
Abstract
Purpose
To understand the role of different surfactants, incorporated into amorphous solid dispersions (ASDs) of ritonavir and copovidone, in terms of their impact on release, phase behavior and stabilization of amorphous precipitates formed following drug release.
Methods
Ternary ASDs with ritonavir, copovidone and surfactants (30:70:5 w/w/w) were prepared by rotary evaporation. ASD release performance was tested using Wood’s intrinsic dissolution rate apparatus and compared to the binary drug-polymer ASD with 30% drug loading. Size measurement of amorphous droplets was performed using dynamic light scattering. Solid state characterization was performed using attenuated total reflectance-infrared spectroscopy, differential scanning calorimetry and scanning electron microscopy.
Results
All surfactant-containing ASDs showed improvement over the binary ASD. Span 85 and D-α-tocopheryl polyethylene glycol succinate (TPGS) showed complete release with no evidence of AAPS or crystallization whereas Span 20 and Tween 80 showed < 50% release with amorphous amorphous phase separation (AAPS). Span 20 also induced solution crystallization. Sodium dodecyl sulfate (SDS) showed very rapid, albeit incomplete (~ 80%) release. AAPS was not observed with SDS. However, crystallization on the dissolving solid surface was noted. Span 20 and TPGS formed the smallest and most size-stable droplets with ~ 1 µm size whereas coalescence was noted with other surfactants.
Conclusions
Surfactants improved the release performance relative to the binary ASD. Different surfactant types impacted overall performance to varying extents and affected different attributes. Overall, Span 85 showed best performance (complete release, no crystallization/AAPS and small droplet size). Correlation between physicochemical properties and surfactant performance was not observed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With a large number of poorly water soluble drugs already in marketed products and the continued increase of such molecules in the development pipeline, pharmaceutical companies rely on enabling strategies to formulate these molecules to enhance their oral bioavailability (1). Amorphous solid dispersion (ASD) is one such widely employed strategy (2). An ASD ideally consists of a drug(s) molecularly dispersed in a polymer matrix, rendered as an amorphous blend by techniques such as rapid solvent evaporation or melt quenching (3). The amorphous drug possesses a higher thermodynamic activity than the crystalline form which translates into a transient enhancement in solubility, leading to a supersaturated solution, and an increase in diffusive flux across a membrane (4–6). These advantages result in increases in oral bioavailability compared to non-supersaturating formulations (7–10).
The role of the polymer in the ASD is at least twofold: 1) crystallization inhibition of amorphous drug and 2) release rate enhancement (11–13). Since the amorphous drug in the ASD, as well as the supersaturated solutions generated upon dissolution, are metastable relative to the thermodynamically stable crystalline state, such formulations have a propensity to crystallize over time into a less soluble crystalline form, losing the bioavailability advantage (14). Polymers added to the ASD can inhibit crystallization in the solid ASD by kinetic (reduction in molecular mobility) and thermodynamic (reduction in activity of drug in the presence of the polymer) mechanisms (15–18). In the solution state, polymers can inhibit both nucleation and growth of the crystal from supersaturating solutions. The mechanism is mainly purported to be inhibition or reduction in attachment of incoming solute molecules at the nascent solid-solution interface (19). This occurs due to polymer adsorption via non-covalent interactions leading to a physical barrier of polymer chains near the growing solute surface (20).
Enhancement in release of poorly aqueous soluble compounds from ASDs due to molecular level mixing with a hydrophilic polymer is well documented (21, 22). The release rate has been shown to be impacted by the polymer type as well as drug loading in the ASD (23–25). In certain cases, typically at low drug loading, rapid release of ASD components can occur which results in an apparent solution concentrations higher than the amorphous solubility of the drug (23). Drug concentrations higher than the amorphous solubility lead to liquid–liquid phase separation (LLPS) (26). The resultant two-phase system consists of a solution phase with concentration equal to the amorphous solubility of the drug and a dispersed phase comprised of drug present in drug-rich nanodroplets. These nanodroplets can redissolve rapidly thereby serving as a reservoir of drug available to sustain the maximum diffusive flux across a membrane (27). The beneficial effects of LLPS have also been confirmed in vivo wherein it was demonstrated that formulations that lead to generation of nanodroplets showed higher oral bioavailability compared to formulations that did not undergo LLPS (28, 29). Purohit et al. have also demonstrated that solid phase transformations in the ASD can have a dramatic impact on the release profiles (23). Here it was observed that ASDs that underwent water-induced drug-polymer demixing (amorphous-amorphous phase separation, AAPS) showed impeded release and did not form nanodroplets whereas ASDs that remained miscible during the course of dissolution dissolved rapidly and generated nanodroplets. In another study on ritonavir (RTV)-polyvinylpyrrolidone/vinyl acetate (PVPVA) ASD systems, a closer look at the release rate of both drug and polymer showed that ASDs that dissolved rapidly (~ 300 fold faster compared to amorphous drug alone) and underwent LLPS showed congruent (simultaneous) release of drug and polymer (30). The limit of congruency (LoC), which is the maximum drug loading at which congruent release is observed, for RTV-PVPVA ASDs was found to be ~ 25% drug loading (DL). Similar to observations made by Purohit et al., solid state characterization revealed that ASDs above 25% DL underwent AAPS resulting in poor dissolution performance. It is thus posited that rapid release of ASD components with a release rate faster than that observed for neat amorphous drug, accompanied by the formation of nanodroplets, occurs when the drug in the ASD remains sufficiently well dispersed with the polymer throughout the hydration and dissolution process. In this release regimen, the release rate of the ASD components is controlled by the polymer and the drug is carried into the bulk solution due to its association with the polymer.
A low LoC has been observed for a variety of systems prepared with PVPVA as the ASD polymer: 5% for ledipasvir (31), 10% for nilvadipine (32), 15% for cilnidipine (32), and 20% for itraconazole (33). Because DLs above the LoC lead to a rapid decline in release performance, it is desirable to formulate an ASD with a DL below the LoC. However, if a given drug has a low LoC, the resultant low DL in the ASD required for optimum release performance ultimately results in large tablet size or a high pill burden, thus, negatively impacting patient compliance. Hence, strategies to increase LoC are essential to formulate ASDs of all but the most potent compounds, allowing higher drug loadings. In this work, incorporation of surface active agents to increase drug loading/LoC without compromising dissolution performance, as compared to the corresponding binary ASD, was explored. In a previous study, RTV-PVPVA ASDs showed incongruent and very slow drug release at DLs of 30% or above. Using this model system, a hydrophilic additive, polyethylene glycol 3350 (PEG 3350), and surfactants with varying hydrophilic-lipophilic balance (HLB) values were incorporated into the ASD to study their impact on release at a 30% DL. The size and solid-state properties of precipitates present in solution following release were evaluated. Solid state characterization of the partially dissolved tablet surface was also carried out to correlate the observed dissolution performance to the properties of the ASD surface.
