
Abstract
Purpose
Paclitaxel (PTX)-loaded genipin-crosslinked gelatin microspheres (GP-MS) are a prolonged IP delivery system under development for the treatment of peritoneal minimal residual disease (pMRD). Here, we show the use of a pharmacokinetic-pharmacodynamic (PKPD) modelling approach to inform the formulation development of PTX-GP-MS in a mice pMRD model.
Methods
PTX blood concentrations and survival data were obtained in Balb/c Nu mice receiving different single IP doses (7.5 and/or 35 mg/kg) of PTX-ethanolic loaded GP-MS (PTXEtOH-GP-MS), PTX-nanosuspension loaded GP-MS (PTXnano-GP-MS), and immediate release formulation Abraxane®. A population PK model was developed to characterize the PTX blood concentration pattern and to predict PTX concentrations in peritoneum. Afterwards, PKPD relationships between the predicted peritoneal or blood concentrations and survival were explored using time-to-event modelling.
Results
A PKPD model was developed that simultaneously describes the competing effects of treatment efficacy (driven by peritoneal concentration) and toxicity (driven by blood concentration) of PTX on survival. Clear survival advantages of PTXnano-GP-MS over PTXEtOH-GP-MS and Abraxane® were found. Simulations of different doses of PTXnano-GP-MS demonstrated that drug-induced toxicity is high at doses between 20 and 35 mg/kg.
Conclusions
The model predicts that the dose range of 7.5-15 mg/kg of PTXnano-GP-MS provides an optimal balance between efficacy and safety.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer is the eighth most common cancer among women (1), and is associated with the highest mortality across gynecological cancers (2). As a clinically silent cancer, more than two-thirds of women receive a diagnosis of advanced disease (International Federation of Gynecology and Obstetrics stage III or IV) that has been widely disseminated within the peritoneal cavity (3).
The standard treatment for advanced ovarian cancer is maximal cytoreductive surgery (CRS) to remove macroscopic tumors in the abdominal cavity followed by intravenous platinum/taxane-based adjuvant therapy to kill residual microscopic disease (4). Despite an initial response rate of 60%-80% to this standard of care (5), the prognosis remains poor with 3-year relapse rate greater than 75% and mean 5-year survival rate less than 25% (6,7,8). The recurrence is generally thought to be due to insufficient killing of undetectable residual cancer cells by first-line chemotherapy (9,10). Therefore, there is a critical need to develop novel strategies for treating peritoneal minimal residual disease (pMRD) with the goal of improving patient survival and preventing disease recurrence.
As the disease is predominantly confined to the peritoneal cavity, the use of intraperitoneal chemotherapy (IPC) (postoperative or intraoperative) is an increasingly popular regional treatment strategy for treating pMRD, which has been explored in numerous clinical studies (3). IPC can enhance drug concentrations in the abdominal cavity while at the same time limit systemic drug exposure, thus potentially achieving improved efficacy with less systemic drug-induced toxicity (11,12,13,14,15). Although IPC has demonstrated survival advantages compared to standard intravenous chemotherapy (16,17,18), the disease recurrence still remains quite high with a 3-year relapse rate greater than 50% (16,19). The intraperitoneal delivery of rapidly absorbed chemotherapeutic solutions with single-dose intraoperative IPC or intermittent doses of postoperative IPC can only provide short-term drug exposure for the residual disease, potentially leading to incomplete cancer cell killing and recurrence of the disease.
Locoregional delivery of sustained release chemotherapeutics to maintain a prolonged drug exposure in the peritoneal cavity might be a promising therapeutic strategy. Paclitaxel (PTX) is a front-line agent for ovarian cancer, and is thought to be a good candidate for IP treatment because of its low peritoneal clearance compared with systemic clearance and also significant first-pass effect, resulting in high area under the concentration-time curve ratio of peritoneal/plasma(>1000) (20). Some previous work has shown survival benefits of sustained PTX IP formulations (e.g. PTX loaded polymeric microparticles and implantable paclitaxel drug delivery system) over solution-based PTX products (e.g. Taxol®) in peritoneal tumor-bearing mice (21,22). Recently, we developed a novel IP delivery system of paclitaxel-loaded genipin-crosslinked gelatin microspheres (PTX-GP-MS) specifically designed for treating pMRD (De Clercq et al., submitted to Scientific Reports). The developed PTX-GP-MS showed prolonged PTX release over at least two weeks both in vitro and in vivo and demonstrated a significantly better survival outcome over a marketed PTX nanoparticle-based drug delivery system, Abraxane® (PTX-loaded albumin nanoparticles, Nab-PTX), in a microscopic peritoneal carcinomatosis mouse model mimicking the residual disease state after CRS. However, toxicity phenomena and/or lethality were observed at high doses of PTX-GP-MS, and the best dose with optimal survival outcome and minimal toxicity were left unidentified in this descriptive study.
Our present model-based analysis study builds on these efforts and aims to investigate the relationships between drug exposure and treatment response using pharmacokinetic-pharmacodynamic (PKPD) modelling to find the optimal dose of PTX-GP-MS to better inform further formulation optimization.
Materials and Methods
Paclitaxel-Loaded Genipin-Crosslinked Gelatin Microspheres
Genipin-crosslinked gelatin microspheres (GP-MS) with an average size of 50 μm were prepared as described in De Clercq et al., 2016 (23). The preparation of PTX-GP-MS with an (optimized) 40% crosslinking degree was described below.
