
Abstract
Optimal development of the embryo and the fetus depends on placental passage of gases, nutrients, hormones, and waste products. These molecules are transferred across the placenta via passive diffusion, carrier-mediated cellular uptake and efflux, and transcytosis pathways. The same mechanisms additionally control the rate and extent of transplacental transfer of drugs taken by the pregnant mother. Essentially all drugs cross the placenta to a certain extent, and some accumulate in the placenta itself at levels that can even exceed those in maternal plasma. Hence, even drugs that are not efficiently transferred across the placenta may indirectly affect fetal development by interfering with placental function. In this article, we describe key properties of the placental barrier and their modulation by medications. We highlight implications for pharmacotherapy and novel approaches for drug delivery in pregnant women and their fetuses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extensive evidence for the permeability of the placenta existed long before the thalidomide disaster. As long ago as 1904, JW Ballantyne, a teratologist in Edinburgh, listed “varieties of fetal morbid states”, including alcoholism, lead-poisoning and morphine-poisoning (1). Sulfonamide drug transfer to the fetus was reported in the late 1930s (2,3), followed by studies in the 1940s and 1950 that demonstrated transplacental transfer of many other drugs (4). The damaging effect of maternal rubella on the fetus was proven by NM Gregg in 1941 (1). However, until the 1960s drugs were prescribed freely during pregnancy, based on the belief that the placenta is a perfect barrier against harmful influences in the environment (1). Moreover, studies on the placental transfer of hormones and nutrients in pregnant women utilized radiolabeled compounds as substrates (5,6,7).
The placenta plays a significant role in fetal and adult health, and placental pathology is implicated in all common obstetric complications, including preeclampsia and intrauterine growth restriction (8,9,10). When pregnant women are treated with medications, the placenta is not only the fetal gatekeeper but is also by itself a potential target to their therapeutic and adverse effects. The consequences of fetal (and placental) exposure to medications can be benign or involve structural or behavioral teratogenicity, or even termination of pregnancy, and are often unknown (11). Furthermore, neurodevelopmental and other defects may be detected long after the drug has been on the market. The pregnancy outcomes depend on intrinsic drug effects and the duration, length and extent of exposure.
In this review, we present the current understanding of placental fate and effects of drugs. To paraphrase Alfred Goodman et al., we describe what the placenta does to the drug (placental pharmacokinetics) and what the drug does to the placental barrier (placental pharmacodynamics) (12). The focus is on the human placenta, but we also present studies conducted in animal models.
The Gate: How Substances are Transferred Into and Across the Placenta
The placenta is formed from the zygote at the start of pregnancy and has the fetal genetic composition. During most of the first trimester of pregnancy maternal blood flow to the placenta is limited, and maternal–fetal exchange takes place via the extracoelomic fluid. By 12 weeks of gestation, the exocoelomic cavity disappears, the amniotic cavity fills the entire uterine cavity, and maternal blood perfuses the placenta. At term, maternal blood supply to the placenta is approximately 30% of her cardiac output (13).
The mature human placenta is a discoid organ, weighing approximately 500 g. It is divided into several cotyledons, each representing an independent functional vascular unit. Placental cotyledons contain branched fetal villous trees, bathed by maternal blood (10,14,15). The villous trees are separated from maternal circulation by an epithelium-like layer, the villous trophoblast. At the end of the first trimester of pregnancy, all villi are covered by a two-layered trophoblast epithelium, consisting of the cytotrophoblast and the syncytiotrophoblast (15). As the pregnancy proceeds, the cytotrophoblast layer partially disappears and the syncytiotrophoblast layer is progressively thinned. At term, a single syncytiotrophoblast layer separates maternal blood from fetal capillary endothelium (10,14).
The syncytiotrophoblast is considered the functional part of the placental barrier. A single placenta has a single syncytiotrophoblast, which covers all villous trees and comprises a continuous layer without lateral cell borders (15). The syncytiotrophoblast is a polar layer with a basal membrane facing the fetal side and a microvillous apical membrane in direct contact with maternal blood. The microvilli provide a surface amplification factor of 5–7 which facilitates solute transfer across the placenta (14,15). Yet, the syncytiotrophoblast may function as a diffusional barrier. Carrier proteins and cellular enzymes can either restrict or facilitate substrate transfer across the placenta. A non-fenestrated endothelium which lines the fetal vessels throughout gestation further reduces paracellular transport between mother and fetus (14,15). Three main processes mediate the transplacental passage of nutrients, hormones, waste products and drugs across the intact placental membranes: passive diffusion, carrier-mediated transport and transcytosis.
Passive Diffusion
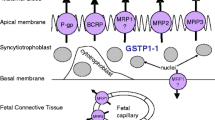
Passive diffusion is the predominant route by which many drugs cross the placenta, and applies mostly to hydrophobic molecules of less than 600 Da. High molecular weight drugs such as insulins and heparins are not transported to the fetus in significant amounts (Fig. 1) (16,17,18). Theoretically, the unbound and unionized concentrations of such drugs should be similar in maternal and fetal circulation (19). However, the total concentrations of drugs that simply diffuse across the placenta (or their metabolites) in fetal blood and tissues may exceed maternal blood levels (Fig. 1). The pH of fetal plasma is lower as compared to that of the mother, by 0.1 of a pH unit, and weakly basic drugs may accumulate in the fetus, especially during fetal distress and acidosis. Drug binding to fetal proteins may form a reservoir from which the drug is slowly released, thereby contributing to longer retention of the drug in the fetal compartment and extended effect on the fetus (20). The diffusion rate of solutes that rapidly diffuse across the placenta such as inhalation anesthetics is influenced by the rate of placental blood flow (“flow-limited”). Placental blood flow increases throughout pregnancy, and, together with the increase in placental surface area and the reduction in the trophoblast’s thickness, can result in accelerated passive diffusion of drugs as pregnancy progresses (21). Placental blood flow can be affected by various conditions, including medications taken by the mother. As an example, some inhalation anesthetics can affect their own transfer due to their effect on maternal respiratory system and circulation (4).

Mechanisms of transplacental transfer of drugs. (a) Schematic diagram showing the major mechanisms contributing to the passage of drugs into and across the syncytiotrophoblast. Drugs and their metabolites can cross the placenta by passive diffusion, carrier-mediate transfer (facilitated diffusion or active transport), or transcytosis. Within the syncytium, drugs can undergo metabolism. (b) Maternal to fetal concentration ratios of representative drugs known to be transferred across the placenta. The drugs are grouped based on known mechanisms of their placental transfer, as shown in (a). Drug transfer was measured ex vivo in the perfused human cotyledon model (fetal to maternal ratio; F:M) and in vivo, in cord and maternal plasma (cord to maternal ratio; C;M). Insulin and low molecular weight heparins are large molecules that are not transferred to fetal blood at significant amounts. Phenobarbital and methohexital rapidly diffuse across the placental membranes. Metformin, valproic acid and cimetidine are predominantly substrates of uptake carriers. Digoxin, cyclosporine, glyburide, indinavir and saquinavir are substrates of P-glycoprotein, the breast cancer resistance protein (BCRP), or both (although uptake transporters are also involved in their transplacental distribution). Values of drugs which are primarily efflux transporter substrates are typically less than unity. IgG and therapeutic IgG-based antibodies are transferred across the placenta by receptor-mediated transcytosis (via FcRn receptors), unless they are based on the AB fragment only (certolizumab). Because transcytosis is a slow process, perfusion studies tend to underestimate their cord:maternal concentration ratios. Note that the transplacental transfer of vedolizumab, natalizumab, adalimumab, ustekinumab and infliximab has been reported only in vivo. LMWH, low molecular weight heparin. The fetal:maternal ratios of insulin lispro, phenobarbital, methohexital, metformin, valproic acid, digoxin, and glyburide were corrected for maternal and fetal/neonatal protein binding and blood pH (16). References are provided in Supplementary Material.
Carrier-Mediated Transport
Placental carriers mediate the uptake of hormones and nutrients from maternal blood into the fetus and the removal of metabolites back to maternal blood, and take part in the homeostasis of placental tissue itself (21). The impact of a carrier on substrate transfer across the placenta would be substantial only if this transporter is the predominant route of placental transfer of the drug (21). Carrier proteins share common features such as saturation kinetics and the ability to be competitively inhibited (14), but may differ from each other with respect to substrate selectivity, placental localization, and transcriptional regulation. Carriers can facilitate the transmembranal transfer of substrate drugs down concentration gradients (facilitated diffusion) or against them (active transport). Uptake carriers are involved in substrate influx into the syncytium and fetal capillary cells, whereas efflux carriers export their substrates from cells. Some carriers can exhibit bidirectional substrate transport. Most placental carrier proteins are members of two major transporter superfamilies, ABC (adenosine triphosphate binding cassette) and SLC (solute carrier) transporters (22,23).
Nutrients and other essential compounds may be transferred across the apical and basolateral membranes of syncytiotrophoblasts by carriers which are relatively substrate-specific, such as those involved in the transfer of folic acid (vitamin B9), including the reduced folate carrier (RFC; SLC19A1) and the proton-coupled folate transporter (SLC26A1) (24). Although such carriers are not likely to be major contributors to transplacental drug transfer, they can become drug targets, as described below. In other cases, placental carriers may be “polyspecific” and significantly contribute to drug disposition (25). For example, substrates of the L-type amino acid transporter (LAT1; SLC7A5) include, in addition to thyroid hormones, gabapentin, melphalan, baclofen, methyldopa, and methyltyrosine (26). Placental carriers have been shown to play key roles in the distribution of many other compounds essential for fetal development and placental function, such as monoamines nucleosides and carnitine, as described below (27).
