
Abstract
This study aimed to gain an in-depth understanding of the pulmonary fate of three experimental fluticasone propionate (FP) dry powder inhaler formulations which differed in mass median aerodynamic diameters (MMAD; A-4.5 µm, B-3.8 µm and C-3.7 µm; total single dose: 500 µg). Systemic disposition parameter estimates were obtained from published pharmacokinetic data after intravenous dosing to improve robustness. A biphasic pulmonary absorption model, with mucociliary clearance from the slower absorption compartment, and three systemic disposition compartments was most suitable. Rapid absorption, presumably from peripheral lung, had half-lives of 6.9 to 14.6 min. The peripherally deposited dose (12.6 µg) was significantly smaller for formulation A-4.5 µm than for the other formulations (38.7 and 39.3 µg for B-3.8 µm and C-3.7 µm). The slow absorption half-lives ranged from 6.86 to 9.13 h and were presumably associated with more central lung regions, where mucociliary clearance removed approximately half of the centrally deposited dose. Simulation-estimation studies showed that a biphasic absorption model could be reliably identified and that parameter estimates were unbiased and reasonably precise. Bioequivalence assessment of population pharmacokinetics derived central and peripheral lung doses suggested that formulation A-4.5 µm lacked bioequivalence compared to the other formulations both for central and peripheral doses. In contrast, the other fomulations were bioequivalent. Overall, population pharmacokinetics holds promise to provide important insights into the pulmonary fate of inhalation drugs, which are not available from non-compartmental analysis. This supports the assessment of the pulmonary bioequivalence of fluticasone propionate inhaled formulations through pharmacokinetic approaches, and may be helpful for discussions on evaluating alternatives to clinical endpoint studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Assessing the bioequivalence (BE) of inhalation drugs is challenging. The US Food and Drug Administration (FDA) currently recommends an aggregate weight of evidence approach [1,2,3,4,5]. With blood being downstream of the pulmonary target site, traditional pharmacokinetic (PK) studies are thought to not capture potential differences in lung performance between test and reference products. Thus, FDA recommends comparative clinical BE studies within the aggregate weight of evidence approach. The associated challenges of this approach are especially pronounced for inhaled corticosteroids which exhibit a flat dose response curve.
Therefore, a less complex approval strategy for inhaled drugs, without the need of clinical endpoint studies, would be desirable [1]. A suitable approach should be sensitive to differences in three pulmonary performance attributes: the pulmonary available dose, the duration a drug stays in the lung, and the regional lung deposition. It is generally accepted that PK is sensitive to the first two attributes (i.e., pulmonary dose and pulmonary residence time) [1]. Previous, systematic simulation studies suggested that PK should be able to provide information on differences in the regional lung deposition for slowly dissolving drugs [6]. This is based on the hypothesis that more centrally deposited, slowly dissolving drug particles are more efficiently removed by mucociliary clearance (MCC) from the central region of the lung. This scenario was predicted to yield a smaller drug exposure (i.e., area under the plasma concentration time curve, AUC) even if two formulations deposit the same total amount of drug into the lung. In addition, the peak plasma concentration (Cmax) should be sensitive to differences in regional lung deposition as peripheral areas of the lung provide higher permeabilities, and therefore faster drug absorption compared to more central lung regions [7].
To test this hypothesis, we attempted previously to develop three experimental fluticasone propionate (FP) dry powder inhaler (DPI) formulations that only differed in their mass median aerodynamic diameter (MMAD) [7]. However, in vitro tests revealed that these formulations also differed in in vitro based lung doses (e.g., average of ex-throat doses obtained from three anatomical mouth-throat models) and dissolution rates, which made the testing of the hypothesis via non-compartmental analyses (NCA) more challenging. The NCA showed that PK can detect differences in the pulmonary available dose (based on AUC data) and pulmonary residence time. The latter was characterized by Cmax and its timing (tmax), which are affected by the formulation dependent dissolution behavior of deposited particles and by factors determining the regional lung deposition (e.g., MMAD).
The NCA approach provided insights into differences in the regional lung deposition. However, due to challenges with precisely determining the pulmonary deposited dose from in vitro experiments, NCA could not provide definitive conclusions [7]. Such in vitro data were required for lung dose normalization. In addition, the formulations differed not only in MMAD but also in their dissolution kinetics. Therefore, a compartmental simulation based on mean data was not conclusive with respect to NCA analysis being sensitive to regional lung deposition differences [7]
One of the reasons for recommending clinical endpoint studies in the BE assessment of inhalation drugs was the belief that PK cannot provide information on the pulmonary fate of inhaled drugs, especially on the regional lung deposition [1]. The main objective of the present population PK analysis for inhaled FP formulations was, therefore, to assess whether this modeling approach can provide insights relevant to the BE asssessment of inhaled FP by providing information on the regional lung deposition. This information was thought to be gained from population PK analysis, if population PK analysis would suggest a biphasic, pulmonary absorption profile with fast and slow absorption occurring from the peripheral and central portions of the lung, respectively. Such an analysis was meant to support and facilitate discussions on the design and use of alternative BE approaches as potential substitute for clinical endpoint studies.
If a biphasic absorption profile could be identified, popPK based BE analysis might (1) be considered as a stand-alone BE method for assessing regional lung deposition for FP. (2) This approach may facilitate the validation of the less direct NCA analysis for detecting differences in regional deposition; thereby further delineating the potential role that PK (i.e. popPK together with NCA) can play in assessing the regional deposition of inhaled corticosteroids. In addition, (3) the data generated by the top-down population PK approach might allow comparison with bottom-up physiologically-based pharmacokinetic (PBPK) approaches in subsequent publications, thereby facilitating potentially a cross-validation of both approaches. As such, the analysis presented in this paper might be central in evaluating complementary approaches for the BE assessment of inhaled corticosteroids as alternative approval pathways for generic inhalation drugs that do not depend on clinical endpoint studies.