PTX-ethanolic loaded GP-MS (PTXEtOH-GP-MS): GP-MS were loaded with PTX by immersion in an ethanolic PTX solution. 200 mg GP-MS with different degrees of crosslinking (7, 25, 40, 60 and 70%) were immersed in 2 ml of a 1, 2.5 or 5 mg/ml PTX (purity of >99%, LC laboratories, Woburn, MA, USA) ethanol/distilled water (75/25, v/v) (absolute ethanol, VWR chemicals, Fontenay-sous-Bois cedex, France) solution under slow magnetic stirring. After 3 hours, GP-MS were vacuum filtered and washed with absolute ethanol (VWR chemicals) to remove unentrapped PTX from the surface. PTX-loaded GP-MS were lyophilised for 24 hours at -50°C and 1 mbar.
PTX-nanosuspension loaded GP-MS (PTXnano-GP-MS): GP-MS were loaded by immersion in an aqueous PTX-nanosuspension. Initially, PTX-nanocrystals were prepared using a wet milling technique. Pluronic® F-127/PTX (Pluronic® F-127, Sigma-Aldrich, Bornem, Belgium) (PTX purity of >99%, LC laboratories) in a 1/4 ratio was transferred in a 20 ml vial containing 5 ml 0.9% sodium chloride solution (Sigma-Aldrich) and 30 g zirconium oxide beads (Netzsch zetabeads, Ghislenghien, Belgium) with a diameter of 0.5 mm as milling pearls. The vials were placed on a roller-mill and grinded at 150 rpm for 60 hours. The nanocrystals were lyophilised for 24 hours at -50°C and 1 mbar. Particle size and polydispersity index of the PTX nanocrystals were determined by dynamic light scattering, using a Zetasizer 3000 (Malvern Instruments, Worcestershire, UK). GP-MS (50, 100 or 200 mg) were immersed for 3 hours in 2 ml aqueous PTX nanosuspension, diluted using 0.9% sodium chloride solution to a concentration of 1, 2.5 or 5 mg PTX/ml. PTX-loaded GP-MS were collected by vacuum filtration and washed using distilled water to remove unentrapped PTX from the surface. PTXnano-GP-MS were lyophilised for 24 hours at -50°C and 1 mbar.
Microscopic Peritoneal Carcinomatosis Model and Intraperitoneal Chemotherapy Treatment
A luciferase positive human ovarian cancer cell line SK-OV-3-Luc IP1 was cultured at 37°C in a 10% CO2 humidified atmosphere in Dulbecco’s Modified Eagle’s Medium (ThermoFisher, Ghent, Belgium), supplemented with 10% fetal bovine serum (Sigma-Aldrich, Diegem, Belgium) and 2% penicillin/streptomycin.
Briefly, six-week old female Balb/c Nu mice (BALB/cOlaHsd-Foxn1nu, Envigo, Horst, The Netherlands) were acclimated to the animal facility one week before the start of the study. After 7 days, mice were intraperitoneally injected with 2 x 106 SK-OV-3-Luc IP1 cells suspended in 1 ml physiological saline. The number of injected cells was optimized in a pilot study for the development of the microscopic peritoneal carcinomatosis model. One day after engraftment with tumor cells (referred to as day 0 of the study), the mice were randomized across several groups, each consisting of 10 to 12 animals: the control group was injected with 2 ml 0.9% saline or 50 mg of blank GP-MS suspended in 2 ml 0.9% saline; PTXnano-GP-MS groups (dosed at 7.5 and 35 mg/kg) represented the relatively slow sustained release PTX delivery system; the PTXEtOH-GP-MS group (7.5 mg/kg) represented the relatively fast sustained release PTX delivery system; while the Nab-PTX group (35 mg/kg) represented the immediate release PTX delivery system. Every dose mentioned in this manuscript describes the PTX dose and not the amount of GP-MS.
Throughout the study, mice were kept in standard housing conditions with water and food ad libitum and a 12 h light/dark cycle. All animal experiments were approved by the Animal Ethics Committee of the Faculty of Medicine at Ghent University (ECD 17/83) and adhered to the “Principles of Laboratory Animal Care” (NIH publication #85-23, revised in 1985).
Collection of Blood Samples
Dried blood spot (DBS) microsampling was performed in our study by withdrawing tiny volumes (< 15 μL at each time-point) of blood from the tail vein. Under a sparse sampling scheme design, blood samples were collected at pre-defined time-points between 0.5 h and 13 days after the start of treatment. Two to five blood samples were taken from each mouse. At each sampling-point, 10 μL of blood was spotted on a blood spot card (PerkinElmer 226 Bioanalysis RUO Card, Perkin Elmer, Greenville, USA).
PTX concentrations of DBS samples were measured using a previously developed UPLC-MS/MS method (24). The lower limit of quantification (LLOQ) of PTX in a DBS sample was 1 ng/ml. The method imprecision was less than 14.6% and bias was less than ± 11.9%.
Recording of Tumor Growth, and Survival
Animals were evaluated weekly from the day of treatment until occurrence of death, the attainment of predefined endpoints or until the end of the study (90 days post-treatment). Mice were euthanised when the following events occurred: excessive weight loss (body weight loss of 20% at any time-point or of 15% maintained for 72 h compared with pre-treatment weight), presence of ascites and signs of abnormal behaviour.
Bioluminescence imaging (BLI) was performed weekly using the IVIS Lumina II system (PerkinElmer) for tumor growth follow-up of each mouse. Luminescence was quantified using the Living Image® 4.3.1 software (PerkinElmer).