Materials
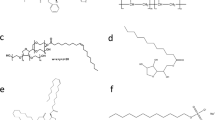
Ritonavir (RTV) was used as provided by AbbVie Inc (North Chicago, IL). Polyvinylpyrrolidone/vinyl acetate (PVPVA) and D-α-tocopheryl polyethylene glycol succinate (TPGS) were supplied by BASF (Ludwigshafen, Germany). Polyethylene glycol 3350 (PEG 3350), sodium dodecyl sulfate (SDS), Tween 80, Span 20, Span 85, dichloromethane and methanol were obtained from Sigma Aldrich Co. LLC (St. Louis, MO). Monobasic sodium phosphate monohydrate and anhydrous dibasic sodium phosphate (Fisher Chemical-Fisher Scientific (Hampton, NH) were used to prepare 50 mM phosphate buffer, pH 6.8 which served as the dissolution medium. 100 mM phosphate buffered saline was prepared using sodium chloride, potassium chloride, anhydrous dibasic sodium phosphate, anhydrous monobasic potassium phosphate (Fisher Chemical-Fisher Scientific (Hampton, NH). Figure 1 and Table 1 present the structures of the materials used and properties of the surfactants employed in this study.

Molecular structures of compounds used in the study
Methods
Preparation of amorphous solid dispersions (ASDs)
Ternary ASDs containing RTV, PVPVA and surfactants were prepared with a ratio of 30:70:5 (RTV:PVPVA:surfactant) by weight. ASD components were dissolved in a dichloromethane:methanol (1:1) mixture at a 20% w/v solids loading and the homogeneous solution was placed in a round bottom flask. ASDs were prepared by solvent evaporation using a Buchi Rotavapor-R210 (New Castle, DE) equipped with a Buchi B-491 water bath maintained at 50 °C and a Buchi V-710 vacuum pump. ASDs were removed from the glassware and secondary dried by placing them overnight in a vacuum oven at room temperature. The ASD powder was then milled and sieved to yield particles in the range of 100 to 150 μm.
Dissolution studies on ASD tablets
Dissolution studies were performed as described in reference (30) using a Wood’s intrinsic dissolution apparatus (IDR) (Agilent technologies, Santa Clara, CA) which consisted of an 8 mm die, a punch, a surface plate and a shaft to mount the die (containing the ASD tablet) onto an overhead stirrer. This setup eliminates variations in surface area by exposing a constant surface area of the dissolving tablet surface to the dissolution medium. 100 mg of powdered ASD was tableted in an 8 mm die by securing it to the surface plate and compressing using a punch with a compression force of 1600 lbs. on a manual Carver press (Carver Inc., Wabash IN). Dissolution studies were performed in 100 mL 50 mM phosphate buffer, pH 6.8 at 37 °C. The die containing the ASD was immersed into the dissolution medium by mounting the die to a shaft, attached to an IKA Eurostar 20 overhead stirrer (IKA Works Inc., Wilmington, NC) with a rotation speed of 100 rpm. 1 mL of dissolution medium was removed at the desired time points to analyze for RTV and PVPVA concentration and replaced with fresh media. No filtration or centrifugation upon sampling was performed because 1) release was observed from an intact tablet due to erosion rather than disintegration into particulates and, 2) to enable assay of drug present in the medium as colloidal species. To understand the effect of wetting by surfactants on ASD dissolution, 50 µg/mL SDS was added to the dissolution medium and an IDR experiment was carried out on a binary ASD with 30% DL.
RTV and PVPVA concentrations were determined by methods described in reference (30). Briefly, reverse phase high performance liquid chromatography was used for RTV. An Agilent 1100 system (Agilent Technologies, Santa Clara, CA) and a 15 cm × 4.6 mm Ascentis® C18 HPLC column (Sigma-Aldrich St. Louis, MO) with 5 μm particle size was used with a mobile phase comprising of 0.1% trifluoroacetic acid in water (aqueous phase, 55%) and acetonitrile (organic phase, 45%) pumped at a flow rate of 1 mL/min. 80 μL was used as the injection volume. RTV was eluted in ~ 3 min and detected using an ultraviolet (UV) detector at a wavelength of 210 nm. A standard curve with a linear regression R2 value of 0.999 was obtained for the concentration range 0.1 to 10 μg/mL. Dilution of the samples with the mobile phase was carried out before HPLC analyses, if necessary, to adjust the concentrations of the analytical samples so that they were within the limits of standard curve.
PVPVA was measured by size exclusion chromatography (SEC) on a 300 × 8 mm A2500 Viscotek column with exclusion limit of 10000 Da for pullulan (Malvern Panalytical, Malvern, UK) using Omnisec Gel Permeation Chromatography (GPC) / Size Exclusion Chromatography (SEC) system (Malvern Panalytical, Malvern, UK). The mobile phase consisted of a 70:30 v/v mixture of 100 mM phosphate buffered saline, pH 7.4 and methanol pumped at a flow rate of 1 mL/min. Under these conditions, PVPVA eluted in ~ 10 min and was detected using an UV detector at a wavelength of 205 nm. A standard curve with linear regression R2 value of 0.999 was obtained for the concentration range 5 to 200 μg/mL. Dilution of the samples with mobile phase was carried out before SEC analysis, if necessary, to adjust the concentrations of the analytical samples so that they are within the limits of standard curve.
Characterization of colloidal solutions upon ASD dissolution
Size measurement by dynamic light scattering (DLS)
The size of the colloidal solutions formed upon dissolution of ASDs during IDR experiments was measured by dynamic light scattering (DLS) technique using a Malvern Zetasizer Nano ZS system (Malvern Instruments Inc., Westborough, MA) equipped with a backscatter detector. The scattering from the particles was collected at 173° angle. Samples were taken based on the dissolution profiles of the ASDs, from the time point when the amorphous solubility of RTV was exceeded.
Optical Microscopy of precipitated phase
Microscope images of the precipitated phase post dissolution was acquired in the case of Tween 80- and Span 20-containing ASDs. Precipitated material was placed on glass slide and observed under 20X magnification using a cross-polarized microscope using an Eclipse E600 POL microscope equipped with a DS Fi1 camera (Nikon Corporation, Tokyo, Japan).
