparison of amylomaltase in Arabidopsis and Escherichia coli. Plant Physiol 142:878–889
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicated genes. Science 290:1151–1155
Meyer K, Stecca KL, Ewell-Hicks K, Allen SM, Everard JD (2012) Oil and protein accumulation in developing seeds is influenced by the expression of a cytosolic pyrophosphatase in Arabidopsis. Plant Physiol 159:1221–1234
Moyroud E, Minguet EG, Ott F, Yant L, Posé D, Monniaux M, Blanchet S, Bastien O, Thévenon E, Weigel D, Schmid M, Parcy F (2011) Prediction of regulatory interactions from genome sequences using a biophysical model for the Arabidopsis LEAFY transcription factor. Plant Cell 23:1293–1306
Nakano K, Fukui T (1986) The complete amino acid sequence of potato a-glucan phosphorylase. J Biol Chem 261:8230–8236
Navarro-De la Sancha E, Coello-Coutiño MP, Valencia-Turcotte LG, Hernández-Domínguez EE, Trejo-Yepes G, Rodríguez-Sotres R (2007) Characterization of two soluble inorganic pyrophosphatases from Arabidopsis thaliana. Plant Sci 172:796–807
Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H, Bakker E, Calabrese P, Gladstone J, Goyal R, Jakobsson M, Kim S, Morozov Y, Padhukasahasram B, Plagnol V, Rosenberg NA, Shah C, Wall JD, Wang J, Zhao K, Kalbfleisch T, Schulz V, Kreitman M, Bergelson J (2005) The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol 3:e196
Nougué O, Corbi J, Ball SG, Manicacci D, Tenaillon MI (2014) Molecular evolution accompanying functional divergence of duplicated genes along the plant starch biosynthesis pathway. BMC Evol Biol 14:103
Osorio S, Nunes-Nesi A, Stratmann M, Fernie AR (2013) Pyrophosphate levels strongly influence ascorbate and starch content in tomato fruit. Front Plant Sci 4:308
Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M (2010) The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327:92–94
Paparelli E, Parlanti S, Gonzali S, Novi G, Mariotti L, Ceccarelli N, van Dongen JT, Kölling K, Zeeman SC, Perata P (2013) Nighttime sugar starvation orchestrates gibberelline biosynthesis and growth in Arabidopsis. Plant Cell 25:3760–3769
Pyl E-T, Piques M, Ivakov A, Schulze W, Ishihara H, Stitt M, Sulpice R (2012) Metabolism and growth in Arabidopsis depend on the daytime temperature but are temperature-compensated against cool nights. Plant Cell 24:2443–24469
Ragel P, Streb S, Feil R, Sahwrawy M, Annunziata MG, Lunn JE, Zeeman S, Mérida Á (2013) Loss of starch granule initiation has a deleterious effect on the growth of Arabidopsis plants due to an accumulation of ADP-glucose. Plant Physiol 163:75–85
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Rathore RS, Garg N, Garg S, Kumar A (2009) Starch phosphorylase: role in starch metabolism and biotechnological applications. Crit Rev Biotechnol 29:214–224
Rogers SO, Bendich AJ (1985) Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol Biol 5:69–76
Roldán I, Wattebled F, Lucas MM, Delvallé D, Planchot V, Jimenez S, Perez R, Ball S, D’Hulst C, Merida A (2007) The phenotype of soluble starch synthase IV defective mutants of Arabidopsis thaliana suggests a novel function of elongation enzymes in the control of starch granule formation. Plant J 49:492–504
Salomé PA, Bomblies K, Fitz J, Laitinen RA, Warthmann N, Yant L, Weigel D (2012) The recombination landscape in Arabidopsis thaliana F2 populations. Heredity 108:447–455
Schulze S, Mant A, Kossmann J, Lloyd JR (2004) Identification of an Arabidopsis inorganic pyrophosphatase capable of being imported into chloroplasts. FEBS Lett 565:101–105
Schwarte S, Tiedemann R (2011) A gene duplication/loss event in the ribulose-1,5-bisphosphate-carboxylase/oxygenase (Rubisco) small subunit gene family among accessions of Arabidopsis thaliana. Mol Biol Evol 28:1861–1876
Schwarte S, Brust H, Steup M, Tiedemann R (2013) Intraspecific sequence variation and differential expression in starch synthase genes of Arabidopsis thaliana. BMC Res Notes 6:84
Scialdone A, Mugford ST, Feike D, Skeffington A, Borrill P, Graf A, Smith AM, Howard M (2013) Arabidopsis plants perform arithmetic division to prevent starvation at night. Elife 2:e00669