Methods
Experimental
Information on the development of three experimental FP DPI formulations (A-4.5 µm, B-3.8 µm and C-3.7 µm), their in vitro evaluation, and details about the clinical PK study (NCT01966692) have been described previously [7]. In brief, the PK study was a four-way crossover in 24 healthy volunteers with one of the three formulations (C-3.7 µm) being repeated (called: CR-3.7 µm) to assess intra-individual variability. In each study period (i.e., occasion), subjects inhaled the content of five capsules each containing 100 µg FP (i.e., total single dose: 500 µg). Subjects inhaled at least twice from each capsule to ensure complete capsule emptying. The FP plasma concentrations were quantified via LC–MS/MS with a lower limit of quantification of 0.5 pg/mL. The bioanalysis, NCA PK and standard BE assessments of AUC and Cmax data have been described previously [7].
Population Pharmacokinetic Modeling
Structural model – systemic disposition
To develop the structural model, a conservative two-step approach was employed. In the first step, mean concentration–time profiles after IV dosing of FP were obtained from the literature [8] to derive the structural model and estimate the systemic disposition parameters of FP. The average reported plasma concentrations were digitized. For the study by Thorssen et al. [8], FP was dosed as a 10 min IV infusion, and the first reported average cocnetration was at 40 min (i.e. 30 min past end of infusion). For another study by Allen et al. [9], FP was infused over 20 min and frequent, early blood samples were obtained. However, the average concentration plot was reported over 32 h and thus early time points had a finite resolution.
Linear models with one, two or three body disposition compartments were evaluated. After identifying the most suitable disposition model, the population mean estimates of the systemic disposition parameters were fixed (i.e. the three clearances and three volumes of distribution). In a separate analysis, we estimated the systemic disposition parameters from the mean concentration–time profile of another study with IV dosing of FP [9] to confirm the structural model and obtain a second set of systemic disposition parameter estimates. The subsequent population PK analyses to estimate the pulmonary absorption kinetics were performed with both sets of fixed systemic disposition parameters, using primarily the data based on Thorsson et al. [8].
Structural model – absorption
In the second step, we employed population PK modeling to characterize the pulmonary absorption, which was modeled to originate from one or two lung compartments (i.e. presumed to represent central and peripheral lung). Drug input was described by five bolus doses (each comprising 100 µg FP) at 0, 1, 2, 3 and 4 min. Actual times of the blood samples were used for population PK modeling.
For each lung compartment, the extent (i.e. the absorbed dose) and the rate of absorption were estimated. The rate of absorption was described by first-order, Michaelis–Menten, or parallel first-order plus Michaelis–Menten kinetics during model development. In the final stages of modeling, MCC was incorporated as a first-order removal process from the central lung compartment. Inclusion of MCC allowed us to estimate the centrally deposited dose and calculate the resulting centrally absorbed dose based on the half-life of MCC (fixed) and that of pulmonary absorption from central lung. A weighted MCC half-life across the first 13 lung generations of 8.44 h was calculated using the MCC rates of individual generations derived from Eq. 4 by Hofmann and Asgharian [10]. The associated first-order rate constant for MCC was fixed to 0.0821 h−1.
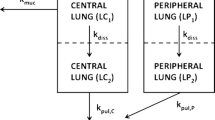
The oral bioavailability of FP is less than 1% [11] and thus much smaller than the bioavailability of FP after inhalation. As orally absorbed FP has a negligible impact on the plasma concentration time profiles [11, 12], oral absorption of swallowed FP was not included and the final model contained two absorption sites and three systemic disposition compartment (Fig. 1).

Population PK model structure which contained a central and peripheral lung compartment, as well as a central, shallow and deep peripheral systemic disposition compartment. All processes followed first-order kinetics. Pulmonary absorption was described by two parallel first-order processes. Mucociliary clearance removed drug from central but not from peripheral lung with a fixed half-life of 8.44 h. Separate parameters were estimated for the bioavailability and absorption half-lives for drug deposited into the central lung (Fc and t1/2,C) and peripheral lung compartment (Fp and t1/2,P). For consistency with in vitro measurements, we report the bioavailability as the deposited doses in μg (based on the total single dose of 500 μg). The rate of absorption from central lung was assumed to be slower than that from peripheral lung. The lung-related parameters were estimated separately for each formulation, whereas the systemic disposition parameters were shared across formulations.
Parameter variability model
For all PK parameters, between subject variability (BSV) and between occasion variability (BOV) were estimated and described by log-normal distributions, with exception of the covariate effect parameter for the peak inspiratory flow rate (PIFR), which was described by a normal distribution. The experimental replication of formulation C-3.7 µm as CR-3.7 µm allowed us to estimate the intra-individual variability (also called BOV). We focused on estimating BOV for lung specific parameters, i.e. the extents and half-lives of absorption from central and peripheral lung. Both BSV and BOV were estimated jointly over all three formulations, since only one formulation was replicated. For systemic disposition parameters, BOV was not included, since these parameters tend to have a small BOV in healthy volunteers.
Covariate effects
We included allometric scaling by total body weight with a reference weight of 70 kg, and fixed exponents of 0.75 for clearances and 1.0 for volumes of distribution [13,14,15,16]. Exploratory covariate analyses were performed by plotting individual PK parameter estimates against covariate values (e.g. for age, sex, height, and observed inhalation parameters).
During each inhalation via the capsule-based DPI device (RS01 Monodose, Plastiape), a complete inhalation profile was recorded [7]. Four parameters were obtained from these inhalation profiles, including the inhalation volume, inhalation time (i.e. duration), peak inspiratory flow rate (PIFR), and time to peak inspiratory flow rate. Informed by empirical covariate effect plots, we evaluated an effect of PIFR on the extents of absorption from central and peripheral lung. The exponents (i.e. PowP for peripheral and PowC for central lung) of these potential covariate effects were estimated using a reference PIFR of 120 L/min (i.e. the median PIFR in the study). The associated covariate effect on the extent of absorption from peripheral lung was calculated as (PIFRi,j / 120 L/min)PowP for the ith subject at the jth occasion (i.e. study period). A positive estimate of PowP indicates a larger extent of absorption from peripheral lung with increasing PIFR, and vice versa.
Estimation and observation model
All FP plasma concentrations were simultaneously fit via population PK modeling (with the mean systemic disposition parameters being fixed) using the importance sampling algorithm (pmethod = 4) in the parallelized S-ADAPT software (version 1.57) [13] facilitated by the SADAPT-TRAN tool [14, 15]. A combined additive plus proportional residual error model was employed.