Solid state characterization
Attenuated total reflectance infrared (ATR-IR) spectroscopy
The IDR experiment was performed as described above for the surfactant-containing ASDs and the tablet was ejected from the die at different time points based on the dissolution-time profiles. Excess water was removed from tablet surface by gently blotting with a Kim wipe. The FT-IR spectrum was obtained using a Bruker Vertex 70 FT-IR spectrophotometer (Bruker Corporation, MA, USA) equipped with an ATR accessory (Golden Gate Mk II model single bounce diamond top-plate ATR from Specac Ltd., Cranston, RI, USA). Each spectrum was a co-addition of 64 scans with a scan range from 400 to 4000 cm−1 and a spectral resolution of 4 cm−1. The FT-IR spectrum of crystalline RTV was also obtained for comparison.
Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) was used to determine glass transition temperature (Tg) of the ASDs. Tg was determined pre- and post-dissolution using a Mettler Toledo DSC1 STARe System (Columbus, OH) equipped with refrigerated cooling assembly. Amongst the surfactant-based ASDs, as significantly impaired dissolution was observed for Tween 80 and Span 20, post dissolution characterization was performed on these system. For this analysis, ASD from the exposed surface was carefully scraped and used for analysis. Approximately 10 mg of sample was weighed into Tzero aluminum pans and pans were used without lids. Samples were purged with nitrogen at a flow rate of 50 mL/min for 5 h before beginning the heating cycle to minimize any artifacts from water. Thermal scans were acquired at a heating rate of 5 °C/min from -60 °C to 150 °C.
Scanning electron microscopy (SEM)
SEM was carried out to observe the microscopic morphological transformations on the tablet surface during dissolution. IDR experiments were carried out and tablets were ejected at different dissolution time points as described in previous section followed by overnight vacuum drying. Platinum coating was applied to the tablets by sputter coating using a Cressington 208HR High Resolution Sputter Coater (Cressington Scientific Instruments, Watford, UK). SEM images were obtained using a FEI Nova nanoSEM field emission scanning electron microscope (FEI Company, Hillsboro, Oregon) equipped with a high resolution through-the-lens detector (TLD). An accelerating voltage of 5 kV was used and the sample was focused at a working distance of ∼5 mm working distance with a spot size of 3.
Results
Release studies on ASD tablets
The release profile of 30% DL RTV-PVPVA ASD is shown in Fig. 2 (adapted from reference (30)) for comparison. Here, a lag phase in the dissolution of the ASD components was observed for ~ 15 min with minimal release of both drug and polymer. This was followed by a period of congruent release for up to next 25 min, where both drug and polymer released simultaneously. Subsequently, separate release of drug and polymer was observed with PVPVA releasing at a faster normalized release rate than RTV. At the end of the dissolution experiment, PVPVA was completely released whereas, RTV release was arrested at ~ 50%.

Release profile of 30:70 RTV:PVPVA binary ASD obtained using Wood’s intrinsic dissolution apparatus. Figure reprinted from reference 30
Figure 3 shows the release data for ASDs containing various additives. To evaluate the additive impact, the RTV:PVPVA ratio was held constant at 30:70, similar to the binary ASD described in Fig. 2. PEG 3350, a hydrophilic additive, slightly reduced the lag time but did not significantly impact the dissolution profile of the ASD when compared to the binary ASD. Among the Span series of surfactants, both the more hydrophilic (Span 20) and the more hydrophobic surfactants (Span 85) eliminated the lag phase and resulted in congruent release of drug and polymer. However, the ASD release with Span 20 plateaued at ~ 40% after 30 min whereas, the ASD with Span 85 underwent near complete release of both the components in ~ 60 min. Tween 80 ASDs showed no lag phase and underwent slow dissolution; however, the release plateaued at 30 to 40% for the drug and polymer respectively. TPGS led to complete release, albeit at a slower rate relative to the SDS and Span 85-containing ASDs. SDS, a negatively charged surfactant, eliminated the initial lag phase observed for the corresponding binary RTV-PVPVA ASD, and resulted in rapid and near complete release of both the components in 20 min. Interestingly, after 20 min, RTV and PVPVA release plateaued at 80%. A release study carried out on a binary ASD with 50 µg/mL SDS dissolved in the medium (corresponds to the amount in the solid compact) did not show any impact on dissolution relative to the binary ASD with no surfactant in the medium (data not shown).


Release profiles of 30:70:5 RTV:PVPVA:additive ASDs obtained using Wood’s intrinsic dissolution apparatus
Size of colloidal species formed upon ASD dissolution
Figure 4 shows the size of the droplets formed upon LLPS over the course of dissolution for various ASDs. The time points were chosen based on the release profiles. In general, the size of the nanodroplets increases as the dissolution experiment progresses and the total concentration of RTV increases. In the absence of surfactant, the size of the nanodroplets when the release extent reaches a plateau (RTV concentration of ~ 140 μg/mL) is around 2000 nm. In the presence of Span 20, where a maximum concentration of ~ 160 μg/mL at ~ 40 min is attained, the nanodroplet size is 1000 nm and increases to 1500 nm over time. ASDs prepared with SDS, TPGS and Span 85 underwent near complete release and are shown as closed symbols in Fig. 4. The arrows indicate the time when the maximum concentration is achieved. At a maximum concentration of ~ 240 μg/mL, with SDS, a nanodroplet size of ~ 1100 nm is noted. These species then grow over time to ~ 2500 nm. Particle size is particularly high with TPGS (4000 nm at the end of dissolution) whereas, Span 85 is seen to maintain the particle size at around 1000 nm. Figure 4B shows size data plotted as a function of RTV concentration. It is evident that particles in presence of TPGS are larger than those formed in absence of any surfactant. Particle size is generally smaller and in the range of 1000 nm with other surfactants.

Size determination of colloidal species formed during ASD release. A Stability of colloidal species over time, B Impact of total concentration on size. Arrows denote the time point at which complete release occurred
Figure 5A shows microscope images of the spherical droplets formed with Tween 80 at the end of the release experiment. Due to their large size, this system could not be evaluated by DLS. The particle size with Tween was in the range of 10 μm. Figure 5B shows an image of precipitate acquired upon dissolution of Span 20 ASD. Needle shaped crystals were observed suggesting solution phase crystallization of ritonavir due to the presence of Span 20.

