
Abstract
Purpose
Reversible self-association (RSA) has long been a concern in therapeutic monoclonal antibody (mAb) development. Because RSA typically occurs at high mAb concentrations, accurate assessment of the underlying interaction parameters requires explicitly addressing hydrodynamic and thermodynamic nonideality. We previously examined the thermodynamics of RSA for two mAbs, C and E, in phosphate buffered saline (PBS). Here we continue to explore the mechanistic aspects of RSA by examining the thermodynamics of both mAbs under reduced pH and salt conditions.
Methods
Dynamic light scattering and sedimentation velocity (SV) studies were conducted for both mAbs at multiple protein concentrations and temperatures, with the SV data analyzed via global fitting to determine best-fit models, interaction energetics, and nonideality contributions.
Results
We find that mAb C self-associates isodesmically irrespective of temperature, and that association is enthalpically driven but entropically penalized. Conversely, mAb E self-associates cooperatively and via a monomer–dimer-tetramer-hexamer reaction pathway. Moreover, all mAb E reactions are entropically driven and enthalpically modest or minimal.
Conclusions
The thermodynamics for mAb C self-association are classically seen as originating from van der Waals interactions and hydrogen bonding. However, relative to the energetics we determined in PBS, self-association must also be linked to proton release and/or ion uptake events. For mAb E, the thermodynamics implicate electrostatic interactions. Furthermore, self-association is instead linked to proton uptake and/or ion release, and primarily by tetramers and hexamers. Finally, although the origins of mAb E cooperativity remain unclear, ring formation remains a possibility whereas linear polymerization reactions can be eliminated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Monoclonal antibodies (mAbs) are vulnerable to an array of protein–protein interactions associated with reduced activity [1]. One such example is reversible self-association (RSA), which we specify to mean the dynamic exchange of monomers with natively folded oligomers. RSA is associated with several problematic outcomes including high drug viscosity, conversion of monomer to irreversible aggregate, phase separation, and opalescence [2,3,4,5]. Even though considerable efforts have gone into minimizing RSA [6, 7], the underlying mechanisms are not entirely clear. Increased thermodynamic insight should prove useful in identifying the molecular forces and linked reactions responsible for self-association, which in turn should benefit protein engineering and formulation efforts.
As desirable as thermodynamic analyses of mAb RSA may be, estimating the underlying interaction parameters is not straightforward. This is in part because of the relatively modest energetics of RSA (Keq), which cause oligomers to form at concentrations in which hydrodynamic nonideality (ks) and thermodynamic nonideality (BM1) become non-negligible. For example, in transport methods such as sedimentation velocity (SV), a self-associating mAb will generate a larger weight-average sedimentation coefficient (S20,w) and wider sedimentation coefficient distribution as mAb concentration is increased. However, at high concentrations (typically greater than several mg/mL), hydrodynamic nonideality will reduce the weight-average sedimentation coefficient and narrow the distribution. Simultaneously, thermodynamic nonideality will cause a slight broadening of the distribution. Collectively, these events are due to the charge, shape and excluded volume effects that underlie the nonideality terms [8, 9]. As a result, nonideality and self-association tend to conceal the presence of each other, thus creating significant challenges in the accurate determination of interaction parameters such as Keq, ks and BM1.
To resolve these issues, we globally fit the SV data to identify the most appropriate interaction models, estimate self-association free energies, and assess contributions from hydrodynamic and thermodynamic nonideality. We previously used this approach to analyze the thermodynamics of two interacting mAbs, C and E, in phosphate buffered saline (PBS) at pH 7.4, finding that the mAbs displayed unique self-association pathways and distinct thermodynamic signatures [10, 11]. Here we investigate the self-association thermodynamics for the same mAbs now under reduced pH and salt concentration (pH 6.0 and 100 mM NaCl). The impetus for the present studies was that mAbs are often formulated under low pH conditions, and that perturbation of solution conditions allows for more detailed insight into the underlying mechanisms of RSA, including the identification of additional reactions that may be linked to self-association. Under the conditions examined here, we find that mAb C follows an isodesmic reaction pathway (i.e., indefinite and noncooperative) for all temperatures. Additionally, the reaction is enthalpically driven and entropically penalized. Conversely, mAb E follows a cooperative monomer–dimer-tetramer-hexamer reaction. Moreover, all self-association steps for mAb E are entropically driven and enthalpically modest or minimal.
The favorable enthalpy and unfavorable entropy for mAb C is traditionally interpreted as arising from Van der Waals interactions and hydrogen bonding [12]. However, we emphasize that the thermodynamics measured here reflect not only the interfacial contacts that directly mediate oligomerization, but also all other reactions that are linked to oligomerization. Noting that our previous studies in PBS (and thus at a higher salt concentration and pH) demonstrated that mAb C also undergoes isodesmic self-association but with stronger energetics than seen here, thermodynamic linkage theory predicts that oligomerization must be linked to net proton release and/or ion uptake.
For mAb E, the signature of a favorable entropy and minimal enthalpy is traditionally viewed as arising from electrostatic interactions [12]. Interestingly, in PBS, mAb E forms only dimers and with comparable energetics to the dimer formation seen here. Thus, in opposition to mAb C, mAb E self-association must be linked to proton uptake and/or ion release, but primarily via formation of tetramers and hexamers. Finally, although a molecular interpretation of mAb E cooperativity is not straightforward, “simple” linear polymerization reactions can be eliminated. Alternative models such as ring formation are discussed.