Model evaluation
Population PK models were compared based on their objective function (negative log-likelihood in S-ADAPT), standard diagnostic plots, and the plausibility of parameter estimates. To assess predictive performance, visual predictive check (VPC) and normalized prediction distribution error (NPDE) plots were used [16, 17].
Parameter uncertainty
Relative standard errors were computed via the importance sampling algorithm [13]. To obtain even more robust uncertainty estimates, a nonparametric bootstrap [18] was performed. In total, 200 datasets were created by random sampling of 24 subjects (with replacement) from the original dataset. Each of these bootstrapped datasets was analyzed using the same approach as that used to estimate the final population PK model. Based on these 200 bootstrap replicates, the median and nonparametric 95% confidence intervals were computed (i.e. 2.5th to 97.5th percentiles) [19, 20].
Validation of biphasic lung absorption model structure
A parametric Monte Carlo simulation estimation study was performed based on the final model with biphasic lung absorption. This simulation included all BSV and BOV terms. This allowed us to assess bias and precision of the model parameters and to determine whether a biphasic absorption could be correctly identified as the true model used during simulations [15, 19]. The distribution of total body weights was simulated via a normal distribution with a mean of 64.56 kg and a standard deviation (SD) of 6.81 kg. The PIFR was simulated with a mean of 120.1 L/min, an SD of 17.6 L/min for BSV, and an SD of 5.7 L/min for BOV.
For each simulated dataset, the objective function and parameter estimates were compared between the final model with biphasic lung absorption and a simpler monophasic lung absorption model. These Monte Carlo simulations (code provided in Supplement) were performed in the Berkeley Madonna software (version 8.3.18, University of California, Oakland, CA). Each simulated dataset contained PK data of 24 healthy volunteers for a four-way crossover study with three DPI formulations using the same design as that in the PK study (i.e., three formulations, with one formulation being repeated, as well as the same sampling times and same doses) [10]. The population means of the systemic disposition parameters were fixed to their true means (obtained from IV PK data). All remaining parameters were estimated simultaneously.
In total, 200 datasets with 24 subjects (and 96 profiles) each were randomly simulated and analyzed by population PK starting with different initial estimates for the absorption parameters compared to those of the final (i.e. true) parameter values. For the systemic disposition parameters, the true (fixed) parameter values were used as initials. In an additional analysis, we assessed the impact of poor initial estimates for 52 of these 200 datasets. This analysis yielded very similar results (not shown). Bias was calculated as the ratio of the mean of the parameter estimates from all 200 replicates divided by the true parameter value used during simulation [21]. Imprecision was computed as the coefficient of variation (CV) of all estimated replicates.
Bioequivalence for central and peripheral lung
The estimated individual extents of drug deposition in central (Fc) and peripheral lung (Fp) were obtained based on the individual subject parameter estimates, while accounting for BSV and BOV, as well as the covariate effects of PIFR on Fp and of WT on clearance and volume of distribution. Based on the pulmonary physiology, MCC is lacking in the peripheral lung and was thus not incorporated in the peripheral lung compartment. Therefore, the extent of absorption from peripheral lung was identical to the extent of drug deposition. However, due to MCC from central lung, the extent of absorption from central lung was calculated according to the MCC half-life and the individual absorption half-lives from central lung for each subject. These individual extents of drug deposition and absorption were used to perform statistical BE analyses. We used the posterior modes (called HOC estimates in the S-ADAPT software) as individual estimates for each subject and occasion.
For average BE testing among all the formulations, linear mixed-effects models were estimated in the Phoenix WinNonlin™ Professional software (Certara, Princeton, NJ, USA). These analysis of variance (ANOVA) models on natural log-scale contained effects for formulation, period, sequence and subject nested within sequence. All pairwise comparisons were performed using the repeat formulation of C-3.7 µm (i.e. CR-3.7 µm) as reference with BE limits of 80 to 125%.
Results
Disposition after IV dosing
When fitting the average concentration profiles after IV dosing from the study by Thorsson et al., a model with three disposition compartments was significantly superior (p < 0.01, likelihood ratio test) to models with one or two disposition compartments. The systemic model parameters were estimated based on either using the entire dataset or using the PK observations up to and including 24 h. For the latter IV dataset, the estimated systemic clearance (72.2 L/h for subjects with 70 kg total body weight) was plausible and close to liver blood flow, volume of distribution at steady-state (i.e. Vdss; 468 L) was within the published range, and the terminal half-life (10.6 h) was close to clinically reported values [8] (Table I). The typical disposition half-lives were 13.4 min for the alpha, 1.30 h for the beta, and 10.6 h for the gamma phase. For the IV dataset from Allen et al. [9], these disposition half-lives were 6.41 min, 1.53 h, and 12.9 h.
Integrating IV deposition parameters into population PK
During population PK modeling, the population means of these systemic disposition parameters (Table I) were fixed to the estimates shown above. However, we allowed the individual subject values to deviate from their population means by including BSV for each systemic disposition parameter. This improved the robustness of the estimation, since only two pulmonary absorption half-lives were estimated and the means of the three disposition half-lives were fixed. When calculated using the individual subject estimates based on the study by Thorsson et al. [8], the alpha half-life was 6.35 min, beta half-life 1.11 h, and gamma half-life 11.6 h. When using the alternate dataset by Allen et al. [9], the three systemic disposition half-lives during population PK modeling were 3.77 min, 1.24 h, and 13.3 h. Thus, consistent across both analyses, population PK estimated a faster alpha half-life compared to that obtained from fitting the mean IV PK profiles, mostly due to a larger estimate for the distribution clearance to the shallow peripheral compartment (CLdshallow). Without fixing any of the mean disposition parameters, population PK modeling estimated half-lives of 6.1 min for the alpha, 0.75 h for the beta, and 11.5 h for the gamma phase. In an additional population PK analysis where CLdshallow was estimated and the other five mean disposition parameters were fixed, the half-life was 5.5 min for the alpha, 1.1 h for the beta, and 11.5 h for the gamma phase. The latter analysis yielded very similar results (not shown) for all absorption parameters compared to those of the final model, suggesting that the formulation comparisons were robust with regard to handling of CLdshallow.