Optical microscope images of precipitated material from ASDs containing (A) Tween 80, (B) Span 20
Solid state characterization
Figure 6 shows SEM image of an SDS-containing ASD tablet surface acquired at 2 min and 15 min post dissolution. At 2 min, the surface of the ASD tablet appears smooth and homogeneous with no evidence of phase transformation whereas the ASD tablet surface at the 15 min timepoint shows needle-shaped crystals. Figure 7 shows the corresponding IR spectra. The spectrum at 2 min is similar to that of the dry ASD suggesting no phase transformation. The spectrum at 15 min differentiates from that of the dry ASD and shows distinct peaks at ~ 1280, 1610 and 1700 cm−1 which correspond to peaks present in crystalline RTV, suggesting crystallization at the surface of the ASD matrix. SEM and IR characterization of the tablet surfaces of ASD with other surfactants did not indicate evidence of crystallization on the ASD surface.

Surface characterization of 30:70:5 RTV:PVPVA:SDS ASD using SEM. A 2 min post dissolution B 15 min post dissolution

Surface characterization of 30:70:5 RTV:PVPVA:SDS ASD using ATR-IR at different timepoints into dissolution. IR spectrum of crystalline RTV is provided for reference. Arrows point to the regions where distinct peaks for crystalline RTV are observed
Figure 8A shows DSC thermograms of dry ASDs before dissolution. All ASDs in dry state showed a single Tg, suggesting miscibility. Figure 8B and C shows the thermogram overlay, before and after exposure to dissolution medium, of Span 20- and Tween 80-containing ASDs respectively. In both cases, a decrease in Tg is observed for the post-dissolution sample. This Tg value is very close to the reported Tg of ritonavir (35). This indicates that phase separation of ritonavir has likely occurred in these ASDs upon contact with water, with the subsequent loss of PVPVA and enrichment of RTV on the surface.


DSC thermograms of ASDs. A dry ASDs, B Span 20 containing ASD before and after dissolution, C Tween 80 containing ASD before and after dissolution
Discussion
Dissolution and phase transformation of ASDs
ASDs are typically comprised of a poorly water-soluble drug dispersed in a hydrophilic polymer matrix. One of the roles of the polymer is to enhance the release rate of the drug from the ASD, relative to the neat amorphous drug. For a binary ASD with non-interacting components, rapid dissolution rates compared to neat amorphous drug alone are typically observed when the drug loading in the ASD is low. In this low drug load regime, the dissolution rate is mainly controlled by the polymer (21, 22, 30), which is advantageous given the large solubility difference between drug and polymer. The highly soluble polymer undergoes rapid dissolution and brings the dispersed drug into solution at the same relative rate, leading to congruent release of the components. In this scenario, the dissolution rate of the drug \(\left({\left(\frac{dC}{dt}\right)}_{drug}\right)\) can be modelled using Eq. 1.
where, \({\left(\frac{dC}{dt}\right)}_{polymer}\) is the dissolution rate of the polymer, \({m}_{drug}\) is the amount of drug and \({m}_{polymer}\) is the amount of polymer in the ASD. As the polymer dissolution rate is faster than the dissolution rate of the drug alone, the resulting apparent concentration of the drug can exceed its amorphous solubility, leading to the formation of drug-rich nanodroplets or amorphous precipitates (LLPS). This was observed for the RTV-PVPVA system. Up to a DL of ~ 25%, polymer-controlled, congruent and complete release of drug was observed (30). The overall release rate of the ASD components was also found to be close to that of neat PVPVA suggesting polymer-driven dissolution. Dissolution from these formulations lead to LLPS and the formation of drug-rich nanodroplets. Further evidence of polymer-controlled release was provided by the observation that initial concentration of drug in the dissolution medium had no impact on the release rate.
In contrast when the drug loading is high, drug-controlled dissolution is observed with a drastically reduced release rate that can be approximated based on the amorphous solubility of the drug using the Nernst-Brunner equation.
In this equation, \({\left(\frac{dC}{dt}\right)}_{drug}\) is the dissolution rate of the drug, that is the change in solution concentration (C) with time (t), Cs is the amorphous solubility of the compound in the medium, C is the concentration attained at time t, D is the diffusion co-efficient, A is the area of the dissolving surface, V is the volume of medium and h is the diffusion layer thickness. The driving force for dissolution is directly dependent on the difference between the amorphous solubility and the bulk concentration. As the bulk concentration reaches the amorphous solubility, drug release is completely halted. This was clearly demonstrated by Indulkar et al., by using RTV-PVPVA ASDs prepared at 10% (polymer-controlled) and 50% (drug-controlled) drug loadings and performing dissolution in a medium saturated at amorphous solubility of the drug. They observed that the polymer-controlled ASD (10% drug loading) underwent complete dissolution and LLPS, whereas the 50% drug-controlled ASD did not release at all (30). Solid state surface characterization of the 50% drug load ASD revealed surface enrichment of the drug. This occurred due to water-induced amorphous-amorphous phase separation (AAPS) in the ASD. This ultimately led to drug-controlled dissolution of both drug and polymer. Due to the poor aqueous solubility of RTV, < 10% of ASD components were released. LLPS was not observed in these ASDs and the dissolution profile followed Eq. 2 (30).
At drug loadings between 30 and 50%, intermediate release behavior was observed for RTV-PVPVA systems. The release was incongruent and incomplete with drug and polymer releasing at separate rates and the observed concentration–time profiles could not be described by the release rate of either neat polymer or neat drug. Similar to the 50% DL ASD, the 30% drug load ASD also underwent AAPS resulting in discrete polymer-rich and drug rich domains. However, the extent of phase separation was not as extensive as for 50% DL ASD. Therefore, the overall dissolution was not completely drug-controlled but instead depended on the composition of individual phases.
Water-induced AAPS is inevitable for hydrophobic drugs in PVPVA especially at high DLs. This occurs due to solubility disparities between the hydrophobic drug and hydrophilic polymer (35–37). Polymer can preferably interact with water, disrupting its interaction with drug, and thus, lead to formation of separate drug and polymer phases (38). Due to its extensive dissociation from polymer, impeded release of large phase separated amorphous drug domains as well as drug crystallization (further decreasing the release rate) can occur (23, 30, 31, 39). Since dissolution, AAPS and crystallization are all kinetic processes that can occur concurrently, their impact on the overall dissolution behavior of an ASD depends on their relative rates. If the release rate is rapid such that the drug is released before extensive AAPS/crystallization can commence, these phase transformations will not impact dissolution. In contrast, if the rate of AAPS is faster than release, incomplete drug release is likely, especially if AAPS leads to contiguous drug-rich domains at the dissolving interface. Thus, the overall dissolution rate of an ASD is a complex balance of these phenomena. To achieve the desirable dissolution performance, an ASD should 1) have a release rate faster than the rate of AAPS and/or crystallization or 2) be resistant to AAPS and crystallization.