Materials and Methods
As noted above, we previously carried out analyses of mAb C and E RSA in PBS at pH 7.4 (10 mM disodium phosphate, 1.8 mM potassium phosphate, 137 mM NaCl, 2.7 mM KCl) [10, 11]. Because there were otherwise no modifications to the present studies beyond changes in buffer conditions, and our methods with complete detail were presented elsewhere [10], we only briefly summarize our approach.
Reagents
Both mAbs were supplied by MedImmune LLC (now part of AstraZeneca) and dialyzed into sodium phosphate buffer (20 mM sodium phosphate, 100 mM NaCl, pH 6.0). Concentrations were estimated via an extinction coefficient of 1.56 mL mg−1 cm−1. For each mAb, the partial specific volume corrected for 2% glycan mass was calculated to be 0.7245 mL/g. SEDNTERP was used to estimate buffer viscosity and density at each temperature [13].
Dynamic Light Scattering
Light scattering studies were conducted using a Zetasizer Nano ZSP DLS instrument (Malvern), with the hydrodynamic diameter (DH) for each mAb being estimated at multiple concentrations and temperatures. Hydrodynamic diameters and mutual diffusion coefficients were determined using the Malvern software. All samples were filtered extensively prior to analysis.
Sedimentation Velocity Analytical Ultracentrifugation
SV experiments were conducted over a range of concentrations and temperatures using a Beckman XLI analytical ultracentrifuge (Beckman Coulter). Samples were sedimented at 42,000 rpm and protein concentrations were 0.3 to 10 mg/mL for mAb C and 0.3 to 13 mg/mL for mAb E. Model-independent analyses were conducted using DCDT + [14]. Global fitting, simulations and statistics were conducted using SEDANAL [15].
Finally, the dependence on concentration for the sedimentation coefficient, s(c), and the diffusion coefficient, D(c), were addressed via Eqs. 1 and 2,
where s0 and D0 are respectively the sedimentation and diffusion coefficients at infinite dilution, and ks and BM1 are respectively hydrodynamic and thermodynamic nonideality.
Results
As Determined by DLS, Both mAbs Undergo Concentration-Dependent RSA, but Only One Shows Temperature Dependence
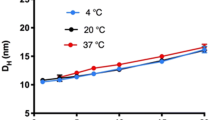
Plotted in Fig. 1A are mAb C hydrodynamic diameters, DH, at multiple protein concentrations and temperatures; the diameters are clearly concentration-dependent and therefore supportive of RSA. Self-association also decreases with increasing temperature. In Fig. 1B are the results for mAb E. This mAb also shows an increase in diameter with concentration, although without a temperature effect. Representative plots of the mutual diffusion coefficient at 4°C versus mAb concentration are shown for mAbs C and E in Fig. 1C and D, respectively. We emphasize that the shapes of the plots reflect contributions from both nonideality and self-association; this is especially evident for mAb C, which shows clear curvature due to the larger role of self-association relative to mAb E. For this reason, diffusion interaction parameters (kd) are not reported.

DLS analysis for mAbs C and E at multiple temperatures. Concentration range covered 0.5 to 15 mg/mL for mAb C (panel A), and 1 to 20 mg/mL for mAb E (panel B). Buffer conditions were 20 mM sodium phosphate, 100 mM NaCl, pH 6.0 and temperatures of 4°C (blue), 20°C (black) and 37°C (red). Examples of the mutual diffusion coefficient versus mAb E and mAb C concentration at 4°C are plotted in panels (C) and (D), respectively.
SV Indicates that the Two mAbs Follow Unique Pathways for Self-Association
To examine the underpinnings of mAb-specific self-association in more detail, we conducted SV analyses over a range of concentrations and temperatures. Data were initially analyzed model-independently to assess the degree of and type of self-association, as well as contributions from hydrodynamic nonideality. (Thermodynamic nonideality is less obvious by visual inspection [16].) In Fig. 2A are examples of normalized g(s*) distributions for mAb C collected at multiple temperatures. Most noticeable, especially the lowest and intermediate temperatures, is that mAb C displays a wide and smooth series of distributions that increase with concentration, suggestive of multiple higher-order species in rapid equilibrium. Our own simulations (see Discussion) and the work of others (Bishop and Correia, submitted) suggest that the shapes of the distributions are indicative of non-cooperative self-association. Also evident is a decrease in self-association at higher temperatures, in line with the DLS studies. Finally, hydrodynamic nonideality is observed most prominently in the 10 and 20°C data as a narrowing of the distributions at the highest concentrations.

Normalized g(s*) analysis for mAbs C and E at multiple temperatures. (A) Concentration-dependent distributions for mAb C at 0.3 (black), 0.5 (green), 1 (blue), 3 (red), 7 (light green), and 10 (light blue) mg/mL in 20 mM sodium phosphate, 100 mM NaCl, pH 6.0 and at 10, 20 and 35°C. (B) Analogous distributions for mAb E at 0.3 (black), 1 (blue), 3 (red), 7 (light green), 10 (light blue), and 13 (pink) mg/mL, also in 20 mM sodium phosphate, pH 6.0 and at 10, 20 and 35°C.
In Fig. 2B are normalized g(s*) results for mAb E, again at multiple temperatures. Like mAb C, a broad concentration-dependence is observed, although the shapes of the distributions are clearly different than mAb C, indicating a distinct self-association pathway. For example, at all temperatures a shoulder at appears ~ 9 S, with the 35°C results in particular showing a nearly bimodal inflection. Again, our simulations and those of Bishop and Correia (submitted) suggest that these shapes reflect cooperative addition of protomers. The distributions also show little temperature dependence as was seen in the DLS studies. Finally, hydrodynamic nonideality is not immediately obvious in the mAb E distributions compared to mAb C. We address this apparent discrepancy below and in the Discussion.