Sharbel TF, Haubold B, Mitchell-Olds T (2000) Genetic isolation by distance in Arabidopsis thaliana: biogeography and postglacial colonization of Europe. Mol Ecol 9:2109–2118
Sim L, Beeren SR, Findinier J, Dauvillée D, Ball S, Henriksen A, Palcic MM (2014) Crystal structure of the Chlamydomonas starch debranching isoamylase ISA1 reveals insights into the mechanism of branch trimming and complex assembly. J Biol Chem 289:22991–23003
Sonnewald U, Kossmann J (2013) Starches - from current models to genetic engineering. Plant Biotechnol J 11:223–232
Spasovski V, Tosic N, Nikcevic G, Stojiljkovic M, Zukic B, Radmilovic M, Karan-Djurasevic T, Srzentic SS, Colovic M, Pavlovic S (2013) The influence of novel transcriptional regulatory element in intron 14 on the expression of Janus kinase 2 gene in myeloproliferative neoplasms. J Appl Genetics 54:21–26
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Stein L (2001) Genome annotation: from sequence to biology. Nat Rev Genet 2:493–503
Streb S, Eicke S, Zeeman SC (2012) The simultaneous abolition of three starch hydrolases blocks transient starch breakdown in Arabidopsis. J Biol Chem 287:41745–41756
Sulpice R, Pyl ET, Ishihara H, Trenkamp S, Steinfath M, Witucka-Wall H, Gibon Y, Usadel B, Poree F, Piques MC, Von Korff M, Steinhauser MC, Keurentjes JJ, Guenther M, Hoehne M, Selbig J, Fernie AR, Altmann T, Stitt M (2009) Starch as a major integrator in the regulation of plant growth. Proc Natl Acad Sci USA 106:10348–10353
Sulpice R, Flis A, Ivakov AA, Apelt F, Krohn N, Encke B, Abel C, Feil R, Stitt M (2014) Arabidopsis coordinates the diurnal regulation of carbon allocation and growth across a wide range of photoperiods. Mol Plant 7:137–155
Sundberg M, Pfister B, Fulton D, Bischof S, Delatte T, Eicke S, Stettler M, Smith SM, Streb S, Zeeman SC (2013) The heteromultimeric debranching enzyme involved in starch synthesis in Arabidopsis requires both isoamylase1 and isoamylase2 subunits for complex stability and activity. PLoS One 8:e75223
Svensson B (1994) Protein engineering in the alpha-amylase family: catalytic mechanism, substrate specificity, and stability. Plant Mol Biol 25:141–157
Szydlowski N, Ragel P, Hennen-Bierwagen TA, Planchot V, Myers M, Mérida Á, D’Hulst C, Wattebled F (2011) Integrated functions among multiple starch synthases determine both amylopectin chain length and branch linkage location in Arabidopsis leaf starch. J Exp Bot 62:4547–4559
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tenorio G, Orea A, Romero JM, Mérida A (2003) Oscillation of mRNA level and activity of granule-bound starch synthase I in Arabidopsis leaves during the day/night cycle. Plant Mol Biol 51:949–958
Tetlow IJ, Wait R, Lu Z, Akkasaeng R, Bowsher CG, Esposito S, Kosar-Hashemi B, Morell MK, Emes MJ (2004) Protein phosphorylation in amyloplasts regulates starch branching enzyme activity and protein-protein interactions. Plant Cell 16:694–708
Tetlow IJ, Beisel KG, Cameron S, Makhmoudova A, Liu F, Bresolin NS, Wait R, Morell MK, Emes JE (2008) Analysis of protein complexes in wheat amyloplasts reveals functional interactions among starch biosynthetic enzymes. Plant Physiol 146:1878–1891
Tomlinson K, Denyer K (2003) Starch synthesis in cereal grains. Adv Bot Res 40:1–61
Utsumi Y, Nakamura Y (2006) Structural and enzymatic characterization of the isoamylase1 homo-oligomer and the isoamylase1-isoamylase2 hetero-oligomer from rice endosperm. Planta 225:75–87
Utsumi Y, Utsumi C, Sawada T, Fujita N, Nakamura Y (2011) Functional diversity of isoamylase oligomers: the ISA1 homo-oligomer is essential for amylopectin biosynthesis in rice endosperm. Plant Physiol 156:61–77
Ventriglia T, Kuhn ML, Ruiz MT, Ribeiro-Pedro M, Valverde F, Ballicora MA, Preiss J, Romero JM (2008) Two Arabidopsis ADP-glucose pyrophosphorylase large subunits (APL1 and APL2) are catalytic. Plant Physiol 148:65–76
Vijn I, Smeekens S (1999) Fructan: more than a reserve carbohydrate? Plant Physiol 120:351–359
Wattebled F, Dong Y, Dumez S, Delvallé D, Planchot V, Berbezy P, Vyas D, Colonna P, Chatterjee M, Ball S, D’Hulst C (2005) Mutants of Arabidopsis lacking a chloroplastic isoamylase accumulate phytoglycogen and an abnormal form of amylopectin. Plant Physiol 138:184–195