The estimated BSV had CVs of 17.4% for total clearance (CL) and 33.9% for the volume of the central compartment (V1, Table I). The BSV for CLd had an estimated CV of 92.2% and was eventually fixed to this value. This improved the robustness of the estimation, potentially because population modeling estimated a faster alpha half-life than that obtained by fitting average profiles after IV dosing. The BSV of the other systemic disposition parameters was small and eventually fixed to a CV of 10% as a pharmacologically plausible value, which did not affect the conclusions.
Population PK model structure and diagnostic plots
Separating the lung into a compartment with slower absorption, presumably from central areas of the lung, and a compartment with rapid absorption from peripheral areas of the lung, significantly improved the log-likelihood (p < 0.01), curve fits and predictive performance (Fig. 1). Inclusion of saturable absorption kinetics from either lung compartment yielded no improvement. We did not consider models with three parallel absorption processes, since the biphasic model provided good fits and was robust. The visual predictive checks (Fig. 2) and normalized prediction distribution error plots (Fig. 3) demonstrated reasonable predictive performance. The observed vs. individual or population fitted concentrations (Figure S1) also supported adequate model performance, keeping in mind that the disposition parameters were fixed to estimates after IV dosing. The individual curve fits were excellent and population fits were good for the more slowly absorbed formulation A-4.5 µm. However, the population fits slightly over-predicted the observed peak concentrations for the other formulation. This occured because the individual estimated alpha half-lives were faster than their fixed population mean (from IV data of another study). This neither affected the individual PK parameter estimates nor the BE analysis, which was based on the individual estimates. We further used NCA to calculate the AUC0-last for the individual model fitted and observed plasma concentrations. The AUC0-last based on the fitted concentrations divided by the AUC0-last based on the observations had a median of 0.99 for all formulations. The individual AUC0-last ratios across all 96 profiles fell between 0.95 and 1.07, indicating a close fit for the drug exposure of all profiles.

Visual predictive checks for fluticasone propionate concentrations in plasma for each formulation. The insets show the first 2 h on linear scale. Ideally, the median of the predicted profiles should match the central tendency of the observations and 10% of the observations should fall outside of the 80% prediction interval (i.e. the 10th to 90th percentiles) on either side. Formulation C-3.7 μm was repeated (as CR-3.7 μm).

Normalized prediction distribution errors (NPDEs) for each formulation (formulation is split into its two replicate doses C and CR). Ideally, the NPDEs should be standard normally distributed with a mean of zero and standard deviation of 1 (i.e. approximately 95% of the NPDEs should fall within -2 and + 2 at each time point). Time past dose is plotted on logarithmic scale to better visualize the absorption phase. The dashed blue line represents a LOESS smoother of the observations and should ideally fall onto the green zero line.
The median parameter estimates from nonparametric bootstrapping were within ± 10% of the estimates from the original dataset for all population mean, BSV and BOV estimates (Table I). The population means of all pulmonary absorption parameters were reasonably precise with relative standard errors (SE) below 30%. Exceptions were an SE of 36.4% for the amount deposited in central lung for A-4.5 µm, 46.9% for the absorption half-life from central lung for B-3.8 µm, and 31.4% for the covariate effect of PIFR. The covariate effect of PIFR on the amount of drug deposition in peripheral lung was significant, because the 95% confidence interval for PowP did not include zero; Table I).
In the Monte Carlo simulation-estimation studies, all 95% confidence intervals for the ratio of the estimated by the true parameter values included 1.0, indicating unbiased estimates (Table II). For all population mean PK parameters (Table III), the mean of the estimated divided by true parameter values were within 0.85 to 1.14 and imprecision was better than 20% (except for 39% for PowP). For the BSV and BOV estimates, these ratios ranged from 0.84 to 1.29. Thus, all model parameter estimates were unbiased and had adequate precision.
When comparing the true model with biphasic to the simplified model with monophasic lung absorption, the -2 × log-likelihood significantly favored the biphasic model by 747 [503 to 940] (median [min–max]; p < 0.001 for all 200 pairwise comparisons; likelihood ratio test). The individual curve fits were significantly better for the biphasic model (proportional residual error, SDsl: median [95% confidence interval] 0.162 [0.154 to 0.171] for the biphasic vs. 0.194 [0.180 to 0.206] for the monophasic model), as shown by the non-overlapping confidence intervals. Moreover, the population fits and the NPDE plots showed systematic bias for the monophasic but not for the biphasic model. Taken together, the biphasic model could be reliably and clearly distinguished from a monophasic absorption model.
Absorption from lung
The final model identified two parallel first-order absorption processes. The slow process was likely associated with absorption from central lung, whereas the fast process was presumably due to absorption from more peripheral areas of the lung. (Fig. 1). The rapid absorption was 38- to 62-fold faster than the slow absorption (Table I), depending on the formulation. For both processes, formulation A-4.5 µm was absorbed more slowly than formulations B-3.8 µm and C-3.7 µm, in agreement with differences observed in the in vitro dissolution rates [7].
Based on the fixed value for MCC, the centrally deposited dose of 61.9 µg for formulation A-4.5 µm was considerably larger than that of formulation B-3.8 µm (40.5 µg) and C-3.7 µm (35.6 µg; Table I). In contrast, the amount of FP deposited and absorbed from the peripheral lung was substantially smaller for A-4.5 µm (12.6 µg) compared to B-3.8 µm (38.7 µg) and C-3.7 µm (39.3 µg; Table I). The total deposited doses were, however, similar for all three formulations (74.5 µg for A-4.5 µm; 79.2 µg for B-3.8 µm and 74.9 µg for C-3.7 µm). Considering the MCC and the absorption half-life from central lung, 48.4% ± 15.4% (average ± SD) of the centrally deposited dose was absorbed into the systemic circulation for formulation A-4.5 µm. These estimates were slightly higher (54.0% ± 15.4% for B-3.8 µm and 54.9% ± 15.2% for C-3.7 µm) due to the faster dissolution rates of the latter formulations [7].