Transporters of the Adenosine Triphosphate Binding Cassette Superfamily
ABC transporters are efflux pumps which depend on energy derived from ATP hydrolysis and function to remove substrates from cells or their membranes. Many placental ABC transporters are located on the maternal-facing membrane of the syncytiotrophoblast, a position suitable for drug removal from the placenta towards the maternal circulation (28). Accordingly, the fetal concentrations of drugs which are predominantly substrates of efflux transporters are usually lower than in maternal plasma (Fig. 1). The most studied placental ABC transporters and likely the most clinically relevant ones are P-glycoprotein (P-gp) and the breast cancer resistance protein (BCRP) (23). Functional activities of several multidrug resistance proteins (MRPs) have also been identified in the human placenta.
P-gp (MDR1)
P-gp is expressed in humans on the apical membrane of syncytiotrophoblasts and on villus maternal and fetal endothelium. P-gp is encoded in humans by the ABCB1 gene (29). In rodents, two homologs genes, Abcb1a and Abcb1b, encode for the corresponding Mdr1a and Mdr1b proteins (30). P-gp substrates are generally lipophilic molecules, of which many are cationic, but several neutral of anionic compounds are also P-gp substrates. Substrate drugs include HIV protease inhibitors, immunosuppressive drugs, and chemotherapeutic compounds such as anthracyclines, vinca alkaloids and tyrosine kinase inhibitors (31). P-gp has been detected in syncytiotrophoblast cells throughout gestation, but expression of its mRNA and protein decreases with gestational age (32,33). Inter-individual variability in P-gp-mediated transport has been suggested to result, as least in part, from single nucleotide polymorphisms of the MDR1 gene, but the importance of such polymorphisms at the human placenta has yet to be established (34,35).
The role of P-gp in fetal protection was first demonstrated in variants of a CF-1 mouse strain containing a defective Mdr1a gene. Following exposure of pregnant mice to an isomer of the pesticide avermectin, cleft palate was observed in all fetuses homozygous for P-gp deficiency and 30% of heterozygous fetuses. The homozygous wild type fetuses were insensitive to avermectin (36). In fetuses devoid of both Abcb1 genes, the concentrations of [3H]digoxin, [14C]saquinavir, and paclitaxel were 2.4, 7, and 16-fold higher, respectively, then in wild-type fetuses of the same dams (37). In the same study, pharmacological inhibition of P-gp in pregnant wild type mice produced a similar increase in drug distribution to that observed with genetic knockout of the Abcb1 genes, indicating that placental P-gp can be completely inhibited in mice by pharmacological means. A lesser extent of placental P-gp inhibition was achieved in pregnant non-human primates using [11C]verapamil as the P-gp substrate and cyclosporine as the inhibitor (38). Further analysis of the data obtained from a larger set of animals demonstrated that, in the absence of functional P-gp, tissue blood flow limits the distribution of [11C]verapamil across the macaque placenta (39). That analysis also highlighted the importance of flow-normalized distribution clearance (K1) as a measure of placental P-gp activity.
In humans, ethical and logistical considerations largely prevent the determination of placental drug transfer until delivery, when a single cord blood sample can be obtained. Typically, the cord-to-maternal concentration ratios of P-gp substrates are below unity, but the time of sampling with regard to drug ingestion by the mother can affect the ratio. Ex vivo and in vitro models which utilize human placentas have been reviewed by Myllynen and Vähäkangas (40). Among these, perfused human placentas provide the only opportunity to investigate placental transfer of substances in organized human placentas (16). Such studies allow measurement over several hours of drug transfer between compartments that represent the maternal and fetal circulations under controlled conditions. The relative contribution of individual transporters to drug transfer can be inferred from comparing drug passage in the presence and the absence of selective transporter inhibitors. The perfusion studies highlighted the importance of P-gp in the human placenta at term, by demonstrating that P-gp inhibition can enhance substrate transfer to the fetal compartment by up to 10-fold (Fig. 2). However, little is known about the role of placental P-gp during the course of 9 months of the human pregnancy.

Effect of P-gp inhibition on drug transfer across perfused human cotyledons. In each study, the maternal-to-fetal transfer was measured at the presence or the absence of selective P-gp inhibitors. a The perfusion study was conducted at physiological albumin concentration (30 and 40 g/L). b Perfusion at low albumin concentration (10 g/L). Data are presented as fold change in substrate distribution to the fetus. References and the inhibitors utilized in each study are listed in Supplementary Material.
The Breast Cancer Resistance Protein
Similar to P-gp, placental BCRP (ABCG2) has been localized to the brush border (apical) membrane of syncytiotrophoblast cells (41). BCRP is expressed in the human placenta throughout pregnancy but its levels may vary (28). In addition, BCRP exhibits polymorphism that can alter its expression and potentially its protective role in the placenta. Kobayashi et al. studied the relationships between ABCG2 polymorphisms and BCRP expression in 100 human placentas. They reported on lower BCRP protein expression in placentas homozygotes for the A421 allele than in those for the C421 allele. The predominant allelic expression pattern of BCRP in placental samples was found to be biallelic (42). In a study in the Han Chinese population that compared the association between ABCG2 polymorphisms and isolated septal defects, more cases than controls were carriers of the 34G > A polymorphism. ABCG2 mRNA and protein expression of the GG genotype was significantly higher than that of the AA genotype in the placentas of the study participants (43). BCRP has additionally been suggested to protect placental trophoblasts themselves against cytokine- and ceramide-induced apoptosis (44).
Many BCRP substrates are sulfate and glucuronide conjugates, but BCRP can also transfer unconjugated drugs, including methotrexate, mitoxantrone, topotecan, dipyridamole, nitrofurantoin and glyburide (28). Drugs such as ritonavir, saquinavir, nelfinavir, and nicadipine and cyclosporine can inhibit BCRP (45,46,47).
The discovery that murine BCRP is an important contributor to the bioluminescence of the luciferase substrate D-luciferin in vivo paved the road for the use of D-luciferin and its derivatives as probes of BCRP activity at the blood-brain barrier (48) and the placenta (49) in transgenic mouse models.
Multidrug Resistance-Associated Proteins
MRPs are predominantly organic anion transporters but in addition transport neutral organic compounds. The substrate and inhibitor selectivity of individual MRPs partially overlaps with that of other MRPs, P-gp, BCRP, and organic anion transporters (28,50). Importantly, folic acid and its antagonist, methotrexate, are substrates of several MRPs expressed in the human placenta (31).
MRP1 (ABCC1) is expressed on fetal blood vessel endothelia and, at lower levels, at the basal membrane of term human syncytiotrophoblasts (51). Some expression has also been detected at the apical layer of syncytiotrophoblasts (52). This pattern of expression suggests that MRP1 primarily removes compounds from fetal blood (35). MRP1 substrates are anionic glutathione, glucuronide and sulfate conjugates of endogenous substances such as bilirubin, 17β estradiol and prostaglandins, but also unconjugated compounds, including folic acid, methotrexate, saquinavir and ritonavir (31). MRP1 levels were found to be significantly higher in term human placentas than in first trimester placental samples (53).
MRP2 (ABCC2), unlike MRP1, is expressed at the apical human syncytiotrophoblast membrane (52), but the substrate specificity of the two transporters is very similar (35). MRP2 mRNA expression at the human placenta is lower than that of MRP1 (54), but may be induced under pathological conditions and related treatments, e.g. in women with intrahepatic cholestasis of pregnancy treated with ursodeoxycholate (55).
The human placenta expresses several other MRPs whose contribution to drug transfer has yet to be clarified, including MRP3 (ABCC3) (56), MRP4 (ABCC4) (55) and MRP 5 (ABCC5). MRP5 substrate selectivity is unique as it includes cyclic nucleotides (57).
Transporters of the Solute Carrier Superfamily
SLCs known to be expressed in the placenta and involved in transplacental drug transfer include organic anion transporting polypeptides, organic anion transporters, organic cation transporters, monoamine transporters and equilibrative nucleoside transporters. The involvement of SLC-mediated transplacental transfer varies across drugs (Fig. 1).
Organic Anion Transporting Polypeptides (OATPs)
Several members of the OATP subfamily are expressed on the placenta. These include OATPs 2B1 (SLCO2B1), 4A1 (SLCO4A1), and, to a lesser extent, 1A2 (SLCO1A2) (54). OATP2B1 is localized at the basal membrane of the syncytiotrophoblast (58), whereas OATP4A1 is predominantly expressed on the apical membrane (59). The placental OATP isoforms are involved in the transmembrane transport of prostaglandins, thyroid hormones, steroid hormone metabolites, and drugs, including fexofenadine, saquinavir, glyburide, imatinib, methotrexate (60) and repaglinide (61). Basolateral OATP2B1 and apical BCRP may act in concert to shuttle steroid sulfates from fetal to maternal circulation (62).
Organic Anion Transporters (OATs)
OAT4 (SLC22A11) is the most abundant OAT on the human placenta, and is expressed on the basal surface of syncytiotrophoblasts (63). OAT4 is involved in the transport of steroid sulfates, steroid precursors for estrogen synthesis and several drugs, including zidovudine (64).
Organic Cation Transporters
Organic cation transporters include the potential-sensitive OCTs and the proton gradient-driven carnitine transporters OCTNs. The predominant OCT in the human placenta is OCT3 (SLC22A3), which mediates substrate uptake at the basolateral side (65). OCT3 exhibits high affinity for monoamines and is considered to be an extraneuronal monoamine transporter (66). It may play a role in catecholamine clearance from fetal circulation (27) and release of acetylcholine from the placenta (67). OCT3 has been suggested to be involved in the transplacental transfer of metformin, a cationic and highly hydrophilic drug (68). However, metformin transport was not shown to be inhibited by the OCT inhibitor cimetidine (69). OCT3 may contribute to vectorial transport of organic cations across the placenta by mediating their uptake from the basolateral membrane (fetal side). Their subsequent removal from cells across the apical membrane may be mediated by other mechanisms, such as efflux by ABC transporters.