Wattebled F, Planchot V, Dong Y, Szydlowski N, Pontoire B, Devin A, Ball S, D’Hulst C (2008) Further evidence for the mandatory nature of polysaccharide debranching for the aggregation of semicrystalline starch and for overlapping functions of debranching enzymes in Arabidopsis leaves. Plant Physiol 148:1309–1323
Weigel D, Mott R (2009) The 1001 genomes project for Arabidopsis thaliana. Genome Biol 10:107
Wolfe KH, Gouy M, Yang YW, Sharp PM, Li WH (1989) Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data. Proc Natl Acad Sci USA 86:6201–6205
Yan HB, Pan XX, Jiang HW, Wu GJ (2009) Comparison of the starch synthesis genes between maize and rice: copies, chromosome location and expression divergence. Theor Appl Genet 119:815–825
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Bio Sci 13:555–556
Yang Z, Nielsen R, Goldman N, Pedersen AMK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yilmaz A, Mejia-Guerra MK, Kurz K, Liang X, Welch L, Grotewold E (2011) AGRIS: the Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res 39(Database issue):D1118–D1122
Zeeman SC, Thorneycroft D, Schupp N, Chapple A, Weck M, Dunstan H, Haldimann P, Bechtold N, Smith AM, Smith SM (2004) Plastidial alpha-glucan phosphorylase is not required for starch degradation in Arabidopsis leaves but has a role in the tolerance of abiotic stress. Plant Physiol 135:849–858
Zeeman SC, Kossmann J, Smith AM (2010) Starch: its metabolism, evolution, and biotechnological modifications in plants. Annu Rev Plant Biol 61:209–234
Zhang X, Myers AM, James MG (2005) Mutations affecting starch synthase III in Arabidopsis alter leaf starch structure and increase the rate of starch synthesis. Plant Physiol 138:663–674
Zhang X, Szydlowski N, Delvallé D, D’Hulst C, James MG, Myers AM (2008) Overlapping functions of the starch synthases SSII and SSIII in amylopectin biosynthesis in Arabidopsis. BMC Plant Biol 8:96
Acknowledgments
The authors thank Thomas Altmann for providing plant seeds and Michael Lenhard for providing access to Realtime PCR equipment. Financial support by the Bundesministerium für Bildung und Forschung (BMBF) within the GoFORSYS initiative and by the University of Potsdam is gratefully acknowledged. MS thanks the Max Planck Institute of Molecular Plant Physiology, Potsdam-Golm, and the University of Guelph for generously permitting the use of the library. We would like to thank the anonymous reviewer for very useful comments and suggestions.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11103_2015_293_MOESM1_ESM.pdf
Fig. S1 Duplications of chromosomal segments and the position of starch-related genes in the A. thaliana genome The chromosomes of A. thaliana (I to V) are shown as grey horizontal bars. The occurrence and positions of segmental duplications (similar sequences; various colors) were taken from Stein (2001; Fig. 3). Duplications occurred within and across chromosomes and could be either unidirectional or inverted (twisted bars). Chromosome length (given in megabases [Mb]) was modified according to TAIR 10.0. Starch-related genes are evenly distributed across the genome. The positions of the genes are given above each chromosome. (PDF 117 kb)
11103_2015_293_MOESM2_ESM.pdf
Fig. S2 Phylogenetic trees of the combined data set of A. thaliana. Rooted maximum likelihood trees among accessions of A. thaliana and A. lyrata as an outgroup based on gene sequences (including introns). The combined data set consists of 105 accessions comprising 26 accessions from our data set and 80 accessions from the 1001 Genome Project (Cao et al. 2011). Accessions that are used for Realtime PCR analyses are highlighted in the same color as in Figs. 3–8. (PDF 7940 kb)
Rights and permissions
About this article

Cite this article
Schwarte, S., Wegner, F., Havenstein, K. et al. Sequence variation, differential expression, and divergent evolution in starch-related genes among accessions of Arabidopsis thaliana . Plant Mol Biol 87, 489–519 (2015). https://doi.org/10.1007/s11103-015-0293-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-015-0293-2