Historically, a quantitative means to visualize and measure hydrodynamic nonideality has been via linearization of Eq. 1:
where in the present case, s(c) represents the weight-average sedimentation coefficient at each mAb concentration as determined from the distributions in Fig. 2. Upon transformation of the weight-average sedimentation coefficients using Eq. 3, a plot of 1/s(C) versus c for a noninteracting mAb would generate a linear or straight-line response with a positive slope, and a fit of the data to Eq. 3 would allow estimation of ks. However, as shown in Fig. 3, representative data for each mAb generate a negative slope, and curvature for at least mAb C. The negative slopes are a result of mAb self-association not being accounted for in Eq. 3, and the curvature in the data for mAb C arises from the more dominant role of hydrodynamic nonideality, ks, relative to mAb E. For both mAbs, if the concentration were further increased, the plotted data would eventually reach a minimum and then increase with a positive, linear slope as hydrodynamic nonideality completely concealed self-association (see Discussion and [16]). Thus, the graphical representation here provides an example of the reciprocal masking of self-association energetics and hydrodynamic nonideality and speaks to the importance of direct boundary fitting to explicitly determine both parameters as described below [11, 16].

Plots of inverse weight-average sedimentation coefficients versus concentration for mAbs C and E. (A) mAb C. (B) mAb E. Weight-average sedimentation coefficients at 10°C and for each mAb concentration were determined from the distributions in Fig. 2 using SEDANAL.
mAb C Self-Association is Isodesmic and Enthalpically Favorable
We next analyzed the sedimentation velocity data to assess the most appropriate self-association models, binding energetics, and contributions from hydrodynamic and thermodynamic nonideality. Specifically, data were analyzed by model-dependent global fitting, guided by our earlier studies [10, 11], the work of others [17,18,19,20], and interpretation of the normalized g(s*) plots in Fig. 2. Parameters were locked (fixed) if we had independent values such as mAb molecular weight, and unless otherwise stated, sedimentation coefficient values were constrained by sn = s1 n2/3, where s1 is the monomer s value, n is stoichiometry of any particular species (e.g. dimer), and sn is the s value for that species [21]. Finally, floating of kinetic rate constants for dissociation returned values of 1 to 0.01 s−1 and without any improvement in fit, thus indicating that that both mAbs are in rapid equilibrium. The rate constants were therefore locked at the default values of 0.01 s−1 and not considered further.
Our earlier studies found that mAb C self-association in PBS is isodesmic in character (2A ⇔ A2; A + A2 ⇔ A3; A + A3 ⇔ A4;…) with the affinity being identical for all reaction steps [10, 11]. This was again the case in the current conditions. A fit of the mAb C data at 20°C using a nonideal isodesmic model is presented in Fig. 4. (All fits also included a parameter for estimating irreversible dimer, typically ~ 2% by mass and otherwise not reported.) This model describes well all of the datasets (RMSD of 0.016 ∆fringes), with a monomer s20,w of 6.30 S, a Keq equal to 1.70e4 M−1, a ks of 19.3 mL/g and a BM1 of 7.8 mL/g. The sedimentation coefficient, interaction affinity, and BM1 term are consistent with expectations; however, ks was unexpectedly high, more typically found to be 4–11 mL/g [8, 9, 22,23,24]. We have observed this anomaly previously and address it in more detail in the discussion. Finally, all other models resulted in poorer fits, consistent with what we reported in our earlier work [10].

Global fitting for mAb C using a nonideal, isodesmic self-association model. SV data were collected in 20 mM sodium phosphate, 100 mM NaCl, pH 6.0 at 0.3, 0.5, 1, 3, 7 and 10 mg/mL and at 20°C. Data (red) presented as difference scans (∆fringes versus radius), with best-fit in green and residuals in blue. 50 to 100 scans were analyzed per concentration, resulting in 25 to 50 difference scans. For clarity, only the first and last difference scans at each concentration are presented. Top row: 0.3, 0.5, and 1 mg/mL mAb C; bottom row: 3, 7 and 10 mg/mL mAb C.
We repeated this analysis for the mAb C data collected at other temperatures, finding that the isodesmic model provided the highest quality fit. Table I tabulates the resultant interaction parameters. We note that relatively weak self-association was observed at 35°C, which as discussed previously, generates increased correlation and poor resolution of Keq and ks [10, 25]. Although one solution to this problem would be to work at even higher mAb concentrations and therefore increase the contribution from the nonideality terms, this is not always possible due to introduction of optical artifacts or if the mAb in question cannot be sufficiently concentrated. ks and BM1 were therefore fixed at 10 mL/g for the 35°C analysis.
We next used the estimated affinities at all temperatures to determine the thermodynamics of mAb C self-association. A van’t Hoff analysis is presented in Fig. 5, with a linear fit (solid line) returned an enthalpy (∆H) of -11.5 ± 1.4 kcal/mol and an entropy (∆S) of -5.8 ± 1.5 kcal/mol at 20°C. For comparative purposes, we also show the analogous results from our previous studies of mAb C self-association in PBS, which show a stronger affinity at all temperatures but with a similar temperature-dependence [10]. Consistent with this, the enthalpy of self-association under this latter condition (-13.3 ± 1.0 kcal/mol), was statistically identical to the value we observe presently, and thus indicates that isodesmic self-association under both conditions is enthalpically driven and entropically penalized.

