
Abstract
Purpose
mRNA has recently emerged as a potent therapeutics and requires safe and effective delivery carriers, particularly prone to address its issues of poor stability and escape from endosomes. In this context, we designed poly(D,L-lactide) (PLA)-based micelles with N-succinimidyl (NS) ester decorated hydrophilic hairy corona to trap/couple a cationic fusogenic peptide and further complex mRNA.
Methods
Two strategies were investigated, namely (i) sequential immobilization of peptide and mRNA onto the micelles (layer-by-layer, LbL) or (ii) direct immobilization of peptide-mRNA pre-complex (PC) on the micelles. After characterization by means of size, surface charge, peptide/mRNA coupling/complexation and mRNA serum stability, carrier cytotoxicity and transfection capacity were evaluated with dendritic cells (DCs) using both GFP and luciferase mRNAs.
Results
Whatever the approach used, the micellar assemblies afforded full protection of mRNA in serum while the peptide-mRNA complex yielded complete mRNA degradation. In addition, the micellar assemblies allowed to significantly reduce the toxicity observed with the peptide-mRNA complex. They successfully transfected hard-to transfect DCs, with a superior efficiency for the LbL made ones (whatever mRNAs studied) showing the impact of the elaboration process on the carrier properties.
Conclusions
These results show the relevance and potential of this new PLA/peptide based micelle platform to improve mRNA stability and delivery, while offering the possibility of further multifunctionality through PLA core encapsulation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
RNA has emerged as a highly potent therapeutics over the last decades [1, 2] whether siRNA/shRNA [3, 4], RNA aptamers [5] and more recently mRNA as vaccines [6,7,8]. However, their rapid degradation by nucleases and difficult escape from endosomes remain major hurdles that still limit their performances. In a very recent review dedicated to nucleic acid vaccines, M.A. Liu states that mRNA stability is one of the key issues that Research and Development efforts have to address to improve potency of the mRNA vaccines [9]. Many research works have been devoted to use of various carriers to protect and deliver RNA [2]. The most currently used approach has relied on RNA encapsulation/complexation in cationic lipid-based nanoparticles, either based on classical lipids such as DOTAP [10,11,12,13] or newly developed ionizable (i.e. pH-sensitive) lipids presenting improved endosomolytic capacity and less potential toxicity [7, 14,15,16,17]. Other reported carriers for RNA complexation and endosomal escape have consisted of cationic compounds/polymers such as polyethyleneimine (PEI) [18, 19], ionizable dendrimers [20], amino-functionalized polyesters [21, 22], histidin/arginin rich amphipatic peptides (e.g., LAH4, RALA) with fusogenic properties [23,24,25], or more recently phosphonium based polymers [26]. However, RNA stability issues still appear as a limiting factor in their biological efficiency. Also, non-degradability, tendency for accumulation in the liver (when injected systemically) and cytotoxicity (related to cationic nature) remain a concern for such carriers [2, 27], especially as they need to be typically used to be used in significant charge excess as compared to nucleic acid for enabling colloidal stability of the complex and successful transfection.
Biocompatible polylactide (PLA) based nanoparticles/micelles have the advantage of safety and degradability and a long history in pharmaceutical use. They remain among the most promising candidates in drug/vaccine delivery, as shown by new formulations still regularly entering clinical development [28]. However, whereas these carriers are highly appropriate and versatile for loading and release of hydrophobic drugs, the delivery of hydrophilic molecules such as DNA/RNA, has remained problematic. The typical approach for loading such drugs in PLA NPs relies on the double emulsion technique (W/O/W). But this technique has limitations, as nucleic acids integrity is significantly affected along the fabrication process (e.g. sonication, water/organic solvent interfaces) and drug loading is quite low [29,30,31,32]. Another problem is that the nucleic acids thus encapsulated loss integrity upon exposure to acidic environment caused by the polymer matrix degradation [30, 33]. Alternative approaches have consisted of post-adsorption of the nucleic acids onto already formed particles after coating with a cationic surfactant/polymer, such as cetyl trimethyl ammonium bromide (CTAB) [34], polylysine [35] or PEI [36]. While avoiding degradation during process, these systems ensure less in vivo protection of surface-adsorbed nucleic acids and can potentially induce its premature release with cationic compound (not covalently bound to the PLA matrix). Original strategies of cationization of PLA for nucleic acid complexation [37] and PLA-DNA/RNA amphiphilic conjugates [38] have been proposed, but possible problems of acid-mediated nucleic acid degradation (upon PLA hydrolysis) and limited nucleic acid protection still remain. Thus, there is still a strong need of developing effective and versatile carriers for delivering RNAs, using solvent-free/water-friendly processes.
In previous works, we have designed a safe, versatile and robust PLA-b-poly(N-acryloxy succinimide-co-N-vinyl pyrrolidone) (PLA-b-P (NAS-co-NVP)) amphiphilic degradable block copolymer platform, able to self-assemble in micelles. The latter allow core/corona functionalization with hydrophobic/hydrophilic molecules, through the hydrophobic PLA core and the presence of N-succinimidyl (NS) ester reactive groups of the NAS units along the corona, while NVP units brought suitable hydrophilic/stealth environment. These nanomicelles afforded core loading with immustimulatory molecules and high surface functionalization density with protein antigens, whose bioactivity was preserved and even improved [39,40,41,42].