Available Data for PKPD Modelling
The final dataset consisted of data collected in the present study and from previous (unpublished) work throughout different study periods. These studies comprised: (i) in a pharmacokinetic study of the developed PTX-GP-MS (40% crosslinking degree), PTX concentrations measured in 24 female Balb/c mice (aged 7 weeks) between 0.5 h and 9 days post IP injection (7.5 and 35 mg/kg) were included for population pharmacokinetic modelling and (ii) in a pilot study for the development of the microscopic peritoneal carcinomatosis model, where five female Balb/c Nu mice were IP injected with 2 x 106 SK-OV-3-Luc IP1 cells suspended in 1 ml physiological saline without any further treatment. The mortality data from this study were included for survival modelling. Finally, mean plasma concentration values in male Balb/c mice (6–8 weeks old) at a single intravenous (IV) dose of 5.0 mg/kg PTX were digitized from a published study by Gao et al. (25). A plasma-to-blood ratio (PBR) of 0.7 was applied to convert plasma concentrations to blood values (26).
PKPD Model Development
The PKPD model building process was carried out in a sequential manner. The population PK model was developed first, and the resulting predicted full PK profile was used as a predictor for survival modelling. The relationship between the potential predictor, BLI based tumor burden, and survival was also examined.
Population Pharmacokinetic Modelling
Non-linear mixed effects modelling was carried out in NONMEM® (version 7.3, Icon Development Solutions, Ellicott City, MD, USA) using the first-order conditional estimation algorithm with interaction. The model was executed through Perl-speaks-NONMEM (PsN, version 4.60, Uppsala University, Uppsala, Sweden) with the Pirana software (version 2.9.6, Pirana Software & Consulting BV) as interface. Data processing and graphical analysis was performed in R (version 3.4.1, R Foundation for Statistical Computing, Vienna, Austria).
To describe the disposition of PTX, both one- and two-compartment disposition models were explored. A peritoneal compartment was implemented in the PK model for obtaining predicted PTX concentrations in peritoneum for IP injected formulations. The overall apparent absorption process was separated into the release of PTX from the administered formulations in the peritoneum and the transmembrane absorption of the dissolved PTX from peritoneum to blood. For the release of PTX from the formulations into the peritoneum, several release models were tested, including single first-order or zero-order release, parallel or sequential first-order release, parallel or sequential zero-order release, and mixed zero- and first-order release (27). PTX transmembrane absorption was assumed to be first-order and the absorption rate constant was fixed to a reported value of 0.95 h-1 (28). The fluid volume of the mice peritoneal cavity was fixed to a physiologically typical value of 0.4 mL (28). The predicted peritoneal PTX concentrations were calculated as: the amount of released PTX in the peritoneum/peritoneal fluid volume. The bioavailability (F) of the IP formulations relative to IV data was estimated (absolute F).
Inter-individual variability (IIV) was modeled using a log-normal distribution with a mean of zero and a variance of ω2. Unexplained residual variability was explored using additive, proportional, and combined (additive + proportional) residual error models. For the measured concentrations between LLOQ and the limit of detection, implementation of an additional residual error model was tested to account for the potentially larger residual variability.
Model development was guided by the objective function value (OFV) between nested models, Akaike’s information criterion (AIC) between non-nested models, and standard goodness of fit (GOF) plots. A ΔOFV criterion of −3.84 corresponded with a significance level of p < 0.05 for one degree of freedom. The predictive performance of the final model was evaluated using prediction-corrected visual predictive checks (pcVPCs) based on 1000 model-based simulations. The sampling importance resampling (SIR) (n=1000) approach was applied on the final model to evaluate model robustness and obtain non-parametric confidence intervals of the estimates (29).
Survival Modelling
A parametric time-to-event approach was utilized to explore the relationships between the predictors (i.e. blood/peritoneal PK profile, and BLI tumor burden) and survival. The peritoneal PK profile was used to describe the treatment effect on survival, while the blood concentration was assumed to be responsible for toxicity-induced death. All the predictor-survival relationships were implemented in a time-varying manner. The time to death event was based on the initiation of IP treatment. Mice who survived at the time of the last day of follow-up (day 90) were right-censored. The dataset was analyzed using the first-order algorithm in NONMEM®.
The probability density f(t) for observing a death event was described as the product of a function S(t) and hazard h(t) at that time, according to equations 1-3.
Where h0 is the baseline hazard, and Drug (t) is a time-varying drug effect. The survival S(t) is an integral function of the hazard h(t) with respect to time, representing the probability of survival (i.e. not having the death event). The same baseline hazard was assumed for all mice since the variability of baseline hazard could not be estimated with survival data.
The base model was developed by exploring different probability density functions for the baseline hazard, including exponential, Weibull, Gompertz, and log-logistic distributions. For the implementation of measured BLI data as a time-varying predictor, linear interpolation was used between adjacent BLI observations. When using the peritoneal PK profile as a time-varying predictor, the drug effect model was optimized by testing a range of different PKPD models such as linear, log-linear, Emax, and sigmoidal Emax relationships (30). The drug effect delay was also evaluated by incorporating an effect compartment model, an indirect response model, and a transit compartment model. Like the implementation of the treatment effect model, various toxicity models using the blood PK profile as a driving force were also tested to account for drug toxicity-induced death.