The variability of the deposited doses was relatively small for an inhaled PK study. The deposited dose in central lung has a small BSV (10% CV; fixed) and a BOV with a CV of 20.2%. For peripheral lung, the BSV was 14.9% and BOV 26.8% (Table I). In contrast, the absorption half-lives were much more variable with a BSV of 70.3% for central lung as well as a BSV of 51.3% and BOV of 52.4% for peripheral lung.
Covariate effects
There was a trend of larger PIFR resulting in larger doses deposited and absorbed from peripheral lung, which was confirmed by population PK modeling. The positive correlation of PIFR with the amount deposited peripherally was described by a positive PowP (estimate: 0.680). The -2 × log-likelihood improved by 7.54 (p = 0.006, likelihood ratio test) and the 95% confidence interval for PowP excluded zero (Table I), indicating a statistically significant covariate effect. Moreover, including PowP explained 45% of the BSV (on variance scale) for the amount deposited in peripheral lung (i.e. the CV decreased from 20.1% without PowP to 14.9% with PowP). The other BSV and BOV estimates were only minimally affected by PowP. In contrast, this covariate effect was lacking for central lung (i.e. PowC was close to zero; results not shown).
Bioequivalence for central and peripheral lung
The BE results based on the individual subject estimates from population PK indicated that the total deposited dose was bioequivalent among all formulations, whereas the total absorbed dose was not (Table IV). When using formulation CR-3.7 µm as reference, formulations B-3.8 µm and C-3.7 µm were bioequivalent for the doses deposited in and absorbed from central and peripheral lung. In contrast, formulation A-4.5 μm was non-bioequivalent for the centrally and peripherally deposited doses, and for the total absorbed dose. The central to peripheral deposition ratio [i.e. C/(C + P)] was considerably larger and non-bioequivalent for formulation A-4.5 μm, and bioequivalent among the other formulations (Table IV).
Discussion
Based on PK data in our previous paper [7], the present population PK analysis provided in-depth insights into the rates and extents of FP deposition and absorption presumably from central and peripheral regions of the lung for three investigational DPI formulations differing in median mass aerodynamic diameter (MMAD).
As the present study lacked data after IV administration of FP, we employed a two-step modeling approach by first obtaining the disposition parameters after IV dosing from two published studies, similar to the approach taken for tiotropium and olodaterol [22, 23]. This provided the population means of the alpha, beta and gamma half-lives after IV dosing of FP. During the second step, we fixed the mean disposition parameters (and thus the three half-lives) during population modeling, but allowed for BSV for all model parameters. Our population PK analysis estimated a faster alpha half-life than that obtained from fitting the average plasma concentrations after IV dosing using the Thorrsson data [12]. This affected the population predictions (Figure S1), but did not impact the lung absorption parameter estimates as shown in additional analyses. The individual curve fits were excellent and all 96 individually fitted vs. observed drug exposures (AUC0-last) were very similar. Thus, the individual absorption parameter estimates, including the pulmonary doses (Table I) used for BE testing (Table IV), were robust. Consistent results were obtained when using an alternative IV PK study [9]. This suggested that the faster alpha half-life and larger CLd did not affect the conclusions and was likely caused by the limited resolution of the reported average plasma concentrations in the IV PK studies [9].
Without integrating IV data into the overall modeling strategy, estimating all five half-lives for a model with biphasic lung absorption resulted in uncertainty in estimating the two absorption and three disposition half-lives for FP, as a slowly absorbed lipophilic drug (similar to flip-flop kinetics). Ideally, an additional IV treatment arm should be included in future cross-over design studies [24], especially if an IV formulation is available. Pulmonary absorption was best described by two parallel first-order processes (Fig. 1 and Table I). The parametric Monte-Carlo simulation estimation study correctly identified the biphasic model as the true one in 200 of 200 cases. Moreover, parameter estimates were unbiased and reasonably precise (Table II). This highlighted the ability of population PK to identify a biphasic lung absorption for FP and provide detailed insights into pulmonary drug absorption. The ability of population PK to distinguish between multiple absorption pathways was in agreement with reports for other inhaled drugs. Bartels et al. identified a slow, moderate, and rapid absorption process from the lung for glycopyronium, in addition to drug absorption from the gastrointestinal tract [24]. Similarly, population PK modeling could identify parallel absorption processes for tiotropium [22, 23] and olodaterol [23]. The lack of reports on the biphasic absorption of FP might be related to these studies focusing on identifying covariates for systemic exposure and characterizing the slow absorption component [25]. Further, this might be related to the systemic rate constants not being restricted during parameter estimations in these publications [26].
We identified a slight positive correlation of PIFR with the amount deposited in peripheral lung. This effect was statistically significant and improved the model performance, but tended to be small, in agreement with the rather homogenous subject population, extensive inhalation training by each subject, and carefully standardized dosing procedures. It might be speculated that increased PIFR results in a more efficient deagglomeration of particles, leading to slightly higher peripheral deposition.
We assumed that the fast absorption of FP is related to absorption from the peripheral lung and that the slow absorption refers to central lung regions. The structural model incorporated a first order removal of centrally deposited drug (Fig. 1) to represent MCC that removed approximately half of the centrally deposited dose. This allowed us to estimate the deposited doses and calculate the absorbed doses from central lung. The MCC half-life was calculated based on Hofmann and Asgharian’s proposed relationship between mucus velocity and airway diameter [10]. The weighted estimates across relevant airway generations agreed with the value of 8.75 h by Boger et al. [27]. The MCC half-life was fixed (without BSV) to support parameter estimation, because MCC and the centrally deposited dose were correlated (as observed in a sensitivity analysis; results not shown). This allowed us to identify the centrally deposited and absorbed doses.
The total deposited pulmonary doses estimated through population PK analysis (Table III) agreed overall with the in vitro estimates (average of VCU, OPC and Alberta throats). However differences between formulations, estimated by population PK were less pronounced than those based on mouth-throat (MT) models [7]. More studies comparing the results from PK based approaches with those derived from in vitro MT models are needed to assess, if in vitro models warrant further refinement. Population PK estimated that formulation A-4.5 µm deposited 82% ± 3.9% of dose centrally, compared to 52% ± 8.0% for B-3.8 µm, 48% ± 8.2% for C-3.7 µm, and 49% ± 9.3% for CR-3.7 µm (Tables III and IV). Thus, the formulation with the largest MMAD deposited more centrally than the other formulations.