van’t Hoff plot for mAb C self-association. Affinities (red circles) were estimated from the global fits of SV data. A linear fit (solid line) resulted in a self-association enthalpy of -11.5 ± 1.4 kcal/mol and entropy of -5.8 ± 1.4 kcal/mol. For comparative purposes, the temperature-dependence of mAb C self-association under our previous conditions (PBS; open black circles) is also shown. For both conditions, the affinities were also fit to a modified expression to estimate an apparent heat capacity change (dashed lines). For some affinity terms, error bars are too small to be seen.
Finally, visual inspection of the data points in the van’t Hoff plot suggests modest curvature in the current buffer. The presence of a nonlinear temperature-dependence indicates that the enthalpy and entropy of self-association are themselves temperature dependent, and that the results could be better described by a modified van’t Hoff expression that allows for a heat capacity change, ∆Cp [26]. Fitting to such an equation (dashed line in Fig. 5) resulted in a visually improved fit and returned an apparent ∆Cp of -0.7 ± 0.3 kcal/mol·K. By comparison, an analogous fit to the data in PBS returned a ∆Cp of -0.5 ± 0.2 kcal/mol·K. We address the possibility and implications of an apparent heat capacity change later.
mAb E Self-Association Follows a Monomer–Dimer-Tetramer-Hexamer Pathway that is Cooperative and Entropically Favorable
We previously showed that mAb E displays nonideal monomer–dimer (2A ⇔ A2) self-association in PBS [10, 11]. Judging by the g(s*) distributions that extend out to nearly 14 S (Fig. 2B), self-association in the present buffer is more extensive than simply monomer–dimer. We therefore examined a wide range of models, with some examples shown in Table II. The best-fit and most parsimonious (shown in bold in Table II) was a cooperative and nonideal monomer–dimer-tetramer-hexamer reaction (2A ⇔ A2; 2A2 ⇔ A4; A2 + A4 ⇔ A6). Shown in Fig. 6 are the results of globally fitting the 20°C data to that model and again allowing for irreversible dimers (RMSD = 0.039 ∆fringes). The resultant parameters were an s20,w of 6.24 S, and Keq values of 3.03e3 M−1 for monomer–dimer formation, 1.19e4 M−1 for dimer-tetramer formation, and 9.40e4 M−1 for tetramer-hexamer formation. Similar results were seen at all other temperatures as summarized in Table III. Finally, the modest Keq for monomer–dimer self-association resulted in strong correlation like that for mAb C, which required locking the nonideality parameters at 10 mL/g.

Global fitting for mAb E using a nonideal, monomer–dimer-tetramer-hexamer model. SV data were collected in 20 mM sodium phosphate, 100 mM NaCl, pH 6.0 at 0.3, 1, 3, 7, 10 and 13 mg/mL and at 10°C. Data (red) presented as difference scans, with best-fit in green and residuals in blue. 50 to 100 scans were analyzed per concentration, resulting in 25 to 50 difference scans. For clarity, only the first and last difference scans at each concentration are presented. Top row: 0.3, 1 and 3 mg/mL mAb E; bottom row: 7, 10 and 13 mg/mL mAb E.
Shown in Fig. 7 is a van’t Hoff plot for mAb E self-association. Unlike mAb C, we see no evidence of curvature in the temperature-dependent affinities, with linear fits returning an enthalpy for self-association of +0.4 ± 1.8 kcal/mol for monomer–dimer formation, + 4.7 ± 2.1 kcal/mol for dimer-tetramer formation, and −0.2 ± 2.2 kcal/mol for tetramer-hexamer formation. The corresponding entropy values were + 5.1 ± 1.9 kcal/mol, + 10.2 ± 4.7 kcal/mol, and + 6.5 ± 2.2 kcal/mol, respectively. Thus, in direct contrast to mAb C, all mAb E self-association reactions are entirely entropically driven and comprised of only a minimal or modest enthalpy contribution.

van’t Hoff plot for mAb E self-association. Affinities for monomer–dimer (red circle), dimer-tetramer (blue squares), and tetramer-hexamer formation (green triangles) estimated from global fits of the SV data. Linear fits (solid lines) resulted in self-association enthalpy of + 0.4 ± 1.8 kcal/mol for monomer–dimer formation, + 4.7 ± 2.0 kcal/mol for dimer-tetramer formation, and -0.2 ± 2.2 kcal/mol for tetramer-hexamer formation. Corresponding entropies were + 5.1 ± 1.9 kcal/mol, + 10.2 ± 4.7 kcal/mol, and + 6.5 ± 2.2 kcal/mol, respectively. For comparative purposes, the temperature-dependence of mAb E monomer–dimer self-association in our previous conditions (PBS; open black circles) are also shown. For some affinity terms, error bars are too small to be seen.
For comparative purposes, we also show the results from our previous studies of mAB E self-association in PBS [10]. This analysis revealed only monomer–dimer formation, and with a slightly weaker affinity than that observed currently (~ twofold) and a similar thermodynamic signature (∆H = −1.7 ± 0.8 kcal/mol, T∆S = + 2.6 ± 0.9 kcal/mol). Collectively then, a conservative interpretation of the results suggests that the reduced pH and salt concentration examined here only mildly influence an already pre-existing dimerization reaction, but dramatically induce tetramer and hexamer formation. Moreover, the increase in affinity from monomer–dimer formation (4e3 M−1 on average) to dimer-tetramer formation (1e4 M−1 on average) to tetramer-hexamer formation (8e4 M−1 on average) suggests that self-association is cooperative in nature, consistent with the interpretation of the model-independent results in Fig. 2B.