OCTN1 (SLC22A4) and OCTN2 (SLC22A5) are expressed at the maternal-facing membrane of syncytiotrophoblasts, where they can facilitate carnitine transfer from maternal blood to the placenta and the fetus (27,70). Drug substrates of OCTN 1 and 2 include verapamil and quinidine (71).
Nucleoside Transporters
Nucleosides are highly hydrophilic molecules, which depend on carriers for crossing membrane barriers. Two families of nucleoside transporters have been described in humans, equilibrative transporters (ENTs; SLC29) and concentrative transporters (CNTs; SLC28), and several members of these families are enriched in the placenta (72,73). Significant expression of ENT1, ENT2, and CNT2 mRNAs and ENT1 and ENT2 proteins was detected in term human placentas. The ENT1 protein was localized to the brush-border membrane with positive staining in endothelial cells of blood vessels. ENT2 staining was less intense than that of ENT1, and was predominantly cytoplasmic (73). The substrates of nucleoside transporters are purine and pyrimidine nucleosides, but also several related anticancer and antiretroviral drugs, including 6-mercaptopurine, cytarabine and gemcitabine (74). In mice, Ent1 affects the rate but not the extent of ribavirin accumulation in the fetus (75).
Monoamine Transporters
The serotonin transporter (SERT; SLC6A4) and the norepinephrine transporter (NET; SLC6A2) are expressed on the maternal-facing membrane of syncytiotrophoblasts and mediate monoamine uptake into the placenta (76,77). Amphetamine and methamphetamine compete with endogenous monoamines for binding to the carrier active site and enter placental cells instead of the monoamine substrates (78). Amphetamine and methylphenidate have nanomolar affinity for NET and micromolar affinity for SERT. Another attention-deficit/hyperactivity disorder medication, atomoxetine, binds both NET and SERT at nanomolar concentrations (79). Whether these drugs are NET or SERT substrates is unknown. Additionally, a variety of antidepressants are established inhibitors of monoamine transporters (78,80). Their effects on placental SERT and NET have yet to be elucidated.
Endocytosis and Exocytosis
Endocytosis consists of several mechanisms, including pinocytosis (fluid-phase endocytosis), phagocytosis (engulfment and destruction of extracellular material), and receptor-mediated endocytosis. The latter involves selective internalization of specific extracellular ligands into cytoplasmic vesicles through their interactions with specific receptors. Particularly, IgG or IgG-based antibodies bind with high affinity to the neonatal Fc receptors in the acidic environment (approximately pH 6) of the endosome. Binding prevents their transfer into the lysosome, and they can be transported in coated vesicles across the trophoblastic layers and released into the fetal circulation (or back into maternal plasma) at physiological pH (81). The rate of endocytosis-mediated transfer is determined by several factors, including membrane fluidity, vesicle mobility in the cytosol, and, in the case of receptor-mediated endocytosis, the rate of receptor turnover (82). Additional examples for placental receptors include folate receptor alpha (FOL1) and megalin. Megalin (LRP2) is most extensively expressed on the apical (lumen-facing) surface of renal proximal tubular epithelial cells, and is involved in the endocytosis of vitamin B12 (83). Megalin-mediated endocytosis has been suggested as the primary mechanism for renal aminoglycoside accumulation and toxicity and was shown to be involved in gentamycin uptake by BeWo cells (84). Hence, endocytosis can contribute to the accumulation of gentamicin in the human placenta (85) and be involved in gentamicin’s transfer to fetal blood. Gentamicin’s transfer across the placenta has been profiled in a recent clinical study (NCT02427932; clinicaltrials.gov). Findings from this study can contribute to understanding of the factors that govern gentamicin’s passage across the placenta. Although, in most cases, transcytosis does not have a significant effect on the extent of transfer of small-molecule drugs, it is relevant for two emerging treatment approaches, namely therapeutic antibodies and drug-containing nanoformulations such as nanoparticles and liposomes.
Antibodies and related products, including antibody–drug conjugates, are the fastest growing class of therapeutic agents (86,87). The most commonly used therapeutic antibodies are of the IgG family (87,88), which are handled by the placenta similarly to naturally occurring maternal IgG antibodies. Transport is mediated by Fc receptors which become functional at week 13 of gestation (89). IgG transport is bidirectional, possibly as a means of achieving homeostasis of IgG within the fetal circulation. Fab’ fragments, which do not bind to Fc receptors, can also be transported across the placenta, but transport is significantly less efficient (88). The rate of transport increases as the pregnancy progresses and is thought to be most efficient in the third trimester, during which 80% of transfer occurs (88,90). In one study, fetal concentrations of IgG at gestational weeks 17–22 were only 5–10% of the maternal levels, but exceeded the maternal levels at term (91). Likewise, the levels of therapeutic antibodies in cord blood may be higher than in the mother at the time of delivery (90,92). For example, in a study of 31 pregnant women with inflammatory bowel disease receiving infliximab, adalimumab, or certolizumab pegol, the concentrations of infliximab and adalimumab (IgG1 antibodies) were higher in infants at birth and their cords than in their mothers (160% and 153%, respectively; drug treatment was stopped weeks before delivery). In comparison, the median cord level of certolizumab, a PEGylated Fab’ fragment of a humanized monoclonal antibody, was only 3.9% of that of the mother (90) (Fig. 1). Its concentrations in fetal circuit were below the limit of quantification in 5 of 6 human placentas perfused ex vivo (93). Similarly, in the perfused human placenta model, minute amounts of abciximab (a Fab fragment of an IgG1 monoclonal antibody) were detected in the fetal circuit and attached to the trophoblastic surface of the placental villi, to maternal platelets and to residual blood platelets found focally in the fetal capillaries. However, it was estimated that abciximab transfer is unlikely to result in biologically significant occupancy of its platelet receptors and pharmacologic inhibition of platelet aggregation in the fetus (94).
Therapeutic antibodies can be detected in the infant serum more than 6 months after birth (88). In one infant, infliximab concentration of 0.03 mg/mL was detectable at 12 months of age (92). It is often advised that pregnant patients on biologic therapy will take their last dose early in the third trimester. In addition, the current recommendation is to wait at least 6 months or until drug levels are undetectable before giving any live vaccines to these neonates exposed in utero to biologics due to potential immune suppression (95).
Nanomaterials are defined as having at least one dimension between 1 and 100 nm, although larger (submicron) structures are often encompassed as well (96). Nanomaterials can be uptaken by the placenta with their content potentially delivered to the fetus. Small liposomes and nanoparticles (<60 nm) may be uptaken into placental trophoblasts and capillary endothelium via coated vesicle-mediated pinocytosis (97,98). In addition, nanoformulations may undergo phagocytosis by a large population of resident macrophages present in the placenta (99). More recently, paracellular passage has been suggested as a transfer mechanism for very small nanoparticles (100). Enhanced uptake and passage of nanoparticles was observed when placental integrity was hampered by inflammation (101). The placental transfer of various types of nanoformulations and their effects on the fetus have been reviewed by Keelan et al. (96) and will be described here in brief.
Liposomes were among the first nanoformulations to be evaluated as a pharmaceutical carrier and as such, have been extensively investigated with regard to transplacental transfer. In the rat placenta, tissue-weight normalized uptake of liposomal inulin was similar to that in the liver (102). Liposomally-entrapped [3H]penicillin, [14C]inulin, [3H]mthotrexate and [3H]rivoflavin were localized much more in the placenta compared with the free substances (5.6-fold, 5.0-fold, 4.6 and 2.9-fold, respectively). No intact liposomes were found to be transported across the placenta, but their content was transferred to the fetus as free molecules. Liposomal [14C]riboflavin, [3H]methotrexate, and [14C]penicillin were transferred to the fetus more than the free molecules (8.8-fold, 7.5-fold, and 4.2-fold, respectively). Placental uptake of liposomes depended on their phospholipid composition, and negative liposomes were localized in rat and rabbit placentas more than neutral and positive liposomes (102). Likewise, anionic liposomes containing carboxyfluorescein were taken up by cultured human term trophoblasts more avidly than neutral and cationic liposomes and the free carboxyfluorescein (103). In perfused human placentas, liposomal thyroxine (T4) crossed the placenta to a greater extent than free thyroxine, which was metabolized in the placenta to T3 (104).
Perfused human placentas have been used to demonstrate placental uptake and transfer to the fetal circulation of additional types of nanoformulations, including polystyrene particles with diameter up to 500 nm (105), silica nanoparticles (106), and fluorescently tagged PAMAM dendrimers (100). Placental transfer ranged with the particle type and size. For example, the concentration of 50 nm polystyrene beads in the fetal circuit was 49% of the concentration in maternal circuit, whereas when 500 nm beads were used, nearly all particles were retained in the maternal circulation or placental tissue. The concentration of the 500 nm beads detected in the fetal circulation was nearly 30-fold lower as compared to that of the 50 nm beads (105).
Placental Drug Metabolizing Enzymes
The human placenta expresses enzymes that catalyze both phase I (drug oxidation, reduction and hydrolysis) and Phase II (conjugation) reactions. Placental cytochrome P450 (CYP) isoform proteins include 1A1, 2E1, 3A4, 3A5, 3A7, 4B1 and 19 (107,108). At the mRNA level, the major isoforms detected in the placenta are placental aromatase (CYP19) and CYP11A (109). Additional phase I enzymes detected at term placentas include alcohol dehydrogenases, epoxide hydrolases and N-acetyltransferases (110). Placental phase II enzymes include uridine diphosphate glucoronosyltransferases (UGTs), glutathione S-transferase, epoxide hydrolase, N-acetyltransferase, and sulphotransferases (110). For instance, in a study of 12 term human placentas, expression of UGT2B4, 2B7, 2B10, 2B11 and 2B15 mRNA was observed in all the placentas, although with great between-placenta variability in expression levels. UGT2B1 and 2B7 were additionally detected at the protein level and were localized to the syncytiotrophoblast (111). Deconjugation reactions can also take place in the placenta, e.g., by glucuronidases, as was demonstrated for the xenoestrogen bisphenol A in a sheep model (112).