Population PK could characterize and estimate multi-phasic pulmonary absorption kinetics. However, this computational approach cannot identify the reasons for biphasic absorption. Considering the physicochemical characteristics of FP and lung physiology, it is likely that the different absorption half-lives are in agreement with the expected fate of a lipophilic drug regarding absorption from more central and peripheral areas of the lung. Rapid absorption is in agreement with dissolution of FP under sink conditions from the peripheral lung, as dissolved drug is efficiently removed across highly permeable and highly perfused alveolar regions [27]. Thus, dissolution is likely rate-limiting for absorption from peripheral lung. Of note, the rank order of in vitro dissolution rates [7] correlated with the absorption half-lives from peripheral lung with formulation A-4.5 µm (14.6 min) showing a slower absorption than the other formulations (6.9 min; Table I).
In contrast, the lack of sink conditions in central lung, where less permeable stuctures and reduced blood flow are observed, is likely the reason for slower dissolution; resulting in 38- to 62-fold slower absorption half-lives from central compared to those from peripheral lung (Table I). However, formulation A-4.5 µm also had the slowest absorption of the three formulations from central lung, suggesting that the dissolution process retained an effect on the overall absorption from central lung. To further corroborate the link between population PK derived absorption half-lives and physiological events, future studies should compare the top-down population PK with bottom-up PBPK modeling.
The early tmax (at 16 to 31 min; from NCA) in relation to the long absorption half-life of the slow absorption process suggests that Cmax is mainly determined by the rate and extent of absorption from peripheral lung. This supports the hypothesis in our original paper [7], that Cmax is likely sensitive to differences in regional deposition. In contrast, the slower absorption from central lung should mainly affect the mean absorption times.
Population PK estimated a small BSV for the amount deposited in central and peripheral lung (< 15% CV). The BOV estimates were larger with CVs of 20.2% for central and 26.8% for peripheral lung (Table I). These were comparable to the inter-subject variability (i.e. BSV) of 33.5% and intra-subject variability (i.e. BOV) of 28.7% for Cmax, and the 24.1% for the inter-subject and 14.6% for the intra-subject variability of AUC from NCA and ANOVA statistics [7]. The centrally and peripherally deposited and absorbed doses both contributed to the variability of AUC and, to some degree, to that of Cmax.
In contrast, population PK estimated CVs of 51.3% for BSV and 52.4% for BOV of the rapid absorption half-life from peripheral lung, and a BSV of 70.3% for the absorption half-life from central lung. These variability estimates were considerably larger than the variability of Cmax from NCA (i.e. 33.5% BSV and 28.7% BOV), suggesting that NCA based Cmax is a more robust parameter for the rate of absorption than the population PK estimated absorption half-lives. Variability in Cmax is a result of multiple PK processes including the drug absorption and distribution kinetics, which may have resulted in a smaller overall variabiltity of Cmax compared to the absorption half-life from peripheral lung. Due to the large variability of the absorption half-lives and the potential issue of distinguishing between absorption and disposition half-lives, our proposed BE strategy only used the individual estimates for the centrally and peripherally deposited doses.
The individual estimates for the centrally and peripherally deposited and absorbed doses from population PK were used to assess the BE of the three formulations. The absolute values of the model-estimated deposited doses may have been affected by the study design (i.e. lack of IV data) and potentially by fixing the disposition half-lives, as well as the MCC to literature estimates. However, the cross-over nature of the PK study ensured that comparisons between formulations were consistent and not affected by these limitations. All dose related parameters (i.e. the centrally and periphally absorbed and deposited doses, total doses, and c/(c + p) ratio; Table IV) were bioequivalent among formulations B-3.8 µm, C-3.7 µm, and CR-3.7 µm. This was in agreement with in vitro and NCA derived PK results (i.e. dissolution behavior, NCA based Cmax and AUC [7]). In contrast, formulation A-4.5 µm was bio-IN-equivalent for all parameters except the total deposited dose (Table IV). For the parameters reflecting the total absorbed doses, BE results agreed with those reported in our previous report for AUC0-inf before dose normalization [7] with formulation A-4.5 µm being bio-IN-equivalent to the other two formulations. This further supported the robustness of the population PK approach for estimating lung doses.
As a top-down approach, population PK characterizes the overall (i.e. rate-limiting) kinetic processes. As an advantage, population PK results are data-driven and not affected by challenges often associated with the validation of bottom-up derived mechanistic input parameters, necessary for PBPK models. Thus, insights from population PK related to the pulmonary fate complement those from PBPK and in vitro approaches, and vice versa. As such, the joint use of in vitro tests [7], PBPK and population PK might provide mutual insights and support cross-validation of the above approaches for FP, and potentially other drugs with similar physicochemical properties. For example, the present study demonstrated the same rank order between the three FP formulations based on population PK regional deposition estimates and the aerodynamic properties.
Standard approaches are available to determine central to peripheral deposition ratios (i.e. c/p ratios) via scintigraphy [28] or deposition modelling [29], which both consider physiological constraints for deriving the c/p ratio. In contrast, population PK modeling approaches estimate c/p ratios by arbitrarily dividing lung absorption into a fast and a slow process. Thus, one would not expect quantitatively matching results between these methods.
The estimated absorption half-lives for the three formulations from central and peripheral lung roughly mirrored the mean dissolution times in vitro, supporting the relevance of dissolution studies for the evaluation of inhalation drugs. This also demonstrated the sensitivity of population PK to detect such differences. Further, the pronounced differences in absorption half-lives from central and peripheral areas of the lung established by population PK indicated that Cmax is predominantly driven by doses deposited in peripheral areas of the lung. Thus, our population PK analysis suggested that NCA derived Cmax estimates should be able to probe for differences in regional lung deposition. This supported our recent hypothesis [7] that traditional NCA-BE analysis should be sensitive to regional lung deposition differences. Similarly, the total deposited lung doses derived from in vitro models (Table III) agreed overall with in vitro estimates; only formulation A-4.5 µm differed by 15% between the two methods (Table III; 64.0 µg vs 75.5 µg), supporting the selected in vitro approach. In addition, future comparisons between population PK parameter estimates and those predicted by PBPK based modeling, as well as the associated in vitro input parameters will be beneficial to cross-validate both modeling approaches.