In this context, our approach aimed at designing a new mRNA delivery nanoplatform combining both PLA based micelles and cationic fusogenic peptide to afford suitable degradability, mRNA stability and endosomolytic properties for translation. Our strategy relies on coupling of RALA peptide on the NS ester functionalized corona of the micelles, for further trapping mRNA through electrostatic interactions. This carrier is shown to fully protect mRNA from serum nuclease degradation, reduce the toxic effect of the cationic peptide, and promote successful transfection of dendritic cells, making it highly attractive for mRNA delivery, particularly in a vaccine context.
Experimental Section
Materials
The poly(D,L-lactide)-b-poly (N-acryloxysuccinimide-co-N- vinylpyrrolidone) (PLA-b-P(NAS-co-NVP)) block copolymer (12,000 g.mol−1 and 9000 g.mol−1 for PLA and P(NAS-co-NVP), respectively; NAS/NVP molar ratio: 53/47, Ð = 1.7) was synthesized from combination of ring-opening polymerization (ROP) and nitroxide mediated polymerization (NMP), as previously described [40]. RALA peptide (N-WEARLARALARALARHLARALARALRACEA-C sequence, molecular weight of 3327.93 g.mol−1, 98.8% of purity) was obtained from GenScript (Piscataway, USA). H2O purified sterile water was purchased to from OTEC (France). Dulbecco’s Phosphate Buffered Saline (DPBS 1x, pH 7.4) was from Gibco. Nuclease-Free Water was provided from Ambion. mRNA were a commercial optimized CleanCap® EGFP mRNA (996 nucleotides) and CleanCap® FLuc mRNA (1921 nucleotides) from TriLink biotechnologies. Immortalized DC 2.4 (a murine bone marrow derived dendritic cell lines) was propagated in complete RPMI-1640 medium with 10% heat inactivated Foetal Bovine Serum (FBS) (10%), 50 μM 2-mercaptoethanol and 10 mM HEPES buffer solution; all of them were purchased from Gibco, ThermoFisher Scientific. Trypsin solution (0.25% trypsin-EDTA) was obtained from Gibco, ThermoFisher.
Micelle Preparation
Copolymer micelles were prepared by nanoprecipitation, as previously described [39]. The PLA-b-P(NAS-co-NVP) copolymer was dissolved in 2.5 ml of acetonitrile (10 mg.ml−1), and the solution was added to 5 ml of H2O OTEC under stirring in a round bottom flask. The acetonitrile in the solution was removed under reduced pressure. The micelle concentration was determined by drying a known volume of micellar solution inside an oven at 56°C for 24 h.
Preparation of the Micellar Assemblies
Layer-by-Layer (LbL) approach: 200 μL micelle solution at 1 or 1.5 mg.mL−1 in PBS (1/2 X) was added to 100 μL of fusogenic peptide (RALA) at 4 mg.mL−1 in nuclease free water, and the solution immediately vortexed. The solution was incubated during 1.5 h for coupling. Then, 100 μL of mRNA solution in nuclease free water (40 μg.mL−1) was added onto the functionalized micelles-peptide solution, and the obtained solution immediately vortexed. The mixture was incubated 30 min for complexation (final concentrations: 500 or 750 μg.mL−1 micelle; 1000 μg.mL−1 peptide; 10 μg.mL−1 mRNA). Micelles-peptide-mRNA thus prepared were referred as “M-P-mRNA LbL”.
Pre-complex approach: The peptide-mRNA pre-complex was first prepared by adding a given volume of mRNA (40 μg.mL−1 in nuclease free water) to the same volume of peptide solution (4 mg.mL−1 in nuclease free water). After vortexing, the mixture was incubated 30 min Then, 200 μL of micelles at 1 or 1.5 mg.mL−1 in PBS (1/2 X) were added to the same volume of pre-complex, and the mixture immediately vortexed. The mixture was incubated for 1.5 h (final concentrations: 500 or 750 μg.mL−1 micelle; 1000 μg.mL−1 peptide; 10 μg.mL−1 mRNA). Micelles-peptide-mRNA thus prepared were referred as “M-P-mRNA PC”.
The different control samples (micelles, mRNA, peptide, peptide-mRNA) were prepared at the same concentrations using exactly the same procedure, by replacement of the solution of compound to omit with the dedicated blank solution (PBS/water), and used for all further evaluations.
Micelle Size and Zeta Potential
The hydrodynamic diameter, polydispersity index (PI) and zeta potential of micelles and formulated micelles were determined by Dynamic Light Scattering (DLS) analysis with the ZetasizerNanoZEN S600 device (Malvern Instrument, UK). The samples were prepared by 1/100 dilution in a solution of NaCl 1 mM. The data were obtained by Zetasizer Software 7.11 (Malvern Instrument, UK). The values were the mean of four measurements.
Peptide Immobilization
The Tricine-SDS-PAGE method was used to assess the peptide coupling onto the micelles. Each sample was diluted at 1/8 in 30 μL of water and with 30 μL of sample buffer which was composed of tricine and β-mercaptoethanol (β ME) (2% v/v). The mixture was heated at 96°C for 7 min and 50 μL of sample was loaded on the gel. The running gel was placed in the device of BioRad and immersed in the running buffer. It was run for 60 min at 100 V and constant intensity. Once the electrophoresis was finished, the gel was cleaned and colored using instant blue solution. Briefly, a sufficient volume of instant blue solution was added to cover the gel, and incubation was performed at room temperature for 60 min with gentle agitation. The gel was rinsed two times and incubated 5 min with ultrapure water. Peptide samples in various concentrations were analyzed in the same conditions for calibration. Analysis was also performed on micelle supernatants. Supernatant samples corresponded to supernatant of micellar formulation (LbL/PC) after 10 min at 10000 g centrifugation. Peptide immobilization on micelles was also assessed by ATR-FTIR analysis (Spectrum Two, Perkin Elmer, equipped with a single reflection diamond ATR accessory) on the pellet recovered after sample centrifugation, rinsing with pure water and drying.