Discussion
mAb C Self-Association is Linked to Additional Reaction Equilibria
Self-association of mAb C has been examined by a number of groups, under a variety of buffer conditions, and described by a range of reaction schemes [10, 11, 17, 18, 27]. In the present buffer and at all temperatures, we find that the best fit model is isodesmic. This finding is in line with our earlier work, which also used SV and was further supported by sedimentation equilibrium studies [10, 11]. The model-independent analyses in Fig. 2A are also in line with this interpretation – as described below and in more detail by Bishop and Correia (submitted), isoenergetic addition of protomers (e.g. via an isodesmic model) will generate smooth distributions without inflections or bimodality, consistent with fundamental Gilbert theory [28].
To better illustrate the above point, we carried out a series of simulations covering a mAb C concentration range from 0.3 to 50 mg/mL. Shown in Fig. 8A are the experimentally determined sedimentation coefficient distributions for mAb C as seen in Fig. 2, but now presented as normalized s*g(s*) plots and in log units to better emphasize the presence of higher-order oligomers. Shown in Fig. 8B are simulated distributions out to 50 mg/mL using the isodesmic model and parameters from Table I, but assuming no contributions from hydrodynamic and thermodynamic nonideality. Although the absence of nonideality is physically unrealistic, it demonstrates how isodesmic self-association generates a broad and smooth distribution without inflections or bimodality regardless of mAb concentration. This is a general consequence of all models in which the Keq values for self-association are identical (i.e., noncooperative). Shown in Fig. 8C is the same simulation except the hydrodynamic and thermodynamic nonideality terms reported in Table I are now included. Nonideality clearly conceals self-association beginning even at 3 mg/mL (compare to the 3 mg/mL distribution in Fig. 8B) and drives the distributions to increasingly smaller s values at concentrations of 25 mg/mL and greater. Note also that the simulated distributions from 0.3 to 10 mg/mL (in grey) closely match the experimental distributions in Fig. 8A once nonideality is included. Finally, shown in Fig. 8D is a plot of mAb C concentration versus inverse weight-average sedimentation coefficient for the experimentally determined data in Fig. 8A using Eq. 3, overlaid with the analogous values from the simulated data in Fig. 8C. Good agreement between experiment and simulation is seen over the range of 0.3 to 10 mg/mL, and analysis out to 50 mg/mL reveals the curvature and eventual increase in 1/S20,w as predicted for a nonideal self-associating system.

Normalized s*g(s*) analysis for mAb C using experimental and simulated data. (A) Normalized s*g(s*) versus log s* for experimental data collected in 20 mM sodium phosphate, 100 mM NaCl, pH 6.0 at 20°C and covering a concentration range of 0.3 to 10 mg/mL. Data are adapted and rescaled from those in Fig. 2 to compare to simulated data below. (B) Normalized s*g(s*) versus log s* plot generated from simulated data using an isodesmic model, the sedimentation coefficient and Keq values in Table I at 20°C and covering a range from 0.3 to 10 mg/mL (grey tones) and 25 to 50 mg/mL (red tones). Ks and BM1 values were set to 0.0 mL/g. Vertical dashed line refers to sedimentation coefficient of mAb C monomer from Table II. (C) Same as panel B, but with ks = 19.3 mL/g and BM1 = 7.8 mL/g. (D) Plot of inverse weight-average sedimentation coefficient versus mAb C concentration. Sedimentation coefficients were determined from experimental data in panel A (black squares) and simulated data in panel C (red circles) using SEDANAL.
Regarding the estimated interaction parameters for mAb C, we note all are in the range of expected values except for the nonideality term, kS, which is greater than typical estimates of 4–11 mL/g reported for monomeric mAbs [8, 9, 22,23,24]. This was especially true at the reduced temperatures where enhanced RSA is occurring and thus where oligomers are increasingly populated. Based on work by Rowe [8], ks can be expressed as follows:
In the present case, \(\overline{\upupsilon }\) refers to the partial specific volume of mAb C (mL/g), Vs refers to the specific volume of mAb C (mL/g), and f/fo refers to the mAb C frictional ratio (unitless). The Vs term describes mAb volume per unit mass, which includes all strongly and weakly bound water molecules (i.e., “solvent entrainment”; [22]). In contrast, the f/fo term reflects the gross structure of the mAb, with values greater than 1 indicating a departure from spherical. Thus, ks represents contributions from two values – mAb solvation (Vs/\(\overline{\upupsilon }\)) and mAb structure (f/fo).
The f/fo values for reversible mAb C oligomers are unknown; however, previous studies have found that irreversible mAb oligomers have f/fo values like that of the monomer (~ 1.5–1.6; [29]). If this relationship holds for the reversible oligomers examined here, and noting that the ks terms in Table I represent an average of ks values over all oligomeric states in solution, then an increase in the average ks must arise from increased solvation of the oligomers relative to the mAb C monomer. Of course, it is also possible that the increase in ks is instead due to larger f/fo values of the oligomers relative to the monomer. In the event that future studies reveal the structure of the oligomers, recent software developments [30] would allow calculation of the ks terms for each stoichiometric species and potential parsing of their solvation and shape contributions.