As noted earlier, for the polymer-controlled release regime, the amorphous solubility of the drug can be exceeded leading to LLPS. The result is the formation of an amorphous precipitate in the form of droplets. These droplets have been shown to serve as depot of drug during absorption, wherein the uptake of the drug occurs from molecularly dissolved drug with a concentration at the amorphous solubility, whereby the droplets, due to their small size and disordered nature, undergo rapid dissolution to maintain the concentration (and consequently, diffusive flux) at amorphous solubility (27–29). The maximum sustained membrane flux can be maintained until the nanodroplets are consumed. However, their nanosize also makes them susceptible to size enlargement through coalescence over time. An alternate mechanism of particle growth is via Ostwald ripening. Ostwald ripening occurs in polydisperse systems where the local solubility varies with particle size. Small particles with diameters less than ~ 200 nm have higher solubility due to high curvature at the particle interface (Kelvin effect) (40, 41). These particles can redissolve in the medium leading to growth of larger particles. Due to redissolution-growth, the mean particle size increases. The particles increase in volume linearly with time (42). One report by Sun et al., on the physical stability of ritonavir droplets suggests coalescence as the main mechanism for growth of RTV droplets over time wherein, individual droplets (‘singlet”) collide first to form doublet followed by collision of these doublets into larger coalesced droplets (43). The impact of droplet size on in vivo absorption was demonstrated in a study by Kesisoglou et al. using anacetrapib as a model compound (44). The authors demonstrated that formulations that formed large particles showed less absorption (lower AUC and Cmax) relative to those that formed smaller particles. Thus, optimized ASDs should not only release completely but also, upon LLPS, maintain the size of colloidal species to realize the maximum absorption advantage of this formulation strategy (45).
Surfactants and dissolution rate of ASDs
Surfactants have been customarily added to ASDs to improve their dissolution performance. Several marketed ASDs contain surfactants. For example, the commercially available ASDs Kaletra (AbbVie Inc.), Venclexta (AbbVie Inc.) and Incivek (Vertex Pharmaceuticals) contain Span 20 (46), Tween 80 (47) and SDS respectively (48). However, surfactant mechanism of action, particularly on phase behavior and dissolution of ASDs is poorly understood. Based on the findings reported by Indulkar et al. (30), 30% DL RTV-PVPVA ASD (Fig. 2) exhibited a lag phase in dissolution, followed by period of congruent release and ultimately, incongruent and incomplete release of RTV. The ASD was shown to undergo AAPS resulting in poor release of RTV. However, as the amount of phase separated RTV was not sufficient to fully cover the surface, PVPVA underwent complete release. 30% DL was therefore chosen in this study to understand the effect of surfactant on the initial lag phase as well as subsequent release of RTV and PVPVA. The hydrophilic non-surface active additive, PEG-3350 did not significantly alter the release profile of the ASD, suggesting that the presence of a hydrophilic additive per se does not enhance release of ASD components. A study carried out on a binary ASD with ~ 50 μg/mL of SDS (surfactant concentration achieved upon complete ASD dissolution) in the dissolution medium did not show any improvement in release performance, suggesting poor wetting of the dissolving surface is unlikely to underly the observed release behavior. Interestingly, all the surfactants incorporated in the ASD eliminated the initial lag phase and showed congruent release for at least 20 min.
To better understand the effect of surfactant on the release performance, consider Fig. 9 which compares the release rate of PVPVA from the surfactant-containing ASDs and the binary ASD at the LoC (25% DL) for the first 20 min. It is evident that the rate of release of PVPVA from the SDS-containing ASD is approximately twice as fast as from than the binary ASD. This suggests that perhaps SDS enhances performance by increasing the dissolution rate to be faster than the rate of AAPS. Interaction between SDS and PVPVA is well documented (49–51). SDS molecules can form micellar aggregates on the PVPVA chain and this complex may dissolve faster than the polymer alone. Both the Spans and TPGS show a polymer release rate similar to that seen from the binary ASD while Tween 80 shows slower polymer dissolution. Span 85 and TPGS show congruent and complete release of the ASDs without enhancing the dissolution rate compared to the binary ASD. This suggests that these surfactants can potentially enhance performance by modifying either the kinetics of the AAPS process and/or the composition/morphology of the phase separated regions. Thus, the two mechanisms noted in this study by which surfactants can influence dissolution of ASD are by enhancement of dissolution rate over polymer alone and by modifying the characteristics of the AAPS process in the ASD matrix.

Comparison of initial release period of PVPVA from surfactant-containing ASDs with a congruently releasing binary ASD (25:75 RTV:PVPVA)
Surfactants and phase transformations
ASDs with SDS, Tween 80, and Span 20 did not undergo complete dissolution. With SDS, ~ 80% of ASD components were released in ~ 15 min. Beyond this point, the dissolution completely ceased. Solid state characterization of SDS-containing ASD (Figs. 6 and 7) revealed crystallization of the drug at the surface of the undissolved ASD matrix, which likely served as a barrier to further drug release, halting dissolution. Thus, incorporation of SDS is a double-edged sword. On one hand, more rapid dissolution relative to in the binary ASD ensures nearly complete release. On the other hand, SDS promotes RTV crystallization, which occurs before ASD dissolution is complete, leading to incomplete release under the non-sink conditions employed herein. The crystallization-inducing ability of surfactants is well documented. Thermodynamically, surfactants can reduce the interfacial surface energy between the nucleating surface and the bulk, thereby reducing the energy penalty that the system incurs during formation of new surfaces (52, 53, 54). Additionally, surfactants can also impact other factors such as rate of solute attachment, molecular ordering at the interface and can also serve as template for nucleation (55, 56, 57). Due to their plasticization effect, surfactants can lower the glass transition temperature of the ASD matrix thereby increasing the mobility of drug molecules (58, 59). This can lead to an increase in probability of solute–solute rearrangement and eventually crystallization. Based on the current data set, it is difficult to ascertain if AAPS in the ASD preceded crystallization or not. A polymer that is effective in the absence of surfactants at inhibiting crystallization, can be rendered completely or partially ineffective in presence of surfactants. Thus, surfactants can increase the tendency to crystallize even when the ASD is homogeneous. This can be particularly true when the surfactant and polymer interact extensively.
With Tween 80 and Span 20, the release rate diminished dramatically after 30–40 min and < 50% of drug was released. DSC data suggest that these ASDs underwent AAPS upon contact with water resulting in a separate ritonavir-rich phase. The time and concentration at which a plateau in the dissolution profile was observed for these ASDs correlates well with corresponding binary ASD. This suggests that these surfactants may improve the initial wetting and dissolution, thereby removing the lag time; however, they are ineffective at preventing AAPS. Unlike SDS, these surfactants do not increase the dissolution rate compared to polymer alone; therefore, the rate of dissolution rate may be lower than the rate of AAPS and the evolution of the drug-rich phase at the surface, and ultimately these ASDs do not fare any better than the binary ASD at 30% drug loading. Span 20 was also found to induce solution crystallization of ritonavir. Although not observed in this study, surfactants may also induce physical instability (crystallization) in the solid ASD during manufacturing or storage; especially when the drug load in the ASD is high (16, 60).