Gel Retardation Assay
The complexation of mRNA onto the micelles was measured with a gel of retardation for electrophoresis. Agarose gel (1%) was prepared in Tris–borate–EDTA (TBE) (1x) buffer with a 2 μl drop of ethidium bromide (EtBr) staining. The gel was loaded with the samples mixed 6× Loading Dye at a concentration of 200 ng of mRNA per well. The electrophoresis process was run for 20 min at 135 mV. Then, the gel was observed at UV-Visible.
mRNA Stability in Serum
In vitro incubation with 10% serum was performed for 30 min at 37°C. In some samples, a heparin/proteinase K treatment was performed in order to separate mRNA from the complexes. Complexes were first treated or not with heparin (Sigma, France) during 30 min at RT, and secondly with proteinase K (NEB, France) at 56°C during 15 min. Samples were analyzed by agarose gel electrophoresis as described above.
Cell Culture Protocol
The cell line used was a murine dendritic cell line (DC 2.4) and has been grown according to typical culture procedure detailed here. The cell culture media is composed of RPMI-1640, Foetal Bovine Serum (FBS) (10%), 2-mercaptoethanol (50 μM) and HEPES buffer solution (10 mM). After aspirating the old culture media, the cells which adhered to the bottom of the flask T75 were washed twice with 10 ml of PBS. Then, 1 mL of trypsin was added to the cells and let for 3 min at 37°C. After 3 min, the trypsin solution containing the cells was mixed with 9 mL of fresh complete culture media. The appropriate amount of cells was re-suspended in 13 ml of fresh culture media in a new flask T75, and let in the CO2 incubator at 37°C. Cells were used with a low passage number (less than 10).
Cell Transfection
One day before transfection, cells were seeded in a 96-well plate at the density of 20,000 cells (in 100 μL of complete medium) per well. After 24 h, transfections were performed with 87.5 ng of mRNA alone (negative control) or in complexes, namely 8.75 μL solution (mRNA alone: 10 μg.mL−1; P-mRNA: 1000–10 μg.mL−1; M-P-mRNA: 500–1000-10 or 750–1000-10 μg.mL−1, i.e. 4.4 or 6.6 μg of polymer and 8.75 μg of peptide) in the presence of 100 μL of medium without additives (serum, antibiotics). 3 h after transfection, supernatants were removed and 100 μL of complete medium was added. Then, cells were incubated at 37°C and 5% CO2 until the analysis. Positive control of transfection was performed with TransIT®-mRNA Transfection Kit (Mirus, Euromedex, France) using the manufacturer’s recommendations. Briefly, 0.5 μg of mRNA was added to 50 μL of serum-free medium, and 1 μL of TransIT mRNA reagent and 1 μL of mRNA Boost Reagent were then added. After 5 min at room temperature, suitable volume of the complex mixture (8.75 μL, i.e. 87.5 ng mRNA) was added to cell in the presence of 100 μL of medium without additives (serum, antibiotics).
For analysis of EGFP expression, cells were kept at 37°C overnight. Cells were imaged and fluorescence excited with the fluorescence inverse microscope (Eclipse Ti-E, Nikon). For analysis of FLuc expression, cells were kept at 37°C overnight. And Bright-GloTM Luciferase Assay was performed. Briefly, 100 μl of reagent (Bright-GloTM Reagent) is added to cells grown in 100 μl of medium. After 2 min, luminescence was detected on Tecan i-Control Infinite M1000. Luminescence was determined as the mean of three replicates and percentage of expression was determined using four independent experiments.
Cytotoxic Studies: Presto Blue
Cytotoxicity of complexes was evaluated by Presto Blue Assay (Thermo Fisher Scientifc) according to the manufacturer’s instructions. Briefly, DC2.4 cells were seeded at a density of 20,000 cells/well into 96-well plates a day prior to the transfection. Transfection with mRNA alone or in complexes was performed as described above (+ naked micelles and naked peptide controls at the same concentration than in formulations), and cytotoxicity was measured 24 h later. After incubation, 11 μL of Presto Blue Reagent was added and plates were incubated 10 min at 37°C. Fluorescence was detected on Tecan i-control Infinite M1000 (560 nm/590 nm; bandwidth 10 nm; gain 91) (Tecan, Swiss). Fluorescence was determined as the mean of three replicates and four independent experiments.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism Version 7.0 software. All of the data are presented as the mean ± SD. Difference between groups was analyzed as described in figure legends. Statistical significances were indicated on the figures.
Results and Discussion
Peptide/mRNA Immobilization on the Micelles
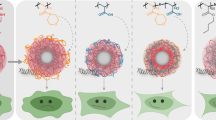
Micelles of the PLA-b-P(NAS-co-NVP) copolymer (12000–9000 g mol−1, Ð = 1.7) were first prepared by the common solvent (nanoprecipitation) method as described in the experimental section and showed a mean size, determined by dynamic light scattering, of 67 nm (polydispersity index (PI) of 0.06). From these obtained micelles, two approaches were adopted for preparing the peptide-mRNA micellar complexes namely (i) sequential immobilization of peptide and mRNA onto the micelles (layer-by-layer, LbL) or (ii) direct immobilization of peptide-mRNA pre-complex (PC) on the micelles (Fig. 1).

Schematic view of the two strategies, Layer-by-Layer (LbL, top) and pre-complex (PC, bottom), to achieve micelle-based mRNA carrier.