The type and the amount of placental drug metabolizing enzymes change during pregnancy. For instance, CYP3A7 in the human placenta is induced during pregnancy (113), whereas UGTs are present at the placenta throughout pregnancy (111). In mice, almost all the studied placental CYP genes were upregulated during mid-pregnancy (gestational day 10) and levels decreased by mid- to late pregnancy (114).
Drugs which have been shown to undergo significant placental metabolism include structural analogues of endogenous compounds, such as zidovudine (115) and dideoxyinosine (116), but also other compounds such as oxcarbazepine (117,118). Placental CYP19 is involved in the metabolism of glyburide (119), buprenorphine (120), and methadone (121). Olanzapine (122) and zidovudine (123) have been shown to be conjugated to N-glucuronide ex vivo, in the perfused cotyledon model. The extent to which the placental metabolic barrier limits fetal exposure to most drugs appears to be minor compared to that of the maternal liver. Yet, it may contribute to the formation of fetotoxic reactive metabolites (124). Nuclear receptors may be involved in upregulation of placental enzyme expression. For example, aryl hydrocarbon receptor ligands induced the expression of CYP1A2 mRNA in human term placental trophoblast cultures (125).
Distribution Kinetics
The intensity and duration of fetal effects of drugs depend the dose taken by the mother, maternal and fetal pharmacokinetics, and individual factors such as pharmacogenetics, concurrent disease states, and concomitantly taken medications. Another key determinant of fetal drug exposure is the rate and extent of drug distribution across the placenta. Some drugs equilibrate rapidly across the placental membranes whereas other have slower distribution, e.g. due to saturable carrier-mediated uptake or endocytosis. Accordingly, the fetus has been suggested by Gerhard Levy to have the pharmacokinetic characteristics of a shallow or deep compartment, respectively (126). In the latter case, the maximum drug concentration in the fetus is lower and the post-distribution fetal: maternal drug concentration ratio is higher than when the drug distributes rapidly across the placenta. Hence, the maternal dosing regimen may significantly affect fetal exposure. After a single, rapid intravenous bolus of the drug, it may not extensively distribute to the fetus before its concentrations decline in maternal plasma. However, when the drug is administered repeatedly or continuously, it can accumulate in the fetus with time, and its effects may be significant for up to a few hours (20). For example, the fetal:maternal concentration ratio of pethidine increase with time and exceed 1 after approximately 4 h, although by this time the concentrations are low in both mother and cord blood (127). Studies in isolated cotyledons may underestimate the in vivo cord blood:maternal blood concentration ratio of the drug if a distributional steady state is not achieved during the perfusion experiment (e.g., cyclosporine and likely therapeutic antibodies, as described above; Fig. 1). Such compounds may also slowly accumulate in placental tissue itself, as was demonstrated by us for indocyanine green in perfused human placenta (Fig. 3) (128). Hydrophilic drugs may never reach distributional equilibrium because the fetus rapidly excretes them into the amniotic fluid. Such compounds are swallowed by mature fetuses and may concentrate in the fetal intestinal lumen. For example, babies born to mothers treated with hexamethonium suffered from paralytic ileus (20).

Time-dependent accumulation of the near infrared marker indocyanine green in human placentas during ex vivo perfusion of placental cotyledons. Indocyanine green was added to the maternal compartment and cotyledons were perfused for up to 3 h. Tissue samples were washed three times in PBS before ex vivo near infrared imaging. Samples are from a cotyledon that was not exposed to indocyanine green (A) or cotyledons exposed for 2 h (B) or 3 h (C).
Compared with abundant data on drug transfer to the fetus, less is known about their concentrations in the placenta. Most of the few studies that evaluated placental drug concentrations were conducted in term placentas, although some utilized placentas obtained during early pregnancy terminations. The available information indicates that some drugs can accumulate within placental tissue and achieve higher concentrations than in maternal plasma. In a study that compared the concentration of gentamicin given to treat chorioamnionitis in maternal blood, cord blood, and placental membranes, placental gentamicin level was about four- and six-fold higher in placental membranes than that in maternal and cord blood, respectively (Fig. 4) (85).

Drug accumulation in the human placenta in vivo. Data are shown as placenta:maternal plasma concentration ratios. Concentrations of antibiotics were measured in women treated for chorioamnionitis. (a) Early pregnancy. (b) Late pregnancy/term. 10-OH-CBZ, 10-hydroxy-10,11-dihydrocarbamazepine. References are provided in Supplementary Material.
The Fate: How Drugs Affect the Placental Barrier
Because placental concentrations of some drugs may be high enough to exert pharmacological activity, their potential direct effects on the placenta cannot be ignored. Specifically, by modulating placental barrier function, a drug can affect the distribution of an essential compound or that of another drug into the fetus or placenta itself. Practically any placental pathway may be affected by medications of various chemical structures and pharmacological classes (Table I). Even drugs that produce very low levels in the fetus such as heparins can affect trophoblast function. For instance, heparins were demonstrated to be beneficial in the prevention of placenta-mediated complications of pregnancy (e.g., by direct proangiogenic effect on the placenta) (139). Examples of drugs that can interfere with the barrier function of the placenta include antiepileptic drugs, corticosteroids and antiretroviral protease inhibitors.
As a group, antiepileptic drugs are among the most common teratogenic medications prescribed to women of childbearing age (140). Harmful effects of exposure to antiepileptic drugs in utero include fetal loss, intrauterine growth restriction, and congenital malformations (141). The risk of major congenital malformations was consistently shown to be two-to-four times as high with the use of valproic acid compared with the use of other antiepileptic drugs such as carbamazepine and lamotrigine (141,142,143). Valproic acid was also associated with impaired cognitive outcomes of the offspring (144,145), increased risk of autism spectrum disorders (146,147) and diagnosis of attention deficit hyperactivity disorder (ADHD) (148). Many antiepileptic drugs cross the placenta by passive diffusion. Valproic acid is exceptional, as its mechanism of placental transfer appears to involve a proton-linked saturable transport system (149,150). Another exception is gabapentin which is transported in JEG-3 cells by LAT1 (151) and has been suggested to act as a placental LAT1 inhibitor (152). Antiepileptic drugs can affect additional placental targets. We recently evaluated the effects of valproic acid, phenytoin, carbamazepine and lamotrigine on carrier expression in BeWo cells (153). Valproic acid-treated cells expressed half the levels of RFC mRNA and protein and up to 2.7-fold higher levels of BCRP mRNA and protein, and displayed greater BCRP activity than cells treated with the vehicle. Changes in transporter expression were also observed in cells treated with phenytoin, carbamazepine, and lamotrigine. Results from pregnant mice treated with valproic acid (200 mg/kg/day for four days) confirmed some of the in vitro findings and pointed to a potential effect of valproic acid on the functionality of the placental barrier (154). Valproic acid and phenytoin, as well as several other antiepileptic drugs, have additionally been shown to significantly inhibit OCTN2-mediated carnitine uptake (Table I) (131,155). This can lead to carnitine deficiency in both the placenta and the fetus. Carnitine supplementation is recommended for certain populations of patients with epilepsy, including infants receiving valproic acid, but is not routinely given to pregnant women treated with antiepileptic drugs (132,156).
Glucocorticoids are administered to women at risk of preterm birth to accelerate fetal lung maturation (157). Because of the clinical need and the inverse correlation between placental sensitivity to glucocorticoids and placental P-gp expression, several studies evaluated the effects of glucocorticoids on the expression of P-gp and other ABC transporters. In guinea pigs betamethasone (1 mg/kg) given on gestational days 40/41 and 50/51 (length of gestation 59–72 days (158)) resulted in a significant decrease in placental Abcb1 mRNA and protein expression (159). However, the same group later observed in mice up-regulation by dexamethasone (0.1 mg/kg or 1 mg/kg, given daily for one week) of placental Abcb1a mRNA and P-gp protein expression. These changes were not accompanied by reduced exposure of the “fetal unit” (the fetus, amniotic fluid and intact fetal membranes) to [3H]digoxin (160). In another study in rats, dexamethasone (repeated administration of 1 mg/kg for 9 days beginning on day 11 after conception) induced Abcb1 expression in fetal liver and brain but not in the placenta (161). In primary human cytotrophoblasts isolated from full term placentas, dexamethasone and betamethasone significantly induced ABCB1 mRNA expression by around 4-fold but did not affect the expression of ABCG2 and ABCC 1–5, 10, and 12 (162). P-gp expression was also studied in placentas collected from 53 women presenting with threatened preterm labor. In that study, P-gp mRNA and protein expression were not affected by the timing of antenatal glucocorticoids but expression was lower in placenta from small-for-gestational age infants than in appropriately grown infants (163). In another study, in placentas derived from female fetuses, high-dose (1 mg/kg) dexamethasone significantly downregulated Abcg2 mRNA expression and inhibited Bcrp1 function (164). Other reported effects of glucocorticoids on the placenta include lower placental CYP19 mRNA content and aromatase activity in betamethasone-treated women (165) and reduced placental system A amino acid transporter activity ex vivo in women who were at risk of preterm labor and received glucocorticoids but delivered at term (166).