Models were selected based on OFV (nested models) and AIC (non-nested models), in combination with simulation-based diagnostics (Kaplan–Meier plots) based on 100 simulations of the model. For the final model, Kaplan–Meier visual predictive check plots based on 1000 simulations were used for internal model validation. The uncertainty (95% confidence intervals) of the parameter estimates was checked through the SIR approach (n=1000).
Dose Simulation of PTXnano-GP-MS
From the observed survival data, PTXnano-GP-MS is the most promising IP delivery system in mice for treating microscopic peritoneal carcinomatosis disease, and the optimal dose falls between 7.5 and 35 mg/kg. With the final PKPD model, survival probabilities (within 90 days) were simulated for different PTXnano-GP-MS dose levels (7.5, 10, 12.5, 15, 20, 25, and 35 mg/kg). 1000 typical subjects (mice with population estimates) were simulated for each scenario. The Kaplan–Meier survival curve and hazard over time were constructed, and the 95% confidence intervals of simulated survival curves at day 40 and 90 were reported.
Results
Exploratory Data Analysis
Figure 1 shows the individual blood concentrations and trends for the different formulations and injection routes applied. As shown in the plot, Nab-PTX displayed a many-fold higher exposure immediately after administration but was eliminated much more rapidly compared to PTX-GP-MS. A burst peak was observed during the first hours for PTX-GP-MS, followed by a prolonged PTX release over several days resulting in relatively stable concentrations. Therefore, dual release rate models were investigated for PTX-GP-MS in order to describe the biphasic PK profile. In total, 195 PTX blood concentrations were included for the population pharmacokinetics analysis. A summary of all collected data is given in Table 1.

Available paclitaxel (PTX) blood concentration data for population pharmacokinetic analysis. A base 10 logarithmic scale was used for the y-axis, and the x-axis was shown using the square root scale. The grey dashed line represents the lower limit of quantification (1 ng/ml) of the analytical method. A Loess smoothing solid line was added to the plot for each formulation.
BLI based tumor growth profiles and Kaplan–Meier survival curves from mice with microscopic peritoneal carcinomatosis disease receiving different IP treatments are presented in supplementary Figure S1 and Fig. 2, respectively. The groups of mice with microscopic peritoneal disease treated with 2 ml physiological saline or 50 mg blank GP-MS suspended in 2 ml saline or without any treatment (from the pilot study) showed comparable survival curves. Therefore, these were merged together as the control group (in total n=26 mice, Fig. 2). The PTXnano-GP-MS groups at doses of 7.5 and 35 mg/kg showed the best survival outcomes; however, some of the mice in the 35 mg/kg dose group of PTXnano-GP-MS experienced toxicity-induced death (indicated by the arrows in Fig. 2, indicating severe hepatotoxicity observed by histopathological analysis). As can be seen in Figure S1, in this dose group, BLI profiles demonstrated no tumor development (except for one mouse) thus good cancer cell killing effect, as indicated by the baseline BLI signals. These observations suggest a toxicity effect on survival that needs to be considered separate from the beneficial treatment effect. During the survival model development, we thus attempted to implement a toxicity model on the hazard to account for the toxicity effect for all PTX delivery systems.

Kaplan-Meier survival curves of the mice with microscopic peritoneal disease after intraperitoneal treatment. Toxicity-related death events are indicated by the arrows.
Population Pharmacokinetics
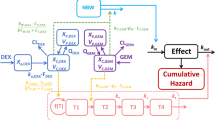
The PK model development was started with first-order release of the formulations into the peritoneum, followed by first-order transmembrane absorption and one-compartment disposition. Introducing a peripheral compartment led to a significant drop in the OFV (ΔOFV of -18.8) which was thus selected for further model development. Among all of the tested release models, the parallel first-order release model (burst + sustained release) gave the lowest AIC for PTX-GP-MS, and the single first-order release model was optimal for Nab-PTX. Owing to sparseness of the data, IIVs could only be identified for the dose fraction parameters (i.e. FORM2F1 and FORM34F1, Table 2) of PTXETOH-GP-MS and PTXnano-GP-MS via burst/sustained release, and the absolute bioavailability parameter of PTXnano-GP-MS (ΔOFV of -86.4 in total). Addition of an extra proportional residual error term for the extrapolated concentrations below LLOQ further improved the model fit (ΔOFV of -55.2). A schematic representation of the final PK model is depicted in Fig. 3. The parameter estimates of the final PK model and associated 95% confidence intervals obtained by the SIR approach are presented in Table 2.

Schematic representation of the final PKPD model. The PK model is represented by the black blocks, while the PD model is displayed by the grey blocks.
The GOF plots in supplementary Figure S2 indicate that the final PK model adequately describes the observed data. The central tendency and spread of the observed PTX blood concentrations were also well captured by the model, as demonstrated in the supplementary pcVPCs plot for all PTX-formulations (Figure S3).
Survival Modelling
In our initial modelling strategy of treatment effect on survival, we first attempted to establish a PKPD relationship between the peritoneal PK profile and BLI tumor burden, and then link the predicted BLI tumor burden to survival. Unfortunately, it was difficult to integrate the PK profile and BLI data both for tumor developing and non-tumor developing mice in a plausible biologically inspired model. As a straightforward evaluation of the predictiveness of BLI tumor burden on survival, the measured BLI data (excluding the 35 mg/kg PTXnano-GP-MS dose group due to obvious toxicity-induced death) were tested as a time-varying covariate on the hazard rate with linear interpolation between adjacent BLI observations. As shown in the supplementary Figure S4, the BLI based survival model gave marked misfits for the control group and the 7.5 mg/kg PTXnano-GP-MS group.