For LbL strategy (Fig. 1, top), the micelles (M) were in a first step allowed to react with the RALA peptide (P) in various concentrations in PBS pH 7.4 for 1.5 h. Control micelles in the coupling conditions (PBS) but with no added peptide showed a negative zeta potential value due to partial hydrolysis of the NS activated esters into carboxylates, as previously reported (−27 mV). To note, these micelles also presented a higher diameter (90 nm from DLS) than that of native micelles in water (67 nm), as a result of the improved hydrophilic character of the corona and repulsion effects upon the formation of these carboxylates by hydrolysis. As expected, the surface charge was reversed along with increasing peptide/micelle ratios, from negative to positive value, going through an instable colloidal domain close to charge neutrality (~200–1000 μg of peptide per mg copolymer) due to insufficient repulsive electrostatic forces for colloid stabilization (Fig. 2). Beyond this domain, positively charged colloids with well defined nanometric size (about 130 nm) and distribution (PI~0.19) were obtained. Based on this preliminary screening, we focused on two stable colloids prepared from micelle concentrations of 750 μg.mL−1 and 500 μg.mL−1, each with a peptide concentration of 1000 μg.mL−1 (i.e. peptide/micelle of 1330 and 2000 μg.mg−1, respectively, Table 1). These colloids showed a higher mean size that of the reference micelles (90 nm), supporting peptide immobilization, and were selected for further complex mRNA encoding EGFP. It is well documented that encapsulation of nucleic acids in RALA complexes and their transfection are more effective when increasing RALA/nucleic acid ratio [25, 43]. In addition, the micelles will undergo flocculation in case of insufficient peptide excess, the positive zeta potential decreasing close to zero value following RNA adsorption. We thus used a quite significant RALA/mRNA ratio, namely 100/1 in weight. In these conditions, minor changes were observed after mRNA fixation regarding mean size (Table 1). The zeta potential slightly decreased, indicating contribution of RNA at the interface (Table 1), which was later shown to be totally complexed on the peptide-colloid (see RNA complexation part). The same RALA/mRNA weight ratio was used in all further studies.

Physico-chemical colloid characteristics (mean size (empty circles) from DLS and zeta potential (black circles)) along RALA peptide coupling on the micelles at various concentrations.
Then, the second approach (PC, Fig. 1, bottom); which lies on the immobilization of pre-mixed peptide/RNA on the micelles, was investigated, in the same amount conditions as for the LbL strategy. This procedure was expected to ensure better burying and protection of the mRNA in the corona, as mRNA is already bound to peptide when performing immobilization on the micelles. As shown in Table 1, stable formulations were achieved with typically similar sizes as those obtained from LbL, but slightly higher polydispersity index, particularly at the lowest peptide/micelle ratio (1333 μg.mg−1, i.e. 1000 μg.mL−1 peptide and 750 μg.mL−1 micelles). These data suggest a less controlled immobilization process when performed in simultaneous presence of peptide and mRNA.
Peptide Coupling Assessment
The covalent character of the peptide coupling on the micelles (through amide bond formation between peptide terminal amine and NS ester functions) was assessed by Tricine SDS-PAGE analysis. This technique is relevant as it breaks only the non-covalent interactions involving peptide. Such a feature was clearly highlighted in Fig. 3A. Indeed, as expected, the peptide-mRNA (P-mRNA) pre-complex gave the same intensity band as the control peptide (P) alone (1000 μg.mL−1, i.e. the concentration at which coupling is performed), because complexation of the peptide with mRNA (further attested by agarose gel electrophoresis) is only mediated by non-covalent interactions (mainly electrostatic). In contrast, the M-P-RNA (here, LbL sample from micelles at 500 μg.mL−1) gave a weak intensity band, indicating that most of peptide had been involved in covalent bonding with the micelles, making it unable to migrate through the gel and observable at the start of the well (see arrows, also further in Fig. 3B). For a suitable quantitative analysis of the couplings, a calibration range in peptide was further used (0 to 1000 μg.mL−1) in the Tricine SDS-PAGE analysis. For the LbL approach (Fig. 3B, left gel), the micelle-peptide-mRNA samples showed a weak free peptide band as a result of peptide coupling on micelles, whatever the micelle concentration used (500 or 750 μg.mL−1). The non covalently bound (“free”) peptide could be roughly quantified based on the intensity of the peptide references and was shown far less than 200 μg.mL−1, i.e. an estimated coupling yield of about 90% for both samples (500 and 750 μg.mL−1 micelle concentration), which corresponds to 11.3 and 7.5 RALA molecules coupled per polymer chain, respectively. It is to mention that this remaining fraction of “free” peptide observed still may be associated on the micelles in a non-covalent manner, i.e. electrostatically (as SDS-PAGE analysis disrupts all the non-covalent interactions). To assess this, we centrifuged the micelle-peptide mixtures after coupling step, and analyzed by SDS-PAGE the supernatant, which revealed negligible peptide amounts. This indicates that the fraction of “free” peptide observed on the gel (i.e. non covalently bound) was in fact associated to the micelles, most probably through electrostatic interactions with the anionic surface of the micelles. Peptide immobilization was also assessed by ATR-FTIR analysis on the pellet obtained after centrifugation (sample micelle-peptide M750-P, LbL, Fig. S1, Supplementary Information). The spectrum clearly showed the presence of both peptide and copolymer, as compared to peptide and copolymer references, as well as apparition of a new peak at 1574 cm−1, indicative of amide formation following covalent coupling. In addition, relative peak areas were consistent with high immobilization of peptide on copolymer, corroborating the SDS-PAGE analysis.