Regarding the molecular forces driving mAb C RSA, we find that self-association is enthalpically driven and entropically penalized. Qualitatively similar thermodynamics have been seen previously [17]. Keeping in mind the limitations of interpreting thermodynamic parameters from van’t Hoff plots [31,32,33], classical interpretations of the above thermodynamics indicate that van der Waals interactions and/or hydrogen bonding are critical in mediating mAb C self-association [12]. Yet we emphasize that the thermodynamics determined here reflect both direct interactions associated with oligomerization and all reactions indirectly linked to self-association (e.g., protonation events, conformational isomerization, or buffer-specific interactions). Noting that mAb C also engages in isodesmic RSA at the higher salt concentration and pH of PBS but with stronger energetics, linkage thermodynamics predict that self-association must be coupled to net proton release and/or ion uptake events [34, 35]. Although the number of net protons and/or ions linked to self-association cannot be determined with the present results, in part due to differences in buffer composition and the limited data from only two solution conditions, this interpretation is in line with earlier work showing that mAb C self-association is influenced by pH, salt concentration, and specific ion types [17]. However, these studies also showed that monovalent cation type had no influence on RSA, suggesting that at least Na+ and K+ are not playing differential roles. Noting that the interaction energetics change in the pH range of 6.0 to 7.4, our results suggest that net proton release is occurring at histidine sidechains, which are being perturbed to more acidic pKas in the oligomers. Of note, histidine residues were previously identified as being at the interface of mAb C oligomers using hydrogen exchange mass spectrometry [19]; however, our thermodynamic interpretation does not preclude the possibility of residues outside of interfacial regions playing a role in proton and/or ion binding events. Therefore, caution should be observed in assigning specific histidines (or any other residues) to the present results.
Finally, we note that the van’t Hoff data show curvature (Fig. 5), suggestive of an apparent heat capacity change upon oligomerization. This behavior was observed regardless of how we analyzed the data and is often interpreted as arising from burial of hydrophobic residues due to the hydrophobic effect. However, apparent heat capacity changes can be observed for a variety of reasons, including the hydrophobic effect, but also molecular bond vibrations or additional linked reactions with differing thermodynamic contributions [35,36,37]. Thus, even though hydrophobic interactions have been identified as playing a role in mAb C RSA [17], considerable more work will be needed to confirm the apparent heat capacity change seen here (e.g. by using sedimentation equilibrium to independently determine the interaction energetics). Subsequent studies would then be needed to assign the change to the hydrophobic effect, linked reactions, or otherwise. With that said, noting that the ∆CP values trend increasingly negative as pH and salt concentration are reduced, one appealing possibility is that the apparent change arises from a proton- or ion-interaction event linked to self-association [31].
Relative to mAb C, Cooperative mAb E Self-Association is Inversely Linked to Net Proton and/or Salt Binding
Under the present conditions, mAb E displays monomer–dimer-tetramer-hexamer RSA. Previous studies suggested a monomer–dimer-tetramer reaction, although that work also showed that mAb E self-association is influenced by concentration of salt, ion type, excipient type, and pH [20]. Noting that the buffer conditions used here are different to those employed previously, it may not be surprising that the best-fit models are different. It is also worth noting that based on the parameters in Table III, the hexamer is only weakly populated even at the highest mAb concentrations (~ 5% by mass), making resolution of this species challenging.
We also note that mAb E self-association is cooperative as judged by the model-dependent increases in affinity for association to hexamer (Table III), and by the model-independent pattern of sedimentation coefficient distributions in Fig. 2. To better emphasize this latter point, we again carried out simulations analogous to those of mAb C. Shown in Fig. 9A are the experimentally determined sedimentation coefficient distributions for mAb E as adapted from Fig. 2. Shown in Fig. 9B are simulated distributions out to 150 mg/mL using the monomer–dimer-tetramer-hexamer model and parameters in Table III, but assuming no hydrodynamic or thermodynamic nonideality. Of note is the significantly different pattern of distributions as compared to the isodesmic simulation in Fig. 8B, specifically the concentration-dependent decrease in maximal signal at the lower concentrations and the inflections that appear at intermediate concentrations. In this example, these differences arise from the increases in Keq values for mAb E association (primarily via dimer and tetramer formation since hexamers are weakly populated) but are also a general feature of other cooperatively interacting systems (Bishop and Correia, submitted). Shown in Fig. 9C are the same simulations but with hydrodynamic and thermodynamic nonideality now added. As was seen for mAb C (Fig. 8C), nonideality conceals self-association at the higher concentrations, even as the distinct pattern associated with cooperativity is maintained at the lower concentrations. Note also that the simulated distributions at lower concentrations again closely match the experimental data (Fig. 9A). Finally, shown in Fig. 9D is the concentration versus inverse weight-average sedimentation coefficient plot for the experimental and simulated data. Both datasets are in good agreement in the experimentally accessible range of 0.3 to 13 mg/mL, with the predicted minimum and eventual positive slope appearing at higher concentrations. Note also how the smaller ks value relative to that of mAb C shifts the minimum to higher mAb concentrations. Overall, the simulations in Figs. 8 and 9 serve a critical role for the visual interpretation of model-independent datasets such as those in Figs. 8A and 9A – in this case suggesting that the data in Fig. 8A support an isoenergetic or noncooperative reaction whereas those in Fig. 9A support a cooperative reaction. See Bishop and Correia (submitted) for a more detailed presentation.

Normalized s*g(s*) analysis for mAb E using experimental and simulated data. (A) Normalized s*g(s*) versus log s* for experimental data collected in 20 mM sodium phosphate, 100 mM NaCl, pH 6.0 at 20°C and covering a concentration range of 0.3 to 13 mg/mL. Data are adapted and rescaled from those in Fig. 2 to compare to simulated data below. (B) Normalized s*g(s*) versus log s* plot generated from simulated data using a monomer–dimer-tetramer-hexamer model, the sedimentation coefficient and Keq values in Table III at 20°C and covering a range from 0.3 to 13 mg/mL (grey tones) and 25 to 150 mg/mL (red tones). Ks and BM1 values were set to 0.0 mL/gL. Left vertical dashed line refers to sedimentation coefficient of mAb E monomer from Table II; right vertical line refers to calculated sedimentation coefficient of mAb E hexamer of 20.53 S. Calculated values of mAb E dimer (9.14 S) and tetramer (15.67 S) are not shown for clarity. (C) Same as panel B, but with ks and BM1 = 10 mL/g. (D) Plot of inverse weight-average sedimentation coefficient versus mAb E concentration. Sedimentation coefficients were determined from experimental data in panel A (black squares) and simulated data in panel C (red circles) using SEDANAL.