Since the BLI data did not well predict the survival from a modelling point of view, we then decided to test peritoneal concentration as a direct predictor of the treatment effect. The model development path of survival modelling using concentration as predictor is summarized in Table 3. Model building started with the control group data, and a log-logistic distribution model (λ, hazard coefficient; γ, shape factor; equation 7) best characterized the underlying baseline hazard, which greatly increased with time (γ=9.67, Table 4). PTX treatment groups (except the 35 mg/kg PTXnano-GP-MS group) were then added into the dataset. A delayed PTX treatment effect on hazard rate was detected, and the indirect response model (IDR) with linear stimulation of the KOUT (equation 6) driven by peritoneal concentration was found optimal. From the VPC plots of this intermediate IDR model, significant misfits were observed for the Nab-PTX group (supplementary Figure S5). Therefore, we decided to first develop a model for all PTX-GP-MS groups. To account for drug-induced toxicity effects on survival that could obviously be observed in the 35 mg/kg PTXnano-GP-MS dose group, a systemic blood concentration-based sigmoid inhibition model on KOUT was incorporated for all PTX-GP-MS groups and led to a clear improvement of the model fit (ΔOFV: -27.6, supplementary Figure S6). Lastly, the Nab-PTX group was added to the dataset, and we implemented a separate treatment efficacy parameter for Nab-PTX (ΔOFV: -26.3) in order to preserve the good model fit of the PTX-GP-MS groups.
The hazard of the final survival model is described in equations 4-7, and the NONMEM control stream of the final PKPD model is provided in the online supplement.
Where EFF is the stimulation effect of the drug treatment on KOUT, and αPTX-GP-MS and αNab-PTX are the efficacy parameters of PTX-GP-MS and Nab-PTX, respectively. TOX is the inhibition effect of drug-induced toxicity on KOUT, and IC50 is the blood concentration at which the KOUT is reduced by 50%. Drug(t) represents the overall effect of drug treatment efficacy and toxicity projected onto the hazard. KIN and KOUT are the zero-order production rate and first-order removal rate of Drug(t), respectively. Hill is the sigmoidicity factor controlling the steepness of the sigmoid function. This factor was fixed to 20 as there was insufficient information in the data to reliably estimate it.
The final survival model is demonstrated as grey blocks in Fig. 3. The parameter estimates and 95% confidence intervals of the final model are shown in Table 4. The Kaplan–Meier plots of the observed survival data overlaid with the simulated 95% confidence interval from the final model are shown in Fig. 4, stratified by treatment group. In general, the control and PTXETOH-GP-MS groups show good agreement between the observed survival curve and the simulated data. For PTXnano-GP-MS groups, an obvious over-prediction between day 70 and 90 was observed at a dose of 7.5 mg/kg, and a less noticeable over-prediction between day 20 and 30 was found at a dose of 35 mg/kg. Similarly, some misfits were also present for Nab-PTX.

Visual predictive checks of the final survival model, stratified by treatment group. The solid lines represent the observed data while the shaded areas represent the 95% confidence intervals of the simulated data.
Dose effects of PTXnano-GP-MS Derived through Simulations
The effect of a dose of PTXnano-GP-MS given to 1000 typical subjects with microscopic peritoneal carcinomatosis disease on the hazard and survival probability over time is illustrated in Fig. 5. The simulated PK profiles in the peritoneal cavity and blood (central compartment), the response in the Drug(t) compartment, and the hazard over time after removal of toxicity effects (i.e. assuming no toxicity effect on the hazard) for the different doses (7.5 to 35 mg/kg) were provided in Figure S7 of Supplement 1. The reader may refer to these details, as well as to Fig. 5, to better understand how the model behaves and how the drug contributes to the efficacy and toxicity effects on the hazard. For doses between 7.5 and 15 mg/kg, the survival probabilities were near 100% before day 40 and all above 80% at day 90, and there were no significant differences in survival curve among these doses (supplementary Table S1). For doses ≥ 20 mg/kg, the survival probability started to decrease at an early time point (e.g. day 25), and the survival outcomes were significantly worse than those of lower doses (7.5 -15 mg/kg), due to the increased hazard caused by the drug-induced toxicity effect (upper plot in Fig. 5). The simulation demonstrated that the dose range of 7.5-15 mg/kg is expected to be optimal for subjects to achieve a good survival outcome while minimizing the drug-induced toxicity effect on the hazard.

Predicted hazard (upper plot) and median survival probability (bottom plot) of a typical subject without PTXnano-GP-MS treatment (no dose as control) or after receiving different single doses of PTXnano-GP-MS (7.5, 10, 12.5, 15, 20, 25, and 35 mg/kg). The Y-axis of the hazard plot is shown as square root scale.