Antiretroviral therapy during pregnancy can treat maternal HIV infection and reduces the likelihood of HIV transmission to the fetus. Recommended treatment regimens for pregnant women include combinations of antiretroviral drugs of several pharmacological classes (167). Among these, atazanavir and lopinavir boosted with ritonavir are currently the first-line protease inhibitors in pregnancy. Indinavir, nelfinavir, and saquinavir exhibit unreliable placental transport (168), but can affect the expression of placental transporters. In human cytotrophoblast cells saquinavir significantly increased both expression (to a 2-fold extent) and functionality (by 18%) of P-gp. In the same study, nelfinavir increased P-gp functionality (by 23%) as a result of its dissociation from caveolin-1. Nelfinavir additionally reduced hCG secretion by 30% (169). Interestingly, P-gp expression in term placentae was greater in HIV-1-infected women receiving pharmacotherapy than in uninfected controls (170).
Outlook
Drugs are usually given to pregnant women to treat the mother, but may be used to manage pathological conditions of the fetus (171) or the placenta (172). Awareness of the principles that govern maternal-fetal pharmacokinetics can minimize unnecessary exposure of the fetus to drugs or help optimize drug delivery to the fetus or the placenta. For instance, when valproic acid is given to pregnant women, it is assumed that “flatter is better”. That is, the teratogenic risk is related to the peak plasma concentration of the drug in maternal plasma (173). Ex vivo, valproic acid readily distributes across the human placenta, reaching distributional equilibrium within 1.5 h (174). Accordingly, it is generally recommended to use a prolonged-release formulation of valproate when given to pregnant women, although this assumption is based solely on a study in mice (175). Alternatively, transnasal administration of valproic acid has been shown in rats to reduce systemic exposure to the drug (176) and may be an optimal delivery route in pregnant women. Carrier inducers or inhibitors can additionally modulate the placental pharmacokinetics of drugs (21), although the effect of the modulator on carriers at drug-eliminating organs may offset those on the placenta (177).
Reduced placental and fetal exposure to drugs, or, in other cases, enhanced drug delivery to the placenta and the fetus may be also achieved by the use of nanosized drug carriers. Even without targeting molecules, nanoparticles may accumulate in the placenta, but effective drug delivery strategies require the development of targeted nanoparticle-based drugs specifically, e.g. for the treatment of trophoblast tumors or infections. For example, doxorubicin-loaded nanocells were constructed with the ability to target trophoblastic tumors via surface decoration with an antibody that binds the epidermal growth factor receptor (EGFR, highly expressed on the placental surface) (178). Although current literature suggests that the risk of fetal exposure and toxicity following maternal use of nanoformulations is low, it is not negligible (96). Ongoing studies evaluate nanoparticles as potential means for drug delivery in pregnant women (NCT02199756 and NCT02720887; clinicaltrials.gov).
The application of the newer ‘placenta on chip’ technologies could allow assessing the in vitro transfer of drugs and drug formulations across the human placental barrier. The model is created in a multilayered microfluidic system that enables co-culture of human trophoblast cells and human fetal endothelial cells that reproduce the formation of microvilli and the syncytialization of trophoblasts (179).
Conclusions
The placenta affects not only pregnancy, but lifelong health. Throughout pregnancy, it provides the varying developmental needs of the embryo and the fetus. To fulfill this function, the placenta is equipped with carriers, enzymes and transcytotic pathways that regulate exchange of essential compounds between the mother and the fetus. Medications are not only transferred to the fetus via these mechanisms, but can also modulate their functional activity, thereby indirectly affecting fetal development. Understanding drug pharmacokinetics and pharmacodynamics in the placenta is a first step towards improving therapeutic regimens in pregnant women and developing strategies that may protect the fetus and the placenta from adverse drug effects. Alternatively, drugs and drug delivery forms can be designed to specifically target the placenta and fetus.
Abbreviations
- ABC:
-
Adenosine triphosphate binding cassette
- BBMVs:
-
Placental brush-border membrane vesicles
- BCRP:
-
Breast cancer resistance protein
- CNT:
-
Concentrative nucleoside transporter
- CYP:
-
Cytochrome P-450
- ENT:
-
Equilibrative nucleoside transporter
- FcRn:
-
Neonatal Fc receptors
- LAT:
-
L-type amino acid transporter
- MDR:
-
Multidrug resistance protein
- MRP:
-
Multidrug resistance-associated protein
- NET:
-
Norepinephrine transporter
- OAT:
-
Organic anion transporter
- OATP:
-
Organic anion transporting polypeptide
- OCT, OCTN:
-
Organic cation transporter
- P-gp:
-
P-glycoprotein
- RFC:
-
Reduced folate carrier
- SERT:
-
Serotonin transporter
- SLC:
-
Solute carrier
- UGT:
-
Uridine diphosphate glucoronosyltransferase
References
Dally A. Thalidomide: was the tragedy preventable? Lancet. 1998;351:1197–9.
Barker RH. Placental transfer of sulfanilamide. N Engl J Med. 1938;41:219.
Speert H. Passage of sulfanilamide through human placenta. Bull Johns Hopkins Hosp. 1938;63:337–9.
Ginsburg J. Placental drug transfer. Annu Rev Pharmacol. 1971;11:387–408.
Grumbach MM, Werner SC. Transfer of thyroid hormone across the human placenta at term. J Clin Endocrinol Metab. 1956;16:1392–5.
Sandler M, Ruthven CR, Contractor SF, Wood C, Booth RT, Pinkerton JH. Transmission of noradrenaline across the human placents. Nature. 1963;197:598.
Abramovich DR, Wade AP. Transplacental passage of steroids: the presence of corticosteroids in amniotic fluid. J Obstet Gynaecol Br Commonw. 1969;76:610–4.
Burton GJ, Jauniaux E. What is the placenta? Am J Obstet Gynecol. 2015;213:S6–8.
Guttmacher AE, Maddox YT, Spong CY. The human placenta project: placental structure, development and function in real time. Placenta. 2014;35:303–4.
Maltepe E, Fisher SJ. Placenta: the forgotten organ. Annu Rev Cell Dev Biol. 2015;31:523–52.
DeVane L, Goetzl LM, Ramamoorthy S. Exposing fetal drug exposure. Clin Pharmacol Ther. 2011;89:786–8.
Goodman AG, Rall TW, Nies AS, Taylor P. Goodman and Gilman’s the pharmacological basis of therapeutics. Eighth ed. New York: Mcgraw-Hill (Tx); 2000.
Tomi M, Nishimura T, Nakashima E. Mother-to-fetus transfer of antiviral drugs and the involvement of transporters at the placental barrier. J Pharm Sci. 2011;100:3708–18.
Burton GJ, Fowden AL. The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci. 2015;370(1663).
Huppertz B. The anatomy of the normal placenta. J Clin Pathol. 2008;61:1296–302.
Hutson JR, Garcia-Bournissen F, Davis A, Koren G. The human placental perfusion model: a systematic review and development of a model to predict in vivo transfer of therapeutic drugs. Clin Pharmacol Ther. 2011;90:67–76.
Bajoria R, Contractor SF. Transfer of heparin across the human perfused placental lobule. J Pharm Pharmacol. 1992;44:952–9.
Holcberg G, Tsadkin-Tamir M, Sapir O, Wiznizer A, Segal D, Polachek H, et al. Transfer of insulin lispro across the human placenta. Eur J Obstet Gynecol Reprod Biol. 2004;115:117–8.
van der Aa EM, Peereboom-Stegeman JH, Noordhoek J, Gribnau FW, Russel FG. Mechanisms of drug transfer across the human placenta. Pharm World Sci. 1998;20:139–48.
Reynolds F. Placental transfer of drugs. Curr Anaesth Crit Care. 1991;2:108–16.
Rubinchik-Stern M, Eyal S. Drug interactions at the human placenta: what is the evidence? Front Pharmacol. 2012;3:126.
Prouillac C, Lecoeur S. The role of the placenta in fetal exposure to xenobiotics: importance of membrane transporters and human models for transfer studies. Drug Metab Dispos. 2010;38:1623–35.
Vähäkangas K, Myllynen P. Drug transporters in the human blood-placental barrier. Br J Pharmacol. 2009;158:665–78.
Prasad PD, Ramamoorthy S, Moe AJ, Smith CH, Leibach FH, Ganapathy V. Selective expression of the high-affinity isoform of the folate receptor (FR-alpha) in the human placental syncytiotrophoblast and choriocarcinoma cells. Biochim Biophys Acta. 1994;1223:71–5.
Staud F, Cerveny L, Ceckova M. Pharmacotherapy in pregnancy; effect of ABC and SLC transporters on drug transport across the placenta and fetal drug exposure. J Drug Target. 2012;20:736–63.
Del Amo E, Urtti A, Yliperttula M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur J Pharm Sci. 2008;35:161–74.
Ganapathy V, Prasad PD. Role of transporters in placental transfer of drugs. Toxicol Appl Pharmacol. 2005;207:381–7.
Ni Z, Mao Q. ATP-binding cassette efflux transporters in human placenta. Curr Pharm Biotechnol. 2011;12:674–85.
Shen DW, Fojo A, Chin JE, Roninson IB, Richert N, Pastan I, et al. Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science. 1986;232:643–5.
Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–33.
Morrissey KM, Wen CC, Johns SJ, Zhang L, Huang SM, Giacomini KM. The UCSF-FDA transportal: a public drug transporter database. Clin Pharmacol Ther. 2012;92:545–6.
Mathias AA, Hitti J, Unadkat JD. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am J Physiol Regul Integr Comp Physiol. 2005;289:R963–9.
Sun M, Kingdom J, Baczyk D, Lye SJ, Matthews SG, Gibb W. Expression of the multidrug resistance P-glycoprotein, (ABCB1 glycoprotein) in the human placenta decreases with advancing gestation. Placenta. 2006;27:602–9.