Surfactants and nanodroplet size
The impact of surfactant type on the droplet size is readily evident from Fig. 4. In the absence of surfactant, the maximum size observed at an RTV concentration of ~ 150 μg/mL is ~ 2000 nm. Based on the observations herein, surfactants can be divided in two categories- surfactant that led to an increase in size > 2000 nm versus those that maintained the droplet size at < 2000 nm. The ASD with Tween 80 reaches a final RTV concentration of ~ 90 μg/mL. However, the droplet size is of the order of 10 μm and greater. TPGS forms fairly large droplets (3–4 μm) at the end of the dissolution experiment. Tween 80 and TPGS could possibly partition into the drug-rich droplets thereby lowering their Tg. These surfactant-loaded drug droplets may then be more susceptible to coalescence than neat drug droplets. SDS-containing ASDs lead to droplets with size ~ 1000 nm at the end of the dissolution experiment which then grow over time to 2400 nm. Both of the relatively hydrophobic surfactants Span 20 and Span 85 maintained the droplet size at ~ 1000 nm. This effect may be due to inhibition of Ostwald ripening (61). These surfactants, due to their poor aqueous solubility, probably mix in the droplet phase and lower the RTV chemical potential, an effect seen for felodipine with the poorly soluble additive, Miglyol (61). As the smaller particles dissolve, the concentration of the surfactant in the droplet increases relative to the concentration in the larger particles. This ultimately results in a balance in the chemical potential or solubilities of different components in the particles of varying sizes and therefore, particle stabilization. Alternatively, Span 20 and 85 may be inhibiting coalescence by steric repulsion.
General comments and relevance
The role of surfactant added to the ASD can be categorized as impact on: 1) release rate 2) AAPS, 3) crystallization and 4) size of nanodroplets formed upon dissolution. Ultimately, all these factors govern release of the drug from the ASD and its subsequent bioperformance. All of the surfactants explored in this study enhanced initial dissolution relative to the surfactant-free ASD. The best initial dissolution rate was obtained with SDS, with the compact reaching near complete dissolution in ~ 15 min whereas ASDs with other surfactants maintained a drug release rate similar to that observed for the surfactant-free ASD. Dissolution in the case of SDS was compromised due to crystallization at a later stage, whereas release from Span 20 and Tween 80 was compromised due to AAPS. The third factor that can also impact absorption is the size of the droplets formed upon LLPS as the ASD dissolves. Although the ASD with TPGS underwent complete dissolution, the droplet size was larger than that obtained using the binary ASD and the largest droplet size was obtained with Tween 80. Amongst all the surfactants, Span 85 fulfils the criteria of an ideal surfactant (resists AAPS and crystallization, and stabilizes the nanodroplets). This ASD undergoes complete dissolution, does not crystallize and is able to maintain a small droplet size. Table 2 summarizes the observed results for the surfactants used in this study.
Conclusions
Congruent and complete release from ASDs is generally observed when the drug loading in the ASD is low. This study demonstrated that addition of a surfactant to an ASD at a drug loading where incongruent and incomplete release was observed had a variable impact on performance. All surfactants eliminated the initial lag phase in release observed with the binary ASD. However, differences were noted in overall release profiles. Ionic surfactants such as SDS may impact ASD dissolution by interacting with the polymer, increasing the overall release rate, whereas non-ionic surfactants may modify phase separation between drug and polymer. Crystallization was promoted in the case of SDS, Tween 80 and Span 20, which led to incomplete drug release from the ASD. Due to the rapid dissolution rate of SDS-containing ASDs, the impact of crystallization was minimal (~ 80% release) compared to Tween 80 and Span 20-containing ASDs (< 50% release). ASDs with TPGS and Span 85 underwent complete and congruent release. Overall, the impact on the release profile was found to be a balance between dissolution rate and the rate/extent of phase transformation (AAPS/crystallization) of the ASDs. The size of the nanodroplets was also influenced by surfactant type. Span-type (hydrophobic) surfactants were seen to maintain nanodroplet size whereas surfactants such as TPGS and Tween 80 led to increased nanodroplet size. The effect of Spans could be related to inhibition of Ostwald ripening whereas the effect of TPGS and Tween 80 may be related to a decrease in glass transition temperature of RTV particles thereby facilitating coalescence.
As demonstrated by this study, drug release and performance of ASD formulations are extremely sensitive to the addition of a small amount of a surface-active excipient. Several mechanisms exist for these surfactants to impact on the formulation performance. They need to be carefully considered and thoroughly examined in order to rationally design formulation with superior performance.
Abbreviations
- ASD:
-
Amorphous Solid Dispersion
- RTV:
-
Ritonavir
- PVPVA:
-
Polyvinylpyrrolidone/vinyl acetate or copovidone
- SDS:
-
Sodium Dodecyl Sulfate
- TPGS:
-
D-α-Tocopheryl Polyethylene Glycol Succinate
- DL:
-
Drug Loading
- IDR:
-
Intrinsic Dissolution Rate
- LoC:
-
Limit of Congruency
- AAPS:
-
Amorphous-Amorphous Phase Separation
- LLPS:
-
Liquid-Liquid phase separation
- DLS:
-
Dynamic Light Scattering (DLS)
- ATR-IR:
-
Attenuated Total Reflectance-InfraRed spectroscopy
- DSC:
-
Differential Scanning Calorimetry
- SEM:
-
Scanning Electron Microscopy
References
Williams HD, Trevaskis NL, Charman SA, Shanker RM, Charman WN, Pouton CW, Porter CJH. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol Rev. 2013;65(1):315–499.
Newman A, Knipp G, Zografi G. Assessing the performance of amorphous solid dispersions. J Pharm Sci. 2012;101(4):1355–77.
Mendonsa N, Almutairy B, Kallakunta VR, Sarabu S, Thipsay P, Bandari S, Repka MA. Manufacturing strategies to develop amorphous solid dispersions: An overview. J Drug Deliv Sci Technol. 2020;55:101459.
Dahan A, Beig A, Ioffe-Dahan V, Agbaria R, Miller JM. The Twofold Advantage of the Amorphous Form as an Oral Drug Delivery Practice for Lipophilic Compounds: Increased Apparent Solubility and Drug Flux Through the Intestinal Membrane. AAPS J. 2013;15(2):347–53.