With respect to the mechanism of mAb E cooperativity, we currently have insufficient information to offer a definitive interpretation; however, linear or noncooperative polymerization models can clearly be eliminated. The enhanced energetics of tetramer formation relative to that of dimers could suggest larger interfaces being formed, followed by a capping mechanism via hexameric ring formation. (Since the fits were not improved by addition of species beyond hexamers (see Table II), there is no need to invoke structures with greater stoichiometries.) Yet the modest contribution of cooperativity in forming hexamers – only an ~ eightfold increase relative to tetramer formation – suggests there are energetic penalties for any putative capping or ring closure event. Whether these penalties arise from topological constraints, structural rearrangements, or otherwise remains to be determined.
Concerning the thermodynamics determined for mAb E, the traditional interpretation is that electrostatics must play a dominant role in self-association [12]. Although this view is consistent with those of previous studies [20], we reiterate that the thermodynamics measured here reflect all reactions linked to self-association, not just those associated with direct interfacial contacts. As we previously reported, mAb E forms only dimers when in PBS [10]. However, in the present buffer, mAb E self-association leads to formation of tetramers and hexamers and with little change in the energetics of dimer formation (Fig. 7). Assuming that the same dimerization reaction is being observed in both PBS and the current conditions, formation of tetramers and hexamers must therefore be preferentially linked to net proton uptake and/or ion release. (To our knowledge and unlike mAb C, the role of monovalent cation in mAb E self-association has not been systematically explored, thus it is unclear if Na+ and K+ play differential roles.) If net proton binding is occurring, such a linkage would again implicate histidine residues. Interestingly – and contrary to mAb C – histidines have not been identified as playing a role in mAb E self-association [20]. This suggests that the pKa’s of other residues are perturbed more dramatically than that of histidine (e.g., cysteines) or that the type of self-association observed in previous studies is different from that observed here. Finally, and from an applied perspective, the near lack of a pH and salt dependence to dimer formation indicates a different mechanism relative to that of tetramer and hexamer formation; therefore, proposed attempts to modulate solution conditions to mitigate RSA may have an impact only on a subset of all reactions [6, 7].
Finally, we note that for both mAbs, the enthalpy changes (∆H) between buffer conditions in PBS and the present conditions (∆∆H) are statistically negligible. This is surprising since proton-and ion-specific binding events are associated with significant enthalpy contributions [9]. Given this lack of change, this raises the possibility of additional and still hidden reactions that thermodynamically compensate for reactions such as proton binding. However, given the limited range of conditions examined so far, more work will be necessary to confirm this interpretation and identify such reactions.
Conclusions
In sum, the present studies demonstrate that mAbs C and E follow distinct self-association pathways; that the thermodynamics of self-association for each mAb point to unique driving forces; that one mAb associates non-cooperatively while the other associates cooperatively; and that even for an individual mAb (mAb E), the mechanisms of successive self-association appear to be distinct. Moreover, relative to our previous studies [10], mAb C self-association is linked to proton release and/or ion uptake whereas mAb E shows the opposite linkage. Future work should continue to focus on the rigorous analysis of how interaction parameters such as Keq, ks and BM1, are modulated under varying solution conditions. Such an approach will reveal the binding equilibria linked to self-association and result in a more predictive and quantitative framework for interpreting mAb-specific RSA.
Data Availability
The data used the current study are available from the corresponding author upon reasonable request.
References
Samaranayake H, Wirth T, Schenkwein D, Räty JK, Ylä-Herttuala S. Challenges in monoclonal antibody-based therapies. Ann Med. 2009;41(5):322–31.
Philo JS, Arakawa T. Mechanisms of protein aggregation. Curr Pharm Biotechnol. 2009;10(4):348–51.
Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004;93(6):1390–402.
Alford JR, Kendrick BS, Carpenter JF, Randolph TW. High concentration formulations of recombinant human interleukin-1 receptor antagonist: II Aggregation kinetics. J Pharm Sci. 2008;97(8):3005–21.
Nishi H, Miyajima M, Nakagami H, Noda M, Uchiyama S, Fukui K. Phase separation of an IgG1 antibody solution under a low ionic strength condition. Pharm Res. 2010;27(7):1348–60.
Wei JY, Bou-Assaf GM, Houde D, Weiskopf A. Technical decision-making with higher order structure data: detecting reversible concentration-dependent self-association in a monoclonal antibody and a preliminary investigation to eliminate it. J Pharm Sci. 2015;104(11):3984–9.
Geoghegan JC, Fleming R, Damschroder M, Bishop SM, Sathish HA, Esfandiary R. Mitigation of reversible self-association and viscosity in a human IgG1 monoclonal antibody by rational, structure-guided Fv engineering. MAbs. 2016;8(5):941–50.
Rowe AJ. The concentration dependence of transport processes: A general description applicable to the sedimentation, translational diffusion, and viscosity coefficients of macromolecular solutes. Biopolymers. 1977;16(12):2595–611.
Tanford C. Physical chemistry of macromolecules. New York: Wiley; 1961.