Tricine SDS PAGE analysis of peptide coupling on the micelles. A: Comparison of the P-RNA and M-P-RNA with control peptide at the concentration used for coupling (1000 μg.mL−1); B: gels for the LbL made micellar formulations (left) and for the pre-complex made ones (right) approach; (top gel: micelles at 500 μg.mL−1; bottom gel: micelles at 750 μg.mL−1). Arrows indicate peptide coupled to micelles, unable to migrate into the gel.
The SDS-PAGE analysis of the micellar assemblies formed from the PC approach was also performed (Fig. 3B, right gel). It indicated a slightly less efficient coupling (about 85% for both 500 and 750 μg.mL−1 samples, meaning about 10.7 and 7 RALA molecules coupled per polymer chain, respectively), which was expected due to counteractive effect of the polyanionic mRNA complexed with the peptide during the coupling process. Also, the supernatant analysis showed that non-negligible amounts of free peptide could be detected, as compared to the LbL approach.
Such amount of coupled peptide is quite important and not surprising considering both the high density of NS ester functions present on the hydrophilic corona and the strongly cationic character of RALA peptide favoring its approach to the negatively charged micelles (through electrostatic interactions) for coupling reaction. The degree of coupling was indeed in the same range as that observed in our previous study regarding the coupling of a cationic lysine tagged IL-1β peptide on the same copolymer used at the surface of PLA nanoparticles (~7 peptides per copolymer chain) [44].
mRNA Complexation
The complexation of EGFP mRNA was assessed by agarose gel electrophoresis (Fig. 4, lanes referred as (−), representative of the samples incubated in vitro with or without serum). Whatever the LbL or PC approach used, mRNA was totally complexed, as no free mRNA was observed on the gel (Fig. 4A). Moreover, interestingly, reference starting micelles (negatively charged) could be observed as a slight smear at high molar masses (probably due to electrostatic interaction with ethidium bromide, positively charged, Fig. 4B), while no more smearing was observed for the micelle-peptide-RNA samples (Fig. 4A). This clearly evidenced that the micelles had been totally involved in charge inversion upon peptide immobilization, corroborating the previous zeta potential and SDS PAGE analyses. To note, mRNA was also totally complexed with the peptide in the reference P-mRNA pre-complex (Fig. 4B).

Agarose gel electrophoresis analysis of micelle assemblies (M-P-GFP mRNA) (A) and dedicated control samples (B) after 30 min incubation with or without serum at 37°C (150 ng/μl of mRNA per well), without (−) or with (+) mRNA desorption treatment. For desorption, samples were incubated for 30 min with heparin and then 15 min at 56°C with proteinase K.
mRNA Serum Stability
Integrity of the mRNA complexed with the peptide-PLA micelles was assessed following desorption upon heparin/proteinase K treatment after in vitro incubation (Fig. 4, lanes referred as (+)). In absence of serum, integrity was preserved for all the micellar assemblies, as well as for the peptide-mRNA pre-complex and naked mRNA. In presence of serum, free mRNA was totally degraded (Fig. 4B), as expected. mRNA from the pre-complex with peptide (P-mRNA) was also totally degraded despite total complexation with the peptide (Fig. 4B). Therefore, while RALA was reported to efficiently protect DNA against serum degradation in several studies [25, 45, 46], it was obviously not the case for mRNA, which is well known to be less stable and much more prone to enzymatic degradation than DNA. To date, very few studies were dedicated to RALA as a complexation agent for RNA. Bennett et al. has focused on this peptide for complexing siRNA, namely siFKBPL, but its serum stability was not investigated, contrary to that of pFKBPL (DNA), which was found good when complexed to RALA [45]. Later, Udhayakumar et al. have studied RALA complexed with eGFP and ovalbumin mRNAs in a vaccine context but their serum stability were not investigated either [47].
Contrary to P-mRNA, very interestingly, the mRNA from the micellar assemblies remained very stable, whatever LbL or PC approach used (Fig. 4A). Furthermore, the intensity of the mRNA band was more intense when copolymer amount in the nano-formulations was improved (750 μg.mL−1 vs. 500 μg.mL−1), supporting the beneficial impact of the micelles for mRNA stability. These results show that, even when the mRNA was immobilized as the last layer onto the peptide previously coupled on the micelles (LbL approach), it was sufficiently buried in the corona for resisting to serum degradation. Still, the gel data suggested that mRNA was better protected when formulations were prepared using the PC approach (M750-P-mRNA sample), supporting our assumption of better entrapment of mRNA through this procedure. These last observations have however to be considered carefully, as intensity of the bands can be subject to variations for various reasons. Anyway, to sum up, our results showed that in contrast to simple mRNA complexation with the peptide, the micelles were highly efficient for protecting mRNA in serum, a parameter of prime importance in the perspective of in vivo application. This protection was most probably due to suitable environment provided by the hydrophilic NVP based hairy corona. Koh et al. have very recently reported the relevance and positive impact of PNVP for RNA integrity in the context of mRNA loaded dissolvable microneedles (RNApatch) [48]. In addition, this polymer is known to reduce RNAse contamination during the RNA extraction/isolation processes.
Cytotoxicity and Transfection Capabilities
The cytotoxicity and transfection efficiency of the above prepared micellar assemblies were assessed using murine dendritic cell line (DC 2.4). Naked micelles did not show toxicity as compared to cells alone, contrary to the naked peptide (Fig. 5a-d). Peptide-mRNA complex exhibited significant toxicity (i, top), which most probably arose from the inherent peptide toxicity, as naked mRNA was innocuous (j, top). More precisely, significant cell death was observed close to the center of the well, while remaining viable cells tended to position at well sides. Such RALA toxicity has been previously evidenced (even at lower doses), and attributed to sequence similarity to cationic antimicrobial peptides [27]. The micelle-peptide-RNA showed reduced toxicity, with the cells remaining homogeneously distributed in the wells, whatever the used strategy (LbL or PC, Fig. 5e-h, top), suggesting the positive impact of the micelles for counteracting the toxicity of the peptide. This could be attributed to the micelle hydrophilic corona able to densely trap and partially mask the peptide, while also mitigating its cationic character.