Hutson JR, Koren G, Matthews SG. Placental P-glycoprotein and breast cancer resistance protein: influence of polymorphisms on fetal drug exposure and physiology. Placenta. 2010;31:351–7.
Joshi AA, Vaidya SS, St-Pierre MV, Mikheev AM, Desino KE, Nyandege AN, et al. Placental ABC transporters: biological impact and pharmaceutical significance. Pharm Res. 2016;33:2847–78.
Lankas GR, Wise LD, Cartwright ME, Pippert T, Umbenhauer DR. Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod Toxicol. 1998;12:457–63.
Smit JW, Huisman MT, van Tellingen O, Wiltshire HR, Schinkel AH. Absence or pharmacological blocking of placental P-glycoprotein profoundly increases fetal drug exposure. J Clin Invest. 1999;104:1441–7.
Eyal S, Chung FS, Muzi M, Link JM, Mankoff DA, Kaddoumi A, et al. Simultaneous PET imaging of P-plycoprotein inhibition in multiple tissues in the pregnant non-human primate. J Nucl Med. 2009;50:798–806.
Ke AB, Eyal S, Chung FS, Link JM, Mankoff DA, Muzi M, et al. Modeling cyclosporine A inhibition of the distribution of a P-glycoprotein PET ligand, 11C-verapamil, into the maternal brain and fetal liver of the pregnant nonhuman primate: impact of tissue blood flow and site of inhibition. J Nucl Med. 2013;54:437–46.
Myllynen P, Vahakangas K. Placental transfer and metabolism: an overview of the experimental models utilizing human placental tissue. Toxicol in Vitro. 2013;27:507–12.
Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH, et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001;61:3458–64.
Kobayashi D, Ieiri I, Hirota T, Takane H, Maegawa S, Kigawa J, et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos. 2005;33:94–101.
Wang C, Xie L, Li H, Li Y, Mu D, Zhou R, et al. Associations between ABCG2 gene polymorphisms and isolated septal defects in a Han Chinese population. DNA Cell Biol. 2014;33:689–98.
Evseenko DA, Murthi P, Paxton JW, Reid G, Emerald BS, Mohankumar KM, et al. The ABC transporter BCRP/ABCG2 is a placental survival factor, and its expression is reduced in idiopathic human fetal growth restriction. FASEB J. 2007;21:3592–605.
Gupta A, Dai Y, Vethanayagam RR, Hebert MF, Thummel KE, Unadkat JD, et al. Cyclosporin A, tacrolimus and sirolimus are potent inhibitors of the human breast cancer resistance protein (ABCG2) and reverse resistance to mitoxantrone and topotecan. Cancer Chemother Pharmacol. 2006;58:374–83.
Gupta A, Zhang Y, Unadkat JD, Mao Q. HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2). J Pharmacol Exp Ther. 2004;310:334–41.
Pollex E, Lubetsky A, Koren G. The role of placental breast cancer resistance protein in the efflux of glyburide across the human placenta. Placenta. 2008;29:743–7.
Bakhsheshian J, Wei BR, Chang KE, Shukla S, Ambudkar SV, Simpson RM, et al. Bioluminescent imaging of drug efflux at the blood-brain barrier mediated by the transporter ABCG2. Proc Natl Acad Sci U S A. 2013;110:20801–6.
Kumar JS, Wei BR, Madigan JP, Simpson RM, Hall MD, Gottesman MM. Bioluminescent imaging of ABCG2 efflux activity at the blood-placenta barrier. Sci Rep. 2016;6:20418.
Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–65.
Nagashige M, Ushigome F, Koyabu N, Hirata K, Kawabuchi M, Hirakawa T, et al. Basal membrane localization of MRP1 in human placental trophoblast. Placenta. 2003;24:951–8.
St-Pierre MV, Serrano MA, Macias RI, Dubs U, Hoechli M, Lauper U, et al. Expression of members of the multidrug resistance protein family in human term placenta. Am J Phys. 2000;279:R1495–503.
Pascolo L, Fernetti C, Pirulli D, Crovella S, Amoroso A, Tiribelli C. Effects of maturation on RNA transcription and protein expression of four MRP genes in human placenta and in BeWo cells. Biochem Biophys Res Commun. 2003;303:259–65.
Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2005;20:452–77.
Azzaroli F, Mennone A, Feletti V, Simoni P, Baglivo E, Montagnani M, et al. Clinical trial: modulation of human placental multidrug resistance proteins in cholestasis of pregnancy by ursodeoxycholic acid. Aliment Pharmacol Ther. 2007;26:1139–46.
Zeng H, Liu G, Rea PA, Kruh GD. Transport of amphipathic anions by human multidrug resistance protein 3. Cancer Res. 2000;60:4779–84.
Jedlitschky G, Burchell B, Keppler D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J Biol Chem. 2000;275:30069–74.
Ugele B, St-Pierre MV, Pihusch M, Bahn A, Hantschmann P. Characterization and identification of steroid sulfate transporters of human placenta. Am J Physiol Endocrinol Metab. 2003;284:E390–8.
Sato K, Sugawara J, Sato T, Mizutamari H, Suzuki T, Ito A, et al. Expression of organic anion transporting polypeptide E (OATP-E) in human placenta. Placenta. 2003;24:144–8.
Obaidat A, Roth M, Hagenbuch B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu Rev Pharmacol Toxicol. 2012;52:135–51.
Tertti K, Petsalo A, Niemi M, Ekblad U, Tolonen A, Rönnemaa T, et al. Transfer of repaglinide in the dually perfused human placenta and the role of organic anion transporting polypeptides (OATPs). Eur J Pharm Sci. 2011;44:181–6.
Grube M, Reuther S, Meyer Zu Schwabedissen H, Köck K, Draber K, Ritter CA, et al. Organic anion transporting polypeptide 2B1 and breast cancer resistance protein interact in the transepithelial transport of steroid sulfates in human placenta. Drug Metab Dispos. 2007;35:30–5.
Ugele B, Bahn A, Rex-Haffner M. Functional differences in steroid sulfate uptake of organic anion transporter 4 (OAT4) and organic anion transporting polypeptide 2B1 (OATP2B1) in human placenta. J Steroid Biochem Mol Biol. 2008;111:1–6.
Takeda M, Khamdang S, Narikawa S, Kimura H, Kobayashi Y, Yamamoto T, et al. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp Ther. 2002;300:918–24.
Sata R, Ohtani H, Tsujimoto M, Murakami H, Koyabu N, Nakamura T, et al. Functional analysis of organic cation transporter 3 expressed in human placenta. J Pharmacol Exp Ther. 2005;315:888–95.
Jonker JW, Schinkel AH. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J Pharmacol Exp Ther. 2004;308:2–9.
Wessler I, Roth E, Deutsch C, Brockerhoff P, Bittinger F, Kirkpatrick CJ, et al. Release of non-neuronal acetylcholine from the isolated human placenta is mediated by organic cation transporters. Br J Pharmacol. 2001;134:951–6.
Kovo M, Kogman N, Ovadia O, Nakash I, Golan A, Hoffman A. Carrier-mediated transport of metformin across the human placenta determined by using the ex vivo perfusion of the placental cotyledon model. Prenat Diagn. 2008;28:544–8.
Tertti K, Ekblad U, Heikkinen T, Rahi M, Rönnemaa T, Laine K. The role of organic cation transporters (OCTs) in the transfer of metformin in the dually perfused human placenta. Eur J Pharm Sci. 2010;39:76–81.
Shekhawat PS, Yang HS, Bennett MJ, Carter AL, Matern D, Tamai I, et al. Carnitine content and expression of mitochondrial beta-oxidation enzymes in placentas of wild-type (OCTN2(+/+)) and OCTN2 Null (OCTN2(−/−)) Mice. Pediatr Res. 2004;56:323–8.
Yabuuchi H, Tamai I, Nezu J, Sakamoto K, Oku A, Shimane M, et al. Novel membrane transporter OCTN1 mediates multispecific, bidirectional, and pH-dependent transport of organic cations. J Pharmacol Exp Ther. 1999;289:768–73.
Errasti-Murugarren E, Diaz P, Godoy V, Riquelme G, Pastor-Anglada M. Expression and distribution of nucleoside transporter proteins in the human syncytiotrophoblast. Mol Pharmacol. 2011;80:809–17.
Govindarajan R, Bakken AH, Hudkins KL, Lai Y, Casado FJ, Pastor-Anglada M, et al. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1809–22.
Griffiths M, Beaumont N, Yao SY, Sundaram M, Boumah CE, Davies A, et al. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med. 1997;3:89–93.
Endres CJ, Moss AM, Ishida K, Govindarajan R, Unadkat JD. The role of the equilibrative nucleoside transporter 1 on tissue and fetal distribution of ribavirin in the mouse. Biopharm Drug Dispos. 2016;37:336–44.
Bzoskie L, Yen J, Tseng YT, Blount L, Kashiwai K, Padbury JF. Human placental norepinephrine transporter mRNA: expression and correlation with fetal condition at birth. Placenta. 1997;18:205–10.
Prasad PD, Hoffmans BJ, Moe AJ, Smith CH, Leibach FH, Ganapathy V. Functional expression of the plasma membrane serotonin transporter but not the vesicular monoamine transporter in human placental trophoblasts and choriocarcinoma cells. Placenta. 1996;17:201–7.
Ganapathy V. Drugs of abuse and human placenta. Life Sci. 2011;88:926–30.
Madras BK, Miller GM, Fischman AJ. The dopamine transporter and attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1397–409.
Velasquez JC, Goeden N, Bonnin A. Placental serotonin: implications for the developmental effects of SSRIs and maternal depression. Front Cell Neurosci. 2013;7:47.