Hate SS, Reutzel-Edens SM, Taylor LS. Insight into Amorphous Solid Dispersion Performance by Coupled Dissolution and Membrane Mass Transfer Measurements. Mol Pharm. 2019;16(1):448–61.
Hancock BC, Parks M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm Res. 2000;17(4):397–404.
Kennedy M, Hu J, Gao P, Li L, Ali-Reynolds A, Chal B, Gupta V, Ma C, Mahajan N, Akrami A, Surapaneni S. Enhanced Bioavailability of a Poorly Soluble VR1 Antagonist Using an Amorphous Solid Dispersion Approach: A Case Study. Mol Pharm. 2008;5(6):981–93.
Newman A, Nagapudi K, Wenslow R. Amorphous solid dispersions: a robust platform to address bioavailability challenges. Ther Deliv. 2015;6(2):247–61.
Onoue S, Sato H, Ogawa K, Kawabata Y, Mizumoto T, Yuminoki K, Hashimoto N, Yamada S. Improved dissolution and pharmacokinetic behavior of cyclosporine A using high-energy amorphous solid dispersion approach. Int J Pharm. 2010;399(1):94–101.
Shah N, Iyer RM, Mair H-J, Choi D, Tian H, Diodone R, Fahnrich K, Pabst-Ravot A, Tang K, Scheubel E, Grippo JF, Moreira SA, Go Z, Mouskountakis J, Louie T, Ibrahim PN, Sandhu H, Rubia L, Chokshi H, Singhal D, Malick W. Improved Human Bioavailability of Vemurafenib, a Practically Insoluble Drug, Using an Amorphous Polymer-Stabilized Solid Dispersion Prepared by a Solvent-Controlled Coprecipitation Process. J Pharm Sci. 2013;102(3):967–81.
Baghel S, Cathcart H, O’Reilly NJ. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J Pharm Sci. 2016;105(9):2527–44.
Knopp MM, Nguyen JH, Becker C, Francke NM, Jørgensen EB, Holm P, Holm R, Mu H, Rades T, Langguth P. Influence of polymer molecular weight on in vitro dissolution behavior and in vivo performance of celecoxib:PVP amorphous solid dispersions. Eur J Pharm Biopharm. 2016;101:145–51.
Konno H, Handa T, Alonzo DE, Taylor LS. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm. 2008;70(2):493–9.
Purohit HS, Trasi NS, Osterling DJ, Stolarik DF, Jenkins GJ, Gao W, Zhang GGZ, Taylor LS. Assessing the Impact of Endogenously Derived Crystalline Drug on the in Vivo Performance of Amorphous Formulations. Mol Pharm. 2019;16(8):3617–25.
Mistry P, Mohapatra S, Gopinath T, Vogt FG, Suryanarayanan R. Role of the Strength of Drug-Polymer Interactions on the Molecular Mobility and Crystallization Inhibition in Ketoconazole Solid Dispersions. Mol Pharm. 2015;12(9):3339–50.
Rumondor ACF, Stanford LA, Taylor LS. Effects of Polymer Type and Storage Relative Humidity on the Kinetics of Felodipine Crystallization from Amorphous Solid Dispersions. Pharm Res. 2009;26(12):2599.
Konno H, Taylor LS. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci. 2006;95(12):2692–705.
Bhugra C, Pikal MJ. Role of Thermodynamic, Molecular, and Kinetic Factors in Crystallization from the Amorphous State. J Pharm Sci. 2008;97(4):1329–49.
Warren DB, Benameur H, Porter CJH, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J Drug Target. 2010;18(10):704–31.
Raghavan SL, Trividic A, Davis AF, Hadgraft J. Crystallization of hydrocortisone acetate: influence of polymers. Int J Pharm. 2001;212(2):213–21.
Simonelli AP, Mehta SC, Higuchi WI. Dissolution Rates of High Energy Polyvinylpyrrolidone (PVP)-Sulfathiazole Coprecipitates. J Pharm Sci. 1969;58(5):538–49.
Corrigan OI. Mechanisms of Dissolution of Fast Release Solid Dispersions. Drug Dev Ind Pharm. 1985;11(2–3):697–724.
Purohit HS, Taylor LS. Phase Behavior of Ritonavir Amorphous Solid Dispersions during Hydration and Dissolution. Pharm Res. 2017;34(12):2842–61.
Alonzo DE, Zhang GGZ, Zhou D, Gao Y, Taylor LS. Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution. Pharm Res. 2010;27(4):608–18.
Jackson MJ, Kestur US, Hussain MA, Taylor LS. Dissolution of Danazol Amorphous Solid Dispersions: Supersaturation and Phase Behavior as a Function of Drug Loading and Polymer Type. Mol Pharm. 2016;13(1):223–31.
Ilevbare GA, Taylor LS. Liquid-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst Growth Des. 2013;13(4):1497–509.
Indulkar AS, Gao Y, Raina SA, Zhang GGZ, Taylor LS. Exploiting the Phenomenon of Liquid-Liquid Phase Separation for Enhanced and Sustained Membrane Transport of a Poorly Water-Soluble Drug. Mol Pharm. 2016;13(6):2059–69.
Wilson V, Lou X, Osterling DJ, Stolarik DF, Jenkins G, Gao W, Zhang GGZ, Taylor LS. Relationship between amorphous solid dispersion in vivo absorption and in vitro dissolution: phase behavior during dissolution, speciation, and membrane mass transport. J Control Release. 2018;292:172–82.
Stewart AM, Grass ME, Brodeur TJ, Goodwin AK, Morgen MM, Friesen DT, Vodak DT. Impact of Drug-Rich Colloids of Itraconazole and HPMCAS on Membrane Flux in Vitro and Oral Bioavailability in Rats. Mol Pharm. 2017;14(7):2437–49.
Indulkar AS, Lou X, Zhang GGZ, Taylor LS. Insights into the Dissolution Mechanism of Ritonavir-Copovidone Amorphous Solid Dispersions: Importance of Congruent Release for Enhanced Performance. Mol Pharm. 2019;16(3):1327–39.
Que C, Lou X, Zemlyanov DY, Mo H, Indulkar AS, Gao Y, Zhang GGZ, Taylor LS. Insights into the Dissolution Behavior of Ledipasvir-Copovidone Amorphous Solid Dispersions: Role of Drug Loading and Intermolecular Interactions. Mol Pharm. 2019;16(12):5054–67.