Our results indicate that population PK analysis might be helpful together with established approaches in BE testing of FP and similar drugs. As a first step, a crossover PK study could be performed to compare the PK profiles of the test and reference products; ideally, PK data should also be obtained in the same subjects after IV dosing (especially if an IV formulation is available or can be created). This study should be analyzed through both standard NCA and population PK analysis. Standard BE statistics could be used to compare the overall extent (AUC0-inf) and rate of pulmonary absorption (Cmax). Population PK would complement NCA analysis by providing information on the drug deposition in central and peripheral lung regions. We do not recommend to include the absorption half-lives into the BE analysis because of the larger variability associated with these half-lives (see above). This approach requires drugs with multi-exponential (i.e. slow and fast) absorption kinetics from the lung. For such drugs, the proposed analysis strategy might be supplemented by additional in vitro data or computational approaches (such as PBPK and/or computational fluid dynamics). This novel, integrated approach might be considered when developing an alternative to clinical endpoint studies.
In summary, the proposed population PK approach could characterize the regional lung deposition by identifying and estimating the absorption of FP presumably from central and peripheral lung. Inclusion of published PK data after IV dosing allowed us to reliably estimate the deposited and absorbed doses in both regions of the lung, which we showed to be unbiased and reasonably precise. Population PK could correctly identify biphasic lung absorption as the true model in 200 of 200 Monte Carlo simulation-estimation study datasets. Population PK based BE results showed bio-IN-equivalence for the formulation with the largest MMAD and BE among all other formulations. This supported the validity of this approach for comparing regional lung deposition between formulations and the ability of population PK based BE to complement standard NCA based BE testing. In addition, the distinct differences in the estimated absorption half-lives of centrally and peripherally absorbed drug further supported our previous hypothesis [7]. As an alternative to the population PK based BE strategy proposed here, NCA based Cmax estimates might be suitable for assessing the BE in regional lung deposition of test and reference products displaying biphasic absorption kinetics.
Data Availability
The raw data for the human PK study were published in AAPS J 2021; 23:48 supported by the US Food and Drug Administration through contracts HHSF223201110117A and HHSF223201610099C and grants 1U01FD004950 and 1U01FD005231. The present population PK analysis did not generate additional experimental data. The population PK modeling andMonte Carlo simulation code is provided in the supplementary materials of the present paper.
References
Hochhaus G, Davis-Cutting C, Oliver M, Lee SL, Lyapustina S. Current Scientific and Regulatory Approaches for Development of Orally Inhaled and Nasal Drug Products: Overview of the IPAC-RS/University of Florida Orlando Inhalation Conference. AAPS J. 2015;17:1305–11.
Lee SL. US regulatory considerations for generic dry powder inhalers. Respiratory drug delivery 2012. Volume 1. In: Dalby RN, Byron PR, Peart J, Suman JD, Young PM, Traini D, editors. River Grove: DHI Publishing; 2012. p. 317–24.
O’Connor D, Adams WP, Chen M-L, Daley-Yates P, Davis J, Derendorf H, et al. Role of pharmacokinetics in establishing bioequivalence for orally inhaled drug products: Workshop summary report. J Aerosol Med Pulm Drug Deliv. 2011;24:119–35.
Adams WP, Ahrens RC, Chen M-L, Christopher D, Chowdhury BA, Conner DP, et al. Demonstrating Bioequivalence of Locally Acting Orally Inhaled Drug Products (OIPs): Workshop Summary Report. J Aerosol Med Pulm Drug Deliv [Internet]. 2010;23:1–29. https://doi.org/10.1089/jamp.2009.0803.
FYs 2013–2017 Regulatory Science Report: Locally-Acting Orally Inhaled and Nasal Drug Products [Internet]. Available from: https://www.fda.gov/drugs/generic-drugs/fys-2013-2017-regulatory-science-report-locally-acting-orally-inhaled-and-nasal-drug-products. Accessed 23 Mar 2023.
Weber B, Hochhaus G. A Systematic Analysis of the Sensitivity of Plasma Pharmacokinetics to Detect Differences in the Pulmonary Performance of Inhaled Fluticasone Propionate Products Using a Model-Based Simulation Approach. AAPS Journal. 2015;17:999–1010 (Springer New York LLC).
Hochhaus G, Chen M-J, Kurumaddali A, Schilling U, Jiao Y, Drescher SK, et al. Can Pharmacokinetic Studies Assess the Pulmonary Fate of Dry Powder Inhaler Formulations of Fluticasone Propionate? AAPS J [Internet]. 2021;23:48. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=33768368&retmode=ref&cmd=prlinks. Accessed 23 Mar 2023.
Thorsson L, Edsbäcker S, Källén A, Löfdahl CG. Pharmacokinetics and systemic activity of fluticasone via Diskus and pMDI, and of budesonide via Turbuhaler. Br J Clin Pharmacol. 2001;52:529–38.
Allen A, Bareille PJ, Rousell VM. Fluticasone furoate, a novel inhaled corticosteroid, demonstrates prolonged lung absorption kinetics in man compared with inhaled fluticasone propionate. Clin Pharmacokinet. 2013;52:37–42.
Hofmann W, Asgharian B. The effect of lung structure on mucociliary clearance and particle retention in human and rat lungs. Toxicol Sci [Internet]. 2003;73:448–56. https://doi.org/10.1093/toxsci/kfg075.
Falcoz C, Oliver R, Mcdowall JE, Ventresca P, Bye A, Daley-Yates PT. Bioavailability of orally administered micronised fluticasone propionate. Clin Pharmacokinet. 2000;39(Suppl. 1):31–9.
Derendorf H, Hochhaus G, Meibohm B, Möllmann H, Barth J. Pharmacokinetics and pharmacodynamics of inhaled corticosteroids. J Allergy Clin Immunol. 1998;101(Supplement):S440–6.
Bauer RJ, Guzy S, Ng C. A survey of population analysis methods and software for complex pharmacokinetic and pharmacodynamic models with examples. AAPS J [Internet]. AAPS J; 2007 [cited 2022 Aug 17];9. Available from: https://pubmed.ncbi.nlm.nih.gov/17408237/. Accessed 23 Mar 2023.