Cytotoxicity (top images, normal phase mode) and transfection (bottom images, fluorescent mode) of the different micellar formulations; transfection was performed with 87.5 ng mRNA/well; peptide: 8.75 μg (peptide/mRNA: 100/1 w/w); analysis was performed 24 h post-transfection.
mRNA alone did not transfect any cells, as expected (Fig. 5j, bottom), and transfection efficiency was improved with the P-mRNA complex (i, bottom), but remained limited as a result of important cell death, with the GFP mainly expressed on the well side, where cells survived. Transfection was enhanced with M-P-RNA, especially with the LbL made micelles (g-h, bottom). Indeed, despite similar cytotoxicity features, the LbL micelles transfected much more DCs than PC made micelles. This was most probably due to the fact that mRNA was less entrapped in the LbL strategy than in PC one, as previously shown, and thus more prone for transfecting the DCs. Therefore, the mode of elaboration of our micellar assemblies had a significant impact on the mRNA accessibility and further transfection capabilities. To note, transfection capacity remained lower than that observed with the TransIT (Fig. 5k), one of the more performant transfection reagent on the market.
To show the versatility of our carrier platform and to confirm these results, we prepared and evaluated in vitro (with DCs) micellar assemblies using another mRNA, namely mRNA encoding luciferase. The micellar assemblies were prepared following exactly the same procedure as for EGFP mRNA. Mean size and zeta potential values were typically similar than that obtained for mRNA EGFP (Table 2). Luciferase mRNA allowed quantifying the cytotoxicity using Presto Blue method (Fig. 6), which was not possible for previously used GFP mRNA due to fluorescence interference with Presto Blue. The results confirmed the previous observations with EGFP mRNA. Indeed, peptide-mRNA exhibited significant cytotoxicity (less than 50% cell viability). In contrast, the micellar assemblies, either prepared through PC or LbL approach, showed reduced cell death, and cell viability tended to increase with the copolymer content in the assembly (M750-P-mRNA vs. M500-P-mRNA), again indicating favorable effect of the micelles on cytocompatibility through peptide incorporation in the corona.

Cytotoxicity, assessed through Presto Blue assay, of the different luciferase mRNA loaded micellar formulations and controls on DC2.4 cells; 87.5 ng mRNA/well; peptide: 8.75 μg (peptide/mRNA: 100/1 w/w, “Peptide” condition is without mRNA), Data represent the mean of four independent experiments, triplicate points being used in each experimental sample. Statistical significance between two groups was determined using a one way Anova test; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
DC transfection efficiency of the different formulations was assessed using TransIT as a positive control (Fig. 7), to which the results were normalized. The reference P-mRNA complex was found about 6-fold less efficient than this transfection reagent. It must be underlined here that TransIT has recently appeared as one of the best ever transfecting agents, outperforming lipofectamine for mostly tested RNA/DNA, while being less toxic [49, 50]. Lipofectamine itself was reported to be more efficient than RALA peptide in some earlier transfection studies [25, 45]. Thus, the transfection efficiency of the RALA-mRNA observed here appeared quite acceptable and fully consistent with previously reported transfection data.

Transfection efficiency of the different luciferase mRNA loaded micellar formulations on DC2.4 normalized to 100% transfection efficiency obtained with TransIT-mRNA (positive control); 87.5 ng mRNA/well; peptide: 8.75 μg (peptide/mRNA: 100/1 w/w). Data represent the mean of four independent experiments, triplicate points being used in each experimental sample. Statistical significance between two groups was determined using a one way Anova test; **, p < 0.01.
Again, LbL made micelles showed higher DC transfection efficiency than PC made ones (Fig. 7), fully confirming the previous results obtained for EGFP mRNA and that less buried mRNAs afforded by LbL protocol were favorable. Transfection was not here found to be improved as compared to that obtained with the peptide-mRNA complex. These results are however consistent with the ones obtained with GFP mRNA, as in this transfection experiment, the toxicity effects (important for peptide-mRNA complex) are not taken into account. Indeed, the luciferase protein expression is assessed in indirect manner through the detection of the oxidized luciferin in the supernatant. Consequently, even if a significant cell death is observed, the luciferase expression of such cells before they died will contribute to the luminescence signal.
Therefore, these combined transfection/toxicity results concerning peptide-RNA highlight both the efficiency of such fusogenic peptide for nucleic acid delivery into cells and its shortcomings regarding toxicity. To this regard, our reactive copolymer based micelles were found highly relevant for reducing the toxicity associated to such complexes while maintaining suitable transfection capacity.
Conclusion
In this work, we have designed PLA-based micelles as a potential carrier for mRNA delivery, through coupling of a cationic cell penetrating peptide. Thanks to their dense NS-based reactive corona, cationic peptide-mRNA functionalized micelles could be easily prepared through two water-friendly based processes (LbL or PC). Whatever the process, the micelles ensured high mRNA protection in serum and limited cytotoxcity, in contrast to peptide-mRNA complex, thanks to the masking effect afforded by the hairy corona. The preparation strategy impacted the obtained transfection properties. In particular, LbL made assemblies were able to transfect DCs in higher efficiency than the PC made ones, which was suspected to arise from less buried mRNA in the LbL based micelles. These results thus demonstrate the potential of this newly engineered PLA-based platform for mRNA delivery. Further studies are ongoing to investigate the “detoxifying” effect of such micelles for other potentially toxic cationic species and their potential in mRNA vaccine/drug delivery, in conjunction with encapsulated hydrophobic ligands or biomolecules of interest.