Saboo S, Mugheirbi NA, Zemlyanov DY, Kestur US, Taylor LS. Congruent release of drug and polymer: A “sweet spot” in the dissolution of amorphous solid dispersions. J Control Release. 2019;298:68–82.
Bhardwaj V, Trasi NS, Zemlyanov DY, Taylor LS. Surface area normalized dissolution to study differences in itraconazole-copovidone solid dispersions prepared by spray-drying and hot melt extrusion. Int J Pharm. 2018;540(1):106–19.
Rowe RC, Sheskey P, Quinn M. Handbook of pharmaceutical excipients. London: Pharmaceutical Press and American Pharmacissts Association; 2009. p. 551,652,678,764.
Rumondor ACF, Wikström H, Van Eerdenbrugh B, Taylor LS. Understanding the Tendency of Amorphous Solid Dispersions to Undergo Amorphous-Amorphous Phase Separation in the Presence of Absorbed Moisture. AAPS PharmSciTech. 2011;12(4):1209–19.
Purohit HS, Taylor LS. Phase Separation Kinetics in Amorphous Solid Dispersions Upon Exposure to Water. Mol Pharm. 2015;12(5):1623–35.
Qian F, Huang J, Hussain MA. Drug-Polymer Solubility and Miscibility: Stability Consideration and Practical Challenges in Amorphous Solid Dispersion Development. J Pharm Sci. 2010;99(7):2941–7.
Marsac PJ, Rumondor ACF, Nivens DE, Kestur US, Stanciu L, Taylor LS. Effect of temperature and moisture on the miscibility of amorphous dispersions of felodipine and poly(vinyl pyrrolidone). J Pharm Sci. 2010;99(1):169–85.
Rumondor ACF, Marsac PJ, Stanford LA, Taylor LS. Phase Behavior of Poly(vinylpyrrolidone) Containing Amorphous Solid Dispersions in the Presence of Moisture. Mol Pharm. 2009;6(5):1492–505.
Knapp LF. The solubility of small particles and the stability of colloids. Trans Faraday Society. 1922;17:457–65.
Skinner LM, Sambles JR. The Kelvin equation—a review. J Aerosol Sci. 1972;3(3):199–210.
Kabalnov AS, Shchukin ED. Ostwald ripening theory: applications to fluorocarbon emulsion stability. Adv Colloid Interface Sci. 1992;38:69–97.
Sun Y, Deac A, Zhang GGZ. Assessing Physical Stability of Colloidal Dispersions Using a Turbiscan Optical Analyzer. Mol Pharm. 2019;16(2):877–85.
Kesisoglou F, Wang M, Galipeau K, Harmon P, Okoh G, Xu W. Effect of Amorphous Nanoparticle Size on Bioavailability of Anacetrapib in Dogs. J Pharm Sci. 2019;108(9):2917–25.
Taylor LS, Zhang GGZ. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv Drug Del Rev. 2016;101:122–42.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021251s052_021906s046lbl.pdf. Accessed 10 Apr 2020.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Accessed 10 Apr 2020.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203188s022l_207925s003lbl.pdf. Accessed 10 Apr 2020.
Ruckenstein E, Huber G, Hoffmann H. Surfactant aggregation in the presence of polymers. Langmuir. 1987;3(3):382–7.
Nagarajan R. Thermodtnamics of surfactant-polymer interactions in dilute aqueous solutions. Chem Phys Lett. 1980;76(2):282–6.
Shirahama K, Tsujii K, Takagi T. Free-boundary Electrophoresis of Sodium Dodecyl Sulfate-Protein Polypeptide Complexes with Special Reference to SDS-Polyacrylamide Gel Electrophoresis. The Journal of Biochemistry. 1974;75(2):309–19.
König A, Emons H-H. Crystallization kinetics of MgSO4 7 H2O in presence of tenside. Cryst Res Technol. 1988;23(3):319–26.
Sangwal K. Effects of impurities on crystal growth processes. Prog Cryst Growth Charact Mater. 1996;32(1):3–43.
Dugua J, Simon B. Crystallization of sodium perborate from aqueous solutions: II. Growth kinetics of different faces in pure solution and in the presence of a surfactant. J Cryst Growth. 1978;44(3):280–6.
Canselier JP. The effects of surfactants on crystallization phenomena. J Dispersion Sci Technol. 1993;14(6):625–44.
Ginde RM, Myerson AS. Effect of impurities on cluster growth and nucleation. J Cryst Growth. 1993;126(2):216–22.
Rauls M, Bartosch K, Kind M, Kuch S, Lacmann R, Mersmann A. The influence of impurities on crystallization kinetics – a case study on ammonium sulfate. J Cryst Growth. 2000;213(1):116–28.
Ghebremeskel AN, Vemavarapu C, Lodaya M. Use of Surfactants as Plasticizers in Preparing Solid Dispersions of Poorly Soluble API: Stability Testing of Selected Solid Dispersions. Pharm Res. 2006;23(8):1928–36.
Solanki NG, Gumaste SG, Shah AV, Serajuddin ATM. Effects of Surfactants on Itraconazole-Hydroxypropyl Methylcellulose Acetate Succinate Solid Dispersion Prepared by Hot Melt Extrusion. II: Rheological Analysis and Extrudability Testing. J Pharm Sci. 2019;108(9):3063–73.
Baghel S, Cathcart H, O’Reilly NJ. Investigation into the Solid-State Properties and Dissolution Profile of Spray-Dried Ternary Amorphous Solid Dispersions: A Rational Step toward the Design and Development of a Multicomponent Amorphous System. Mol Pharm. 2018;15(9):3796–812.
Lindfors L, Skantze P, Skantze U, Rasmusson M, Zackrisson A, Olsson U. Amorphous Drug Nanosuspensions. 1. Inhibition of Ostwald Ripening. Langmuir. 2006;22(3):906–10.
Acknowledgements
The authors would like to acknowledge AbbVie Inc. for providing an internship opportunity to Anura Indulkar and research funding for this project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
AbbVie and Purdue University jointly participated in study design, research, data collection, analysis and interpretation of data, writing, reviewing, and approving the publication. Anura S. Indulkar was a graduate student at Purdue University. Lynne S. Taylor is a professor at Purdue University. Lynne S. Taylor has no additional conflicts of interest to report. Anura S. Indulkar, Xiaochun Lou and Geoff G. Z. Zhang are employees of AbbVie and may own AbbVie stock.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Indulkar, A.S., Lou, X., Zhang, G.G.Z. et al. Role of Surfactants on Release Performance of Amorphous Solid Dispersions of Ritonavir and Copovidone. Pharm Res 39, 381–397 (2022). https://doi.org/10.1007/s11095-022-03183-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-022-03183-4