Schneider H, Miller RK. Receptor-mediated uptake and transport of macromolecules in the human placenta. Int J Dev Biol. 2010;54:367–75.
Akour AA, Kennedy MJ, Gerk P. Receptor-mediated endocytosis across human placenta: emphasis on megalin. Mol Pharm. 2013;10:1269–78.
Arora K, Sequeira JM, Quadros EV. Maternofetal transport of vitamin B12: role of TCblR/CD320 and megalin. FASEB J. 2017;31:3098–106.
Akour AA, Kennedy MJ, Gerk PM. The role of megalin in the transport of gentamicin across BeWo cells, an in vitro model of the human placenta. AAPS J. 2015;17:1193–9.
Maberry MC, Trimmer KJ, Bawdon RE, Sobhi S, Dax JB, Gilstrap LC 3rd. Antibiotic concentration in maternal blood, cord blood and placental tissue in women with chorioamnionitis. Gynecol Obstet Investig. 1992;33:185–6.
Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37.
Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–52.
Kane SV, Acquah LA. Placental transport of immunoglobulins: a clinical review for gastroenterologists who prescribe therapeutic monoclonal antibodies to women during conception and pregnancy. Am J Gastroenterol. 2009;104:228–33.
Mahadevan U, McConnell RA, Chambers CD. Drug safety and risk of adverse outcomes for pregnant patients with inflammatory bowel disease. Gastroenterology. 2017;152:451–62.e2.
Mahadevan U, Wolf DC, Dubinsky M, Cortot A, Lee SD, Siegel CA, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286–e24.
Malek A. Ex vivo human placenta models: transport of immunoglobulin G and its subclasses. Vaccine. 2003;21:3362–4.
Julsgaard M, Christensen LA, Gibson PR, Gearry RB, Fallingborg J, Hvas CL, et al. Concentrations of adalimumab and infliximab in mothers and newborns, and effects on infection. Gastroenterology. 2016;151:110–9.
Porter C, Armstrong-Fisher S, Kopotsha T, Smith B, Baker T, Kevorkian L, et al. Certolizumab pegol does not bind the neonatal Fc receptor (FcRn): Consequences for FcRn-mediated in vitro transcytosis and ex vivo human placental transfer. J Reprod Immunol. 2016;116:7–12.
Miller RK, Mace K, Polliotti B, DeRita R, Hall W, Treacy G. Marginal transfer of ReoPro (Abciximab) compared with immunoglobulin G (F105), inulin and water in the perfused human placenta in vitro. Placenta. 2003;24:727–38.
Kathpalia P, Kane S, Mahadevan U. Detectable drug levels in infants exposed to biologics: so what? Gastroenterology. 2016;151:25–6.
Keelan JA, Leong JW, Ho D, Iyer KS. Therapeutic and safety considerations of nanoparticle-mediated drug delivery in pregnancy. Nanomedicine (Lond). 2015;10:2229–47.
Ockleford CD, Menon G. Differentiated regions of human placental cell surface associated with exchange of materials between maternal and foetal blood: a new organelle and the binding of iron. J Cell Sci. 1977;25:279–91.
Wiu AE. In transport at the cellular level. Symp Soc Exp Biol. 1974;28:521–46.
Wood GW. Mononuclear phagocytes in the human placenta. Placenta. 1980;1:113–23.
Menjoge AR, Rinderknecht AL, Navath RS, Faridnia M, Kim CJ, Romero R, et al. Transfer of PAMAM dendrimers across human placenta: prospects of its use as drug carrier during pregnancy. J Control Release. 2011;150:326–38.
Tian X, Zhu M, Du L, Wang J, Fan Z, Liu J, et al. Intrauterine inflammation increases materno-fetal transfer of gold nanoparticles in a size-dependent manner in murine pregnancy. Small. 2013;9:2432–9.
Tuzel-Kox SN, Patel HM, Kox WJ. Uptake of drug-carrier liposomes by placenta: transplacental delivery of drugs and nutrients. J Pharmacol Exp Ther. 1995;274:104–9.
Bajoria R, Sooranna SR, Contractor SF. Endocytotic uptake of small unilamellar liposomes by human trophoblast cells in culture. Hum Reprod. 1997;12:1343–8.
Bajoria R, Fisk NM, Contractor SF. Liposomal thyroxine: a noninvasive model for transplacental fetal therapy. J Clin Endocrinol Metab. 1997;82:3271–7.
Wick P, Malek A, Manser P, Meili D, Maeder-Althaus X, Diener L, et al. Barrier capacity of human placenta for nanosized materials. Environ Health Perspect. 2010;118:432–6.
Poulsen MS, Mose T, Maroun LL, Mathiesen L, Knudsen LE, Rytting E. Kinetics of silica nanoparticles in the human placenta. Nanotoxicology. 2015;(Suppl 1):79–86.
Myllynen P, Immonen E, Kummu M, Vähäkangas K. Developmental expression of drug metabolizing enzymes and transporter proteins in human placenta and fetal tissues. Expert Opin Drug Metab Toxicol. 2009;5:1483–99.
Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet. 2004;43:487–514.
Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi. 2003;123:369–75.
Pavek P, Smutny T. Nuclear receptors in regulation of biotransformation enzymes and drug transporters in the placental barrier. Drug Metab Rev. 2014;46:19–32.
Collier AC, Ganley NA, Tingle MD, Blumenstein M, Marvin KW, Paxton JW, et al. UDP-glucuronosyltransferase activity, expression and cellular localization in human placenta at term. Biochem Pharmacol. 2002;63:409–19.
Corbel T, Perdu E, Gayrard V, Puel S, Lacroix MZ, Viguie C, et al. Conjugation and deconjugation reactions within the fetoplacental compartment in a sheep model: a key factor determining bisphenol A fetal exposure. Drug Metab Dispos. 2015;43:467–76.
Schuetz JD, Kauma S, Guzelian PS. Identification of the fetal liver cytochrome CYP3A7 in human endometrium and placenta. J Clin Invest. 1993;92:1018–24.
Shuster DL, Bammler TK, Beyer RP, Macdonald JW, Tsai JM, Farin FM, et al. Gestational age-dependent changes in gene expression of metabolic enzymes and transporters in pregnant mice. Drug Metab Dispos. 2013;41:332–42.
Liebes L, Mendoza S, Lee JD, Dancis J. Further observations on zidovudine transfer and metabolism by human placenta. AIDS. 1993;7:590–2.
Dancis J, Lee JD, Mendoza S, Liebes L. Transfer and metabolism of dideoxyinosine by the perfused human placenta. J Acquir Immune Defic Syndr. 1993;6:2–6.
Pienimäki P, Lampela E, Hakkola J, Arvela P, Raunio H, Vähäkangas K. Pharmacokinetics of oxcarbazepine and carbamazepine in human placenta. Epilepsia. 1997;38:309–16.
Myllynen P, Pienimäki P, Raunio H, Vähäkangas K. Microsomal metabolism of carbamazepine and oxcarbazepine in liver and placenta. Hum Exp Toxicol. 1998;17:668–76.
Zharikova OL, Fokina VM, Nanovskaya TN, Hill RA, Mattison DR, Hankins GD, et al. Identification of the major human hepatic and placental enzymes responsible for the biotransformation of glyburide. Biochem Pharmacol. 2009;78:1483–90.
Deshmukh SV, Nanovskaya TN, Ahmed MS. Aromatase is the major enzyme metabolizing buprenorphine in human placenta. J Pharmacol Exp Ther. 2003;306:1099–105.
Nanovskaya TN, Deshmukh SV, Nekhayeva IA, Zharikova OL, Hankins GD, Ahmed MS. Methadone metabolism by human placenta. Biochem Pharmacol. 2004;68:583–91.
Schenker S, Yang Y, Mattiuz E, Tatum D, Lee M. Olanzapine transfer by human placenta. Clin Exp Pharmacol Physiol. 1999;26:691–7.
Collier AC, Keelan JA, Van Zijl PE, Paxton JW, Mitchell MD, Tingle MD. Human placental glucuronidation and transport of 3'azido-3′-deoxythymidine and uridine diphosphate glucuronic acid. Drug Metab Dispos. 2004;32:813–20.
Pasanen M, Pelkonen O. The expression and environmental regulation of P450 enzymes in human placenta. Crit Rev Toxicol. 1994;24:211–29.
Stejskalova L, Vecerova L, Perez LM, Vrzal R, Dvorak Z, Nachtigal P, et al. Aryl hydrocarbon receptor and aryl hydrocarbon nuclear translocator expression in human and rat placentas and transcription activity in human trophoblast cultures. Toxicol Sci. 2011;123:26–36.
Levy G. Pharmacokinetics of fetal and neonatal exposure to drugs. Obstet Gynecol. 1981;58(5 Suppl):9s–16s.
Tomson G, Garle RI, Thalme B, Nisell H, Nylund L, Rane A. Maternal kinetics and transplacental passage of pethidine during labour. Br J Clin Pharmacol. 1982;13:653–9.
Rubinchik-Stern M, Shmuel M, Bar J, Eyal S, Kovo M. Maternal-fetal transfer of indocyanine green across the perfused human placenta. Reprod Toxicol. 2016;62:100–5.
Cool DR, Liebach FH, Ganapathy V. Interaction of fluoxetine with the human placental serotonin transporter. Biochem Pharmacol. 1990;40:2161–7.
Jayanthi LD, Vargas G, DeFelice LJ. Characterization of cocaine and antidepressant-sensitive norepinephrine transporters in rat placental trophoblasts. Br J Pharmacol. 2002;135:1927–34.