References
Burnett JC, Rossi JJ. RNA-based therapeutics: current Progress and future prospects. Chem Biol. 2012;19:60–71.
Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med [Internet]. 2017 [cited 2018 Jul 3];9. Available from: http://genomemedicine.biomedcentral.com/articles/10.1186/s13073-017-0450-0
Subhan MA, Torchilin VP. Efficient nanocarriers of siRNA therapeutics for cancer treatment. Transl Res [Internet]. 2019 [cited 2019 Sep 3]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S1931524419301392
Resnier P, Montier T, Mathieu V, Benoit J-P, Passirani C. A review of the current status of siRNA nanomedicines in the treatment of cancer. Biomaterials. 2013;34:6429–43.
Kang K-N, Lee Y-S. RNA Aptamers: A Review of Recent Trends and Applications. In: Zhong J-J, editor. Future Trends Biotechnol [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012 [cited 2018 Jul 3]. p. 153–69. Available from: http://springerlink.bibliotecabuap.elogim.com/10.1007/10_2012_136
Midoux P, Pichon C. Lipid-based mRNA vaccine delivery systems. Expert Rev Vaccines. 2015;14:221–34.
Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543:248–51.
Linares-Fernández S, Lacroix C, Exposito J-Y, Verrier B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol Med [Internet]. 2019 [cited 2019 Dec 2]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S1471491419302448
Liu. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines. 2019:7–37.
Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci. 2012;109:14604–9.
Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401.
Pollard C, Rejman J, De Haes W, Verrier B, Van Gulck E, Naessens T, et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol Ther. 2013;21:251–9.
Sayour EJ, De Leon G, Pham C, Grippin A, Kemeny H, Chua J, et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. OncoImmunology. 2017;6:e1256527.
Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed. 2012;51:8529–33.
Pardi N, Secreto AJ, Shan X, Debonera F, Glover J, Yi Y, et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat Commun. 2017;8:14630.
Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell. 2017;168:1114–1125.e10.
Stadler CR, Bähr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med. 2017;23:815–7.
Zhao M, Li M, Zhang Z, Gong T, Sun X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv 2015;1–12.
Wang LL, Sloand JN, Gaffey AC, Venkataraman CM, Wang Z, Trubelja A, et al. Injectable, guest–host assembled Polyethylenimine hydrogel for siRNA delivery. Biomacromolecules. 2017;18:77–86.
Chahal JS, Khan OF, Cooper CL, McPartlan JS, Tsosie JK, Tilley LD, et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci. 2016;113:E4133–42.
Su X, Fricke J, Kavanagh DG, Irvine DJ. In Vitro and in Vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol Pharm. 2011;8:774–87.
Yan Y, Xiong H, Zhang X, Cheng Q, Siegwart DJ. Systemic mRNA delivery to the lungs by functional polyester-based carriers. Biomacromolecules. 2017;18:4307–15.
Kichler A, Leborgne C, März J, Danos O, Bechinger B. Histidine-rich amphipathic peptide antibiotics promote efficient delivery of DNA into mammalian cells. Proc Natl Acad Sci. 2003;100:1564–8.
Lam JKW, Liang W, Lan Y, Chaudhuri P, Chow MYT, Witt K, et al. Effective endogenous gene silencing mediated by pH responsive peptides proceeds via multiple pathways. J Control Release. 2012;158:293–303.
McCarthy HO, McCaffrey J, McCrudden CM, Zholobenko A, Ali AA, McBride JW, et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J Control Release. 2014;189:141–9.
Borguet YP, Khan S, Noel A, Gunsten SP, Brody SL, Elsabahy M, et al. Development of fully degradable Phosphonium-functionalized Amphiphilic Diblock copolymers for nucleic acids delivery. Biomacromolecules. 2018;19:1212–22.
Nouri FS, Wang X, Dorrani M, Karjoo Z, Hatefi A. A recombinant biopolymeric platform for reliable evaluation of the activity of pH-responsive Amphiphile Fusogenic peptides. Biomacromolecules. 2013;14:2033–40.
Multicenter Phase II. Clinical trial of Genexol-PM® with gemcitabine in advanced biliary tract Cancer. Anticancer Res. 2017;37:1467–74.
Ando S, Putnam D, Pack DW, Langer R. PLGA microspheres containing plasmid DNA: preservation of supercoiled DNA via cryopreparation and carbohydrate stabilization. J Pharm Sci. 1999;88:126–30.
Walter E, Moelling K, Pavlovic J, Merkle HP. Microencapsulation of DNA using poly(dl-lactide-co-glycolide): stability issues and release characteristics. J Control Release. 1999;61:361–74.
Cun D, Foged C, Yang M, Frøkjær S, Nielsen HM. Preparation and characterization of poly(dl-lactide-co-glycolide) nanoparticles for siRNA delivery. Int J Pharm. 2010;390:70–5.
Jain AK, Massey A, Yusuf H, Kett VL, McDonald D, McCarthy H. Development of polymeric–cationic peptide composite nanoparticles, a nanoparticle-in-nanoparticle system for controlled gene delivery. Int J Nanomedicine. 2015;7183.