Bulitta JB, Bingölbali A, Shin BS, Landersdorfer CB. Development of a new pre- and post-processing tool (SADAPT-TRAN) for nonlinear mixed-effects modeling in S-ADAPT. AAPS J. 2011;13:201–11.
Bulitta JB, Landersdorfer CB. Performance and robustness of the Monte Carlo importance sampling algorithm using parallelized S-ADAPT for basic and complex mechanistic models. AAPS J [Internet]. AAPS J; 2011 [cited 2022 Aug 17];13:212–26. Available from: https://pubmed.ncbi.nlm.nih.gov/21374103/. Accessed 23 Mar 2023.
Bulitta JB, Okusanya OO, Forrest A, Bhavnani SM, Clark K, Still JG, et al. Population pharmacokinetics of fusidic acid: Rationale for front-loaded dosing regimens due to autoinhibition of clearance. Antimicrob Agents Chemother. 2013;57:498–507.
Brendel K, Comets E, Laffont C, Laveille C, Mentré F. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res [Internet]. Springer; 2006 [cited 2022 Aug 17];23:2036–49. Available from: https://doi.org/10.1007/s11095-006-9067-5
Parke J, Holford NHG, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Methods Programs Biomed [Internet]. Comput Methods Programs Biomed; 1999 [cited 2022 Aug 17];59:19–29. Available from: https://pubmed.ncbi.nlm.nih.gov/10215174/. Accessed 23 Mar 2023.
Jiao Y, Kim TH, Tao X, Kinzig M, Landersdorfer CB, Drescher SK, et al. First population pharmacokinetic analysis showing increased quinolone metabolite formation and clearance in patients with cystic fibrosis compared to healthy volunteers. Eur J Pharm Sci. 2018,123:416–28.
Bulitta JB, Duffull SB, Kinzig-Schippers M, Holzgrabe U, Stephan U, Drusano GL, et al. Systematic comparison of the population pharmacokinetics and pharmacodynamics of piperacillin in cystic fibrosis patients and healthy volunteers. Antimicrob Agents Chemother [Internet]. Antimicrob Agents Chemother; 2007 [cited 2022 Aug 17];51:2497–507. Available from: https://pubmed.ncbi.nlm.nih.gov/17485505/. Accessed 23 Mar 2023.
Jiao Y, Kim TH, Tao X, Kinzig M, Landersdorfer CB, Drescher SK, et al. First population pharmacokinetic analysis showing increased quinolone metabolite formation and clearance in patients with cystic fibrosis compared to healthy volunteers. Eur J Pharma Sci. 2018;123:416–28 (Elsevier B.V.).
Weber B, Borghardt JM, Parra-Guillen ZP, Sharma A, Retlich S, Staab A, et al. Tiotropium pharmacokinetics and pharmacodynamics – What are drivers for systemic levels and local pulmonary responses? Respiratory Drug Delivery 2016. Volume 1. In: Dalby RN, Byron PR, Peart J, Suman JD, Young PM, Traini D, editors. River Grove: DHI Publishing; 2016. p. 45–54.
Borghardt JM, Weber B, Staab A, Kunz C, Formella S, Kloft C. Investigating pulmonary and systemic pharmacokinetics of inhaled olodaterol in healthy volunteers using a population pharmacokinetic approach. Br J Clin Pharmacol. 2016;81:538–52 (Blackwell Publishing Ltd).
Bartels C, Looby M, Sechaud R, Kaiser G. Determination of the pharmacokinetics of glycopyrronium in the lung using a population pharmacokinetic modelling approach. Br J Clin Pharmacol. 2013;76:868–79.
Brindley C, Falcoz C, Mackie AE, Bye A. Absorption Kinetics after Inhalation of Fluticasone Propionate via the Diskhaler ® , Diskus ® and Metered-Dose Inhaler in Healthy Volunteers. Clin Pharmacokinet. 2000;39(Suppl. 1):1–8.
Soulele K, Macheras P, Silvestro L, RizeaSavu S, Karalis V. Population pharmacokinetics of fluticasone propionate/salmeterol using two different dry powder inhalers. European Journal of Pharmaceutical Sciences Elsevier. 2015;80:33–42.
Boger E, Evans N, Chappell M, Lundqvist A, Ewing P, Wigenborg A, et al. Systems Pharmacology Approach for Prediction of Pulmonary and Systemic Pharmacokinetics and Receptor Occupancy of Inhaled Drugs. CPT Pharmacometrics Syst Pharmacol [Internet]. 2016;5:201–10. https://doi.org/10.1002/psp4.12074.
Biddiscombe MF, Meah SN, Underwood SR, Usmani OS. Comparing lung regions of interest in gamma scintigraphy for assessing inhaled therapeutic aerosol deposition. J Aerosol Med Pulm Drug Deliv [Internet]. 2011;24:165–73. https://doi.org/10.1089/jamp.2010.0845.
Asgharian B, Hofmann W, Bergmann R. Particle deposition in a multiple-path model of the human lung. Aerosol Sci Technol [Internet]. 2001;34:332–9.
Acknowledgements
We thank Drs. Denise Conti, Oluwamurewa (Murewa) Oguntimein, Renishkumar Delvadia, and other FDA colleagues for many fruitful discussions and their many contributions while executing the PK, bioanalytical, and NCA parts of this project. This work has been in part presented at the Drug Delivery to the Lungs conference, Edinburgh, UK in 2018 and the Respiratory Drug Delivery conference virtually in 2020. The DPI formulation development, dissolution testing and PK study, as described elsewhere [10], were supported by FDA contracts HHSF223201110117A and HHSF223201610099C, and by grants 1U01FD004950 and 1U01FD005231 (to GH and JBB). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. Food and Drug Administration (FDA).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors have no conflict on interest for the work presented in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Drescher, S.K., Jiao, Y., Chen, MJ. et al. Central and peripheral lung deposition of fluticasone propionate dry powder inhaler formulations in humans characterized by population pharmacokinetics. Pharm Res 40, 1177–1191 (2023). https://doi.org/10.1007/s11095-023-03472-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-023-03472-6