Discussion
In this work, first a population PK model was developed to characterize the in vivo concentration-time profiles of different PTX formulations. A peritoneal compartment with a fixed peritoneal fluid volume was added during population PK modelling in order to obtain the predicted peritoneal concentrations. The in vivo release models of the formulations were optimized to give the best description of PTX blood concentrations, and a fixed first-order transmembrane absorption rate constant was assumed for the released PTX regardless of the formulations. During the PK modelling, the use of parameter estimates from the best fitted in vitro release model as input for in vivo release model was also considered, but this didn’t give a good description of PTX blood concentrations. This is because PTX-GP-MS released much faster in vivo compared with in vitro, confirmed by the deconvolution results (data not shown). Because the in vitro dissolution of PTX-GP-MS was not fully indicative for its in vivo behavior, the in vitro release data were not used to identify the in vivo release rate. Due to the difficulty of reliably sampling and measuring the dissolved PTX fraction in peritoneal fluid (excluding the particles), PTX concentrations in peritoneum were not determined. The lack of PTX measurements in the peritoneum did not allow us to separately estimate the actual transmembrane absorption rate for different formulations when the in vivo release rate is also estimated, and thus a reported PTX peritoneal transmembrane rate constant had to be adopted. We acknowledge that this is a simplification of the reality of the physiological absorption process of these formulations. We also acknowledge that the predicted peritoneal PK profile from the PK model may not fully reflect reality.
During the subsequent survival modelling, predicted peritoneal concentrations were used as the time-varying input for treatment effects considering that the disease is limited to the peritoneal cavity, whereas blood concentrations were assumed to be responsible for the systemic toxicity effect on survival. To describe the competing effects of PTX treatment and toxicity on survival, a sigmoidal inhibition model of the toxicity effect on KOUT was found to be optimal. Estimation of the sigmoidicity factor was not supported by the data, it was thus empirically fixed to a high value of 20. A sensitivity analysis on this sigmoidicity factor at values of 5, 10, 20, and 50 was performed to inspect its influence on parameter estimates and model fit. The analysis demonstrated that the parameter estimates (changes ≤ ± 10%) and model fit were not influenced by sigmoidicity factor values between 5 and 50. The IC50 estimate (0.695 ng/ml) is the PTX blood concentration inflection point significantly producing (> 0.695 ng/ml)/not significantly having (< 0.695 ng/ml) a drug-induced toxicity signal on the hazard. Depending on the time period that the blood concentration remains above the IC50, the accumulated toxicity effect on the hazard can be lethal (e.g. 35 mg/kg PTXnano-GP-MS) or non-lethal (e.g. 7.5 mg/kg PTX-GP-MS). This indicates the importance of achieving an appropriate concentration during dose optimization of PTX-GP-MS to avoid a toxicity-lethal hazard, as demonstrated in the hazard plot (Fig. 5) for the dose simulations of PTXnano-GP-MS.
The final survival model indicates that Nab-PTX has a larger efficacy parameter than that of PTX-GP-MS. Indeed, pharmacokinetic studies have demonstrated that, compared with PTX, Nab-PTX is associated with a 9-fold greater penetration of PTX into tissues and also a longer tissue retention, thus potentially a higher activity (31). This is because Nab-PTX consists of nanoparticle albumin-bound paclitaxel, which exploits the natural interactions between albumin and the gp60/caveolin-1 receptor pathway, thus leading to enhanced transport across endothelial cell monolayers and greater delivery of paclitaxel to tumors (32). However, as an immediate release formulation, Nab-PTX only provided a very short drug exposure towards residual disease thus finally resulted in a worse survival outcome compared to a sustained release PTX-GP-MS formulation. We acknowledge that the formulation vehicle effect of Nab-PTX was difficult to be captured in its entirety, hence the estimated αNab-PTX might to some extent be a value to be used with caution.
Our study demonstrates that the local PTX concentration is a better survival predictor than the BLI data. This might be due to the fact that BLI is a rather semi-quantitative method since various endogenous and exogenous factors can impact the signal quantification and might lead to erroneous readouts (33). Even with the final concentration-driven survival model, some discordances were seen in the VPC plots across PTX formulations. This might partly be explained by the sparse blood sampling scheme and small sample size of the survival data. Sparse blood sampling can impede the estimates of the IIV of population PK parameters and may lead to less precise estimates of individual PK parameters, whereas the small sample size can lead to a large uncertainty of the observed Kaplan–Meier survival curve. For example, only the typical concentration-time profile can be predicted for Nab-PTX because only six blood concentrations were available. The difficulty of appropriately modelling the underlying formulation vehicle effect (e.g. Nab-PTX) on PKPD might be an additional explanation.
The main strength of this model is its ability to appropriately describe the competing effects of PTX treatment efficiency and toxicity on survival using a parametric approach. The ability to simultaneously model treatment and toxicity effects on survival allows for a better understanding of the overall drug effect. Also, as demonstrated, a PK driven PD model gives flexibility for using the model to simulate unexplored scenarios to find the optimal dose achieving satisfactory survival and avoiding unwanted lethal toxicity.
Our present study showed a pronounced improvement on survival rate of a sustained release system (i.e. PTX-GP-MS) over an immediate release system (i.e. Nab-PTX) in a microscopic peritoneal carcinomatosis mouse model. PTXnano-GP-MS was identified to be superior to PTXEtOH-GP-MS and needs to provide a suitably sustained concentration to avoid a toxicity-lethal hazard, as indicated by the PKPD model. Further simulations of new treatment regimens of PTXnano-GP-MS indicate that 7.5 – 15 mg/kg seems to be the optimal dose range to provide the optimal balance between efficacy and safety. The use of PKPD modelling and simulation allows us to provide answers on the optimal efficacy and safety of new drug formulation faster and at a lower cost. In the future, a prospective evaluation is warranted to validate the findings from our simulation study. Also, further efficacy and toxicity evaluations of PTX-GP-MS in other animal models (non-rodent species) are planned. Thereafter, we intend to combine all of the obtained pre-clinical knowledge into a modelling framework aimed at deriving a suitable dosage form to be tested in a Phase I clinical trial.