Hopkins MM, Parupudi A, Bee JS, Bain DL. Energetic dissection of mab-specific reversible self-association reveals unique thermodynamic signatures. Pharm Res. 2021;38(2):243–55.
Hopkins MM, Lambert CL, Bee JS, Parupudi A, Bain DL. Determination of interaction parameters for reversibly self-associating antibodies: a comparative analysis. J Pharm Sci. 2018;107(7):1820–30.
Ross PD, Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry. 1981;20(11):3096–102.
Hayes D LT, Philo J. Program SEDNTERP: Sedimentation Interpretation Program. Durham, NH, University of New Hampshire. http://www.jphilo.mailway.com/download.htm. 1995.
Philo JS. Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal Biochem. 2006;354(2):238–46.
Stafford WF, Sherwood PJ. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem. 2004;108(1–3):231–43.
Correia JJ, Wright RT, Sherwood PJ, Stafford WF. Analysis of nonideality: insights from high concentration simulations of sedimentation velocity data. Eur Biophys J. 2020;49(8):687–700.
Esfandiary R, Parupudi A, Casas-Finet J, Gadre D, Sathish H. Mechanism of reversible self-association of a monoclonal antibody: role of electrostatic and hydrophobic interactions. J Pharm Sci. 2015;104(2):577–86.
Parupudi A, Chaturvedi SK, Adão R, Harkness RW, Dragulin-Otto S, Kay LE, et al. Global multi-method analysis of interaction parameters for reversibly self-associating macromolecules at high concentrations. Sci Rep. 2021;11(1):5741.
Arora J, Hickey JM, Majumdar R, Esfandiary R, Bishop SM, Samra HS, et al. Hydrogen exchange mass spectrometry reveals protein interfaces and distant dynamic coupling effects during the reversible self-association of an IgG1 monoclonal antibody. MAbs. 2015;7(3):525–39.
Arora J, Hu Y, Esfandiary R, Sathish HA, Bishop SM, Joshi SB, et al. Charge-mediated Fab-Fc interactions in an IgG1 antibody induce reversible self-association, cluster formation, and elevated viscosity. MAbs. 2016;8(8):1561–74.
Correia JJ. Analysis of weight average sedimentation velocity data. Methods Enzymol. 2000;321:81–100.
Creeth JM, Knight CG. On the estimation of the shape of macromolecules from sedimentation and viscosity measurements. Biochim Biophys Acta. 1965;102(2):549–58.
Yang D, Correia JJ, Stafford Iii WF, Roberts CJ, Singh S, Hayes D, et al. Weak IgG self- and hetero-association characterized by fluorescence analytical ultracentrifugation. Protein Sci. 2018;27(7):1334–48.
Wright RT, Hayes D, Sherwood PJ, Stafford WF, Correia JJ. AUC measurements of diffusion coefficients of monoclonal antibodies in the presence of human serum proteins. Eur Biophys J. 2018;47(7):709–22.
Cole JL, Correia JJ, Stafford WF. The use of analytical sedimentation velocity to extract thermodynamic linkage. Biophys Chem. 2011;159(1):120–8.
LiCata VJ, Liu CC. Analysis of free energy versus temperature curves in protein folding and macromolecular interactions. Methods Enzymol. 2011;488:219–38.
Esfandiary R, Hayes DB, Parupudi A, Casas-Finet J, Bai S, Samra HS, et al. A systematic multitechnique approach for detection and characterization of reversible self-association during formulation development of therapeutic antibodies. J Pharm Sci. 2013;102(1):62–72.
Gilbert GJR. Boundary problems in the sedimentation and electrophoresis of complex systems in rapid equilibrium. Nature. 1956;177:853–4.
Philo JS. Characterizing the aggregation and conformation of protein therapeutics. Am Biotechnol Lab. 2003;3(21):22–6.
Fleming PJ, Correia JJ, Fleming KG. Revisiting macromolecular hydration with HullRadSAS. Eur Biophys J. 2023. https://doi.org/10.1007/s00249-022-01627-8
Horn JR, Brandts JF, Murphy KP. van’t Hoff and calorimetric enthalpies II: effects of linked equilibria. Biochemistry. 2002;41(23):7501–7.
Horn JR, Russell D, Lewis EA, Murphy KP. Van’t Hoff and calorimetric enthalpies from isothermal titration calorimetry: are there significant discrepancies? Biochemistry. 2001;40(6):1774–8.
Winzor DJ, Jackson CM. Interpretation of the temperature dependence of equilibrium and rate constants. J Mol Recognit. 2006;19(5):389–407.
Wyman J. Heme Proteins. Adv Prot Chem. 1948;4:407-531.
Wyman J, Gill SJ. Binding and linkage: functional chemistry of biological macromolecules. Mill Valley, CA: University Science Books; 1990.
Sturtevant JM. Heat capacity and entropy changes in processes involving proteins. Proc Natl Acad Sci U S A. 1977;74(6):2236–40.
Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63.
Acknowledgements
We thank Jack Correia and Karl Maluf for insightful conversations, thank Reid Bishop and Jack Correia for sharing of data prior to publication (recently accepted at European Biophysics Journal), and thank Martin Leon for technical assistance.
Funding
MedImmune, LLC (now part of AstraZeneca) supported the above work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures of Financial Interest
No financial interest is declared by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hopkins, M.M., Antonopoulos, I.H., Parupudi, A. et al. Comparative Thermodynamics of the Reversible Self-Association of Therapeutic mAbs Reveal Opposing Roles for Linked Proton- and Ion-Binding Events. Pharm Res 40, 1383–1397 (2023). https://doi.org/10.1007/s11095-023-03485-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-023-03485-1