Tinsley-Bown AM, Fretwell R, Dowsett AB, Davis SL, Farrar GH. Formulation of poly(d,l-lactic-co-glycolic acid) microparticles for rapid plasmid DNA delivery. J Control Release. 2000;66:229–41.
O’Hagan D, Singh M, Ugozzoli M, Wild C, Barnett S, Chen M, et al. Induction of potent immune responses by cationic microparticles with adsorbed human immunodeficiency virus DNA vaccines. J Virol. 2001;75:9037–43.
Maruyama A, Ishihara T, Kim J-S, Kim SW, Akaike T. Nanoparticle DNA Carrier with Poly( l -lysine) Grafted Polysaccharide Copolymer and Poly( d , l -lactic acid). Bioconjug Chem. 1997;8:735–42 Nanoparticle DNA carrier with poly(L-lysine) grafted polysaccharide copolymer and poly(D,L-lactic acid).
Trimaille T, Pichot C, Delair T. Surface functionalization of poly(d,l-lactic acid) nanoparticles with poly(ethylenimine) and plasmid DNA by the layer-by-layer approach. Colloids Surf Physicochem Eng Asp. 2003;221:39–48.
Chen C-K, Jones CH, Mistriotis P, Yu Y, Ma X, Ravikrishnan A, et al. Poly(ethylene glycol)-block-cationic polylactide nanocomplexes of differing charge density for gene delivery. Biomaterials. 2013;34:9688–99.
Ni Q, Zhang F, Zhang Y, Zhu G, Wang Z, Teng Z, et al. In situ shRNA synthesis on DNA-Polylactide nanoparticles to treat multidrug resistant breast Cancer. Adv Mater. 2018;30:1705737.
Handké N, Lahaye V, Bertin D, Delair T, Verrier B, Gigmes D, et al. Elaboration of glycopolymer-functionalized micelles from an N-vinylpyrrolidone/lactide-based reactive copolymer platform. Macromol Biosci. 2013;13:1213–20.
Handké N, Trimaille T, Luciani E, Rollet M, Delair T, Verrier B, et al. Elaboration of densely functionalized polylactide nanoparticles from N-acryloxysuccinimide-based block copolymers. J Polym Sci Part Polym Chem. 2011;49:1341–50.
Jiménez-Sánchez G, Terrat C, Verrier B, Gigmes D, Trimaille T. Improving bioassay sensitivity through immobilization of bio-probes onto reactive micelles. Chem Commun. 2017;53:8062–5.
Jiménez-Sánchez G, Pavot V, Chane-Haong C, Handké N, Terrat C, Gigmes D, et al. Preparation and in vitro evaluation of Imiquimod loaded Polylactide-based micelles as potential vaccine adjuvants. Pharm Res. 2015;32:311–20.
Nazari M, Zamani Koukhaloo S, Mousavi S, Minai-Tehrani A, Emamzadeh R, Cheraghi R. Development of a ZHER3-Affibody-targeted Nano-vector for gene delivery to HER3-overexpressed breast Cancer cells. Macromol Biosci. 2019;19:1900159.
Handké N, Ficheux D, Rollet M, Delair T, Mabrouk K, Bertin D, et al. Lysine-tagged peptide coupling onto polylactide nanoparticles coated with activated ester-based amphiphilic copolymer: a route to highly peptide-functionalized biodegradable carriers. Colloids Surf B Biointerfaces. 2013;103:298–303.
Bennett R, Yakkundi A, McKeen HD, McClements L, McKeogh TJ, McCrudden CM, et al. RALA-mediated delivery of FKBPL nucleic acid therapeutics. Nanomed. 2015;10:2989–3001.
Ali AA, McCrudden CM, McCaffrey J, McBride JW, Cole G, Dunne NJ, et al. DNA vaccination for cervical cancer; a novel technology platform of RALA mediated gene delivery via polymeric microneedles. Nanomedicine Nanotechnol Biol Med. 2017;13:921–32.
Udhayakumar VK, De Beuckelaer A, McCaffrey J, McCrudden CM, Kirschman JL, Vanover D, et al. Arginine-rich peptide-based mRNA Nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic Organization of the Peptide. Adv Healthc Mater. 2017;6:1601412.
Koh KJ, Liu Y, Lim SH, Loh XJ, Kang L, Lim CY, et al. Formulation, characterization and evaluation of mRNA-loaded dissolvable polymeric microneedles (RNApatch). Sci Rep [Internet]. 2018 [cited 2019 Aug 16];8. Available from: http://www.nature.com/articles/s41598-018-30290-3
McLenachan S, Zhang D, Palomo ABA, Edel MJ, Chen FK. mRNA Transfection of Mouse and Human Neural Stem Cell Cultures. Wu Q, editor. PLoS ONE. 2013;8:e83596.
Tinsley JH, Hawker J, Yuan Y. Efficient protein transfection of cultured coronary venular endothelial cells. Am J Physiol-Heart Circ Physiol. 1998;275:H1873–8.
Acknowledgments and Disclosures
We would like to thank J. Y Exposito and P. Libeau for useful discussions on mRNA aspects of delivery. We are grateful to AMU and CNRS for financial support. Financial support is also gained from ANRS in the framework of HIVERA JTC 2014 (HIV NANOVA), and Euronanomed II (Flunanoair) and from ANR-16-CE20-0002-01 (FishRNAVax) to BV.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 394 kb)
Rights and permissions
About this article

Cite this article
Lacroix, C., Humanes, A., Coiffier, C. et al. Polylactide-Based Reactive Micelles as a Robust Platform for mRNA Delivery. Pharm Res 37, 30 (2020). https://doi.org/10.1007/s11095-019-2749-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11095-019-2749-6