Our results of PTX-GP-MS in a microscopic peritoneal ovarian cancer disease mouse model align with earlier work of other PTX sustained IP delivery formulations in peritoneal cancer xenograft mice models with macroscopic tumor disease (21,22). Vassileva et al. demonstrated that an implantable PTX drug delivery system gave about 30-fold lower tumor burden and a 2-times higher survival rate in comparison to intermittent Taxol® solution therapy in ovarian cancer tumor-bearing mice (22). Similarly, a 2-times longer survival extension of PTX loaded polymeric microparticles over paclitaxel Cremophor®EL solution in mice bearing human pancreatic tumors was reported by Tsai et al (21). Compared with previously reported sustained PTX IP formulations, our developed PTX-GP-MS formulation has a dual application in the treatment of advanced ovarian cancer: 1) the prevention of peritoneal adhesions (pathological attachments between tissues and organs of the abdominal cavity) after surgery (shown in our previous work) (23) and 2) the improvement of the efficacy of IP chemotherapy by providing a prolonged drug delivery of a chemotherapeutic agent. Peritoneal adhesions are a common complication after CRS that compromise quality of life by causing small bowel obstruction, chronic abdominal pain and female infertility (34). The promising preclinical results and the potential dual application of the concept in the treatment modality of advanced ovarian cancers marks the clinical relevance and supports the continued (pre)clinical evaluation of the developed PTX-GP-MS.
A limitation of the study is the small sample size of the survival data and the limited PK samples per mouse, a natural limitation for this animal species. With more data, knowledge of inter-individual variability in PK and uncertainty in survival could be obtained, which would further strengthen our conclusions. Nevertheless, our work also demonstrates the power and advantages of the non-linear mixed effects modelling approach, for pooling and maximizing the utility of this limited amount of data in a way still suitable to inform dose optimization, e.g. in early preclinical trials, even when inherently confronted with sparse data.
Conclusion
In the current work, a PKPD model was developed to simultaneously describe the competing effects of efficacy and toxicity of PTX on survival for PTX-GP-MS and Nab-PTX formulations in mice with microscopic peritoneal carcinomatosis disease. The developed model allows in a flexible way to explore the interplay of positive treatment effects and toxicity of PTX on survival outcome. PTXnano-GP-MS was identified as the most promising IP drug delivery system for treating peritoneal minimal residual disease, and model-based simulations indicate that 7.5 – 15 mg/kg seems to be the optimal dose range.
Abbreviations
- AIC:
-
Akaike’s information criterion
- BLI:
-
Bioluminescence imaging
- CRS:
-
Cytoreductive surgery
- DBS:
-
Dried blood spot
- GOF:
-
Goodness of fit
- GP-MS:
-
Genipin-crosslinked gelatin microspheres
- IIV:
-
Inter-individual variability
- IPC:
-
Intraperitoneal chemotherapy
- LLOQ:
-
Lower limit of quantification
- Nab-PTX:
-
Paclitaxel-loaded albumin nanoparticles
- OFV:
-
Objective function value
- pcVPCs:
-
Prediction-corrected visual predictive checks
- PKPD:
-
Pharmacokinetic-pharmacodynamic
- pMRD:
-
Peritoneal minimal residual disease
- PTX:
-
Paclitaxel
- PTX-GP-MS:
-
Paclitaxel-loaded genipin-crosslinked gelatin microspheres
- PTXEtOH-GP-MS:
-
Paclitaxel-ethanolic loaded genipin-crosslinked gelatin microspheres
- PTXnano-GP-MS:
-
Paclitaxel-ethanolic loaded genipin-crosslinked gelatin microspheres
- SIR:
-
Sampling importance resampling
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Nosov V, Su F, Amneus M, Birrer M, Robins T, Kotlerman J, et al. Validation of serum biomarkers for detection of early-stage ovarian cancer. Am J Obstet Gynecol. 2009;200(6):639. e631–9.
Tummala MK, Alagarsamy S, McGuire WP. Intraperitoneal chemotherapy: standard of care for patients with minimal residual stage III ovarian cancer? Expert Rev Anticancer Ther. 2008;8(7):1135–47.
Du Bois A. Treatment of advanced ovarian cancer. Eur J Cancer. 2001;37:1–7.
McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6.
Ozols R. Treatment goals in ovarian cancer. Int J Gynecol Cancer. 2005;15:3–11.
Berrino F, De Angelis R, Sant M, Rosso S, Lasota MB, Coebergh JW, Santaquilani M, Group EW. Survival for eight major cancers and all cancers combined for European adults diagnosed in 1995–99: results of the EUROCARE-4 study. Lancet Oncol 2007;8(9):773-783.
Du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d'Investigateurs Nationaux Pour les Etudes des Cancers de l'Ovaire (GINECO). Cancer. 2009;115(6):1234–44.
Chefetz I, Alvero A, Holmberg J, Lebowitz N, Craveiro V, Yang-Hartwich Y, et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle. 2013;12(3):511–21.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. nature. 2001;414(6859):105.
Bijelic L, Jonson A, Sugarbaker P. Systematic review of cytoreductive surgery and heated intraoperative intraperitoneal chemotherapy for treatment of peritoneal carcinomatosis in primary and recurrent ovarian cancer. Ann Oncol. 2007;18(12):1943–50.