Lahjouji K, Elimrani I, Lafond J, Leduc L, Qureshi IA, Mitchell GA. L-Carnitine transport in human placental brush-border membranes is mediated by the sodium-dependent organic cation transporter OCTN2. Am J Physiol Cell Physiol. 2004;287:C263–9.
Wu SP, Shyu MK, Liou HH, Gau CS, Lin CJ. Interaction between anticonvulsants and human placental carnitine transporter. Epilepsia. 2004;45:204–10.
Hirano T, Yasuda S, Osaka Y, Asari M, Kobayashi M, Itagaki S, et al. The inhibitory effects of fluoroquinolones on L-carnitine transport in placental cell line BeWo. Int J Pharm. 2008;351:113–8.
Fathe K, Palacios A, Finnell RH. Brief report novel mechanism for valproate-induced teratogenicity. Birth Defects Res A Clin Mol Teratol. 2014;100:592–7.
Keating E, Goncalves P, Campos I, Costa F, Martel F. Folic acid uptake by the human syncytiotrophoblast: interference by pharmacotherapy, drugs of abuse and pathological conditions. Reprod Toxicol. 2009;28:511–20.
Acevedo CG, Rojas S, Bravo I. L-arginine transport at the fetal side of human placenta: effect of aspirin in pregnancy. Exp Physiol. 1999;84:1127–36.
Williams JB, Mallorga PJ, Conn PJ, Pettibone DJ, Sur C. Effects of typical and atypical antipsychotics on human glycine transporters. Schizophr Res. 2004;71:103–12.
He B, Zhang N, Zhao R. Dexamethasone downregulates SLC7A5 expression and promotes cell cycle arrest, autophagy and apoptosis in BeWo cells. J Cell Physiol. 2016;231:233–42.
Kingdom JC, Drewlo S. Is heparin a placental anticoagulant in high-risk pregnancies? Blood. 2011;118:4780–8.
Schwarz EB, Maselli J, Norton M, Gonzales R. Prescription of teratogenic medications in United States ambulatory practices. Am J Med. 2005;118:1240–9.
Tomson T, Battino D. Teratogenic effects of antiepileptic drugs. Lancet Neurol. 2012;11:803–13.
Tomson T, Battino D, Bonizzoni E, Craig J, Lindhout D, Perucca E, et al. Dose-dependent teratogenicity of valproate in mono- and polytherapy: an observational study. Neurology. 2015;85:866–72.
Tomson T, Battino D, Bonizzoni E, Craig J, Lindhout D, Sabers A, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609–17.
Meador KJ, Baker GA, Browning N, Clayton-Smith J, Combs-Cantrell DT, Cohen M, et al. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N Engl J Med. 2009;360:1597–605.
Meador KJ, Baker GA, Browning N, Cohen MJ, Bromley RL, Clayton-Smith J, et al. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurol. 2013;12:244–52.
Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA. 2013;309:1696–703.
Wood AG, Nadebaum C, Anderson V, Reutens D, Barton S, O'Brien TJ, et al. Prospective assessment of autism traits in children exposed to antiepileptic drugs during pregnancy. Epilepsia. 2015;56:1047–55.
Cohen MJ, Meador KJ, Browning N, May R, Baker GA, Clayton-Smith J, et al. Fetal antiepileptic drug exposure: Adaptive and emotional/behavioral functioning at age 6 years. Epilepsy Behav. 2013;29:308–15.
Nakamura H, Ushigome F, Koyabu N, Satoh S, Tsukimori K, Nakano H, et al. Proton gradient-dependent transport of valproic acid in human placental brush-border membrane vesicles. Pharm Res. 2002;19:154–61.
Utoguchi N, Audus KL. Carrier-mediated transport of valproic acid in BeWo cells, a human trophoblast cell line. Int J Pharm. 2000;195:115–24.
Furugen A, Ishiguro Y, Kobayashi M, Narumi K, Nishimura A, Hirano T, et al. Involvement of l-type amino acid transporter 1 in the transport of gabapentin into human placental choriocarcinoma cells. Reprod Toxicol. 2017;67:48–55.
Ohman I, Vitols S, Tomson T. Pharmacokinetics of gabapentin during delivery, in the neonatal period, and lactation: Does a fetal accumulation occur during pregnancy? Epilepsia. 2005;46:1621–4.
Rubinchik-Stern M, Shmuel M, Eyal S. Antiepileptic drugs alter the expression of placental carriers: an in vitro study in a human placental cell line. Epilepsia. 2015;56:1023–32.
Meir M, Bishara A, Mann A, Udi S, Portnoy E, Shmuel M, et al. Effects of valproic acid on the placental barrier in the pregnant mouse: optical imaging and transporter expression studies. Epilepsia. 2016;57:e108–12.
Ohashi R, Tamai I, Yabuuchi H, Nezu JI, Oku A, Sai Y, et al. Na(+)-dependent carnitine transport by organic cation transporter (OCTN2): its pharmacological and toxicological relevance. J Pharmacol Exp Ther. 1999;291:778–84.
Asadi-Pooya AA, Mintzer S, Sperling MR. Nutritional supplements, foods, and epilepsy: is there a relationship? Epilepsia. 2008;49:1819–27.
Roberts D, Brown J, Medley N, Dalziel SR. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2017;3:Cd004454.
Walker N, Filis P, Soffientini U, Bellingham M, O'Shaughnessy PJ, Fowler PA. Placental transporter localization and expression in the human: the importance of species, sex, and gestational age differencesdagger. Biol Reprod. 2017;96:733–42.
Kalabis GM, Petropoulos S, Gibb W, Matthews SG. Multidrug resistance phosphoglycoprotein (ABCB1) expression in the guinea pig placenta: developmental changes and regulation by betamethasone. Can J Physiol Pharmacol. 2009;87:973–8.
Petropoulos S, Gibb W, Matthews SG. Effect of glucocorticoids on regulation of placental multidrug resistance phosphoglycoprotein (P-gp) in the mouse. Placenta. 2010;31:803–10.
Salje K, Lederer K, Oswald S, Dazert E, Warzok R, Siegmund W. Effects of rifampicin, dexamethasone, St. John’s Wort and thyroxine on maternal and foetal expression of Abcb1 and organ distribution of talinolol in pregnant rats. Basic Clin Pharmacol Toxicol. 2012;111:99–105.
Manceau S, Giraud C, Decleves X, Scherrmann JM, Artiguebieille F, Goffinet F, et al. ABC drug transporter and nuclear receptor expression in human cytotrophoblasts: influence of spontaneous syncytialization and induction by glucocorticoids. Placenta. 2012;33:927–32.
Hodyl NA, Stark MJ, Butler M, Clifton VL. Placental P-glycoprotein is unaffected by timing of antenatal glucocorticoid therapy but reduced in SGA preterm infants. Placenta. 2013;34:325–30.
Petropoulos S, Gibb W, Matthews SG. Glucocorticoid regulation of placental breast cancer resistance protein (Bcrp1) in the mouse. Reprod Sci. 2011;18:631–9.
Paakki P, Kirkinen P, Helin H, Pelkonen O, Raunio H, Pasanen M. Antepartum glucocorticoid therapy suppresses human placental xenobiotic and steroid metabolizing enzymes. Placenta. 2000;21:241–6.
Audette MC, Challis JR, Jones RL, Sibley CP, Matthews SG. Synthetic glucocorticoid reduces human placental system a transport in women treated with antenatal therapy. J Clin Endocrinol Metab. 2014;99:E2226–33.
Günthard HF, Saag MS, Benson CA, del Rio C, Eron JJ, Gallant JE, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2016 recommendations of the international antiviral society–USA panel. JAMA. 2016;316:191–210.
McCormack SA, Best BM. Protecting the fetus against HIV infection: a systematic review of placental transfer of antiretrovirals. Clin Pharmacokinet. 2014;53:989–1004.
Beghin D, Forestier F, Noel-Hudson MS, Gavard L, Guibourdenche J, Farinotti R, et al. Modulation of endocrine and transport functions in human trophoblasts by saquinavir and nelfinavir. Eur J Obstet Gynecol Reprod Biol. 2010;152:55–9.
Camus M, Deloménie C, Didier N, Faye A, Gil S, Dauge MC, et al. Increased expression of MDR1 mRNAs and P-glycoprotein in placentas from HIV-1 infected women. Placenta. 2006;27:699–706.
Zoeller BB. Treatment of fetal supraventricular tachycardia. Curr Treat Options Cardiovasc Med. 2017;19:7.
Ilekis JV, Tsilou E, Fisher S, Abrahams VM, Soares MJ, Cross JC, et al. Placental origins of adverse pregnancy outcomes: potential molecular targets: an Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Am J Obstet Gynecol. 2016;215(1 Suppl):S1–s46.
Tomson T, Marson A, Boon P, Canevini MP, Covanis A, Gaily E, et al. Valproate in the treatment of epilepsy in girls and women of childbearing potential. Epilepsia. 2015;56:1006–19.
Fowler DW, Eadie MJ, Dickinson RG. Transplacental transfer and biotransformation studies of valproic acid and its glucuronide(s) in the perfused human placenta. J Pharmacol Exp Ther. 1989;249:318–23.
Nau H. Teratogenic valproic acid concentrations: infusion by implanted minipumps vs conventional injection regimen in the mouse. Toxicol Appl Pharmacol. 1985;80:243–50.
Eskandari S, Varshosaz J, Minaiyan M, Tabbakhian M. Brain delivery of valproic acid via intranasal administration of nanostructured lipid carriers: in vivo pharmacodynamic studies using rat electroshock model. Int J Nanomedicine. 2011;6:363–71.
Bishara A, Meir M, Portnoy E, Shmuel M, Eyal S. Near infrared imaging of indocyanine green distribution in pregnant mice and effects of concomitant medications. Mol Pharm. 2015