Chua TC, Robertson G, Liauw W, Farrell R, Yan TD, Morris DL. Intraoperative hyperthermic intraperitoneal chemotherapy after cytoreductive surgery in ovarian cancer peritoneal carcinomatosis: systematic review of current results. J Cancer Res Clin Oncol. 2009;135(12):1637–45.
Oršolić N, Bevanda M, Kujundžić N, Plazonic A, Stajcar D, Kujundžić M. Prevention of peritoneal carcinomatosis in mice by combining hyperthermal intraperitoneal chemotherapy with the water extract from Burr parsley (Caucalis platycarpos L.). Planta Med. 2010;76(08):773–9.
Bakrin N, Classe J, Pomel C, Gouy S, Chene G, Glehen O. Hyperthermic intraperitoneal chemotherapy (HIPEC) in ovarian cancer. J Visc Surg. 2014;151(5):347–53.
De Bree E, Helm CW. Hyperthermic intraperitoneal chemotherapy in ovarian cancer: rationale and clinical data. Expert Rev Anticancer Ther. 2012;12(7):895–911.
Markman M, Bundy BN, Alberts DS, Fowler JM, Clark-Pearson DL, Carson LF, et al. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J Clin Oncol. 2001;19(4):1001–7.
Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34–43.
Van Driel WJ, Koole SN, Sikorska K, Schagen van Leeuwen JH, Schreuder HW, Hermans RH, et al. Hyperthermic intraperitoneal chemotherapy in ovarian cancer. N Engl J Med. 2018;378(3):230–40.
Spiliotis J, Vaxevanidou A, Sergouniotis F, Lambropoulou E, Datsis A, Christopoulou A. The role of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in the management of recurrent advanced ovarian cancer: a prospective study. J BUON. 2011;16(1):74–9.
Markman M, Rowinsky E, Hakes T, Reichman B, Jones W, Lewis Jr JL, Rubin S, Curtin J, Barakat R, Phillips M. Phase I trial of intraperitoneal taxol: a Gynecoloic Oncology Group study. J Clin Oncol 1992;10(9):1485-1491.
Tsai M, Lu Z, Wientjes MG, Au JL-S. Paclitaxel-loaded polymeric microparticles: quantitative relationships between in vitro drug release rate and in vivo pharmacodynamics. J Control Release. 2013;172(3):737–44.
Vassileva V, Moriyama E, De Souza R, Grant J, Allen C, Wilson B, et al. Efficacy assessment of sustained intraperitoneal paclitaxel therapy in a murine model of ovarian cancer using bioluminescent imaging. Br J Cancer. 2008;99(12):2037.
De Clercq K, Schelfhout C, Bracke M, De Wever O, Van Bockstal M, Ceelen W, et al. Genipin-crosslinked gelatin microspheres as a strategy to prevent postsurgical peritoneal adhesions: In vitro and in vivo characterization. Biomaterials. 2016;96:33–46.
Xie F, De Thaye E, Vermeulen A, Van Bocxlaer J, Colin P. A dried blood spot assay for paclitaxel and its metabolites. J Pharm Biomed Anal. 2018;148:307–15.
Gao Y, Shen JK, Choy E, Zhang Z, Mankin HJ, Hornicek FJ, et al. Pharmacokinetics and tolerability of NSC23925b, a novel P-glycoprotein inhibitor: preclinical study in mice and rats. Sci Rep. 2016;6:25659.
Liu X-R, Wu K-C, Huang Y, Sun J-B, Ke X-Y, Wang J-C, et al. In vitro and in vivo studies on plasma-to-blood ratio of paclitaxel in human, rabbit and rat blood fractions. Biol Pharm Bull. 2008;31(6):1215–20.
Owen JS, Fiedler-Kelly J. Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models: John Wiley & Sons; 2014.
Au JL-S, Guo P, Gao Y, Lu Z, Wientjes MG, Tsai M, et al. Multiscale tumor spatiokinetic model for intraperitoneal therapy. AAPS J. 2014;16(3):424–39.
Dosne A-G, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn. 2016;43(6):583–96.
Upton R, Mould D. Basic concepts in population modeling, simulation, and Model-Based drug development: Part 3—Introduction to pharmacodynamic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2014;3(1):1–16.
Schettini F, Giuliano M, De Placido S, Arpino G. Nab-paclitaxel for the treatment of triple-negative breast cancer: Rationale, clinical data and future perspectives. Cancer Treat Rev. 2016;50:129–41.
Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–24.
Badr CE. Bioluminescence imaging: basics and practical limitations. In. Bioluminescent Imaging: Springer; 2014. p. 1-18.
Van Goor H. Consequences and complications of peritoneal adhesions. Color Dis. 2007;9:25–34.
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by a Concerted Research Action (GOA) grant (project number: 01G02916) from Ghent University. Feifan Xie acknowledges the China Scholarship Council (CSC) for his Ph.D. grant. An Vermeulen is an 80% employee of Johnson and Johnson and owns J&J stock/stock options. She is also a visiting Professor at Ghent University. All other authors declare have no conflicts of interest that are directly relevant to the contents of this manuscript. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. (Animal Ethics Committee of the Faculty of Medicine at Ghent University, permit number: ECD 17/83).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xie, F., De Clercq, K., Vervaet, C. et al. Model-based analysis of treatment effects of paclitaxel microspheres in a microscopic peritoneal carcinomatosis model in mice. Pharm Res 36, 127 (2019). https://doi.org/10.1007/s11095-019-2660-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11095-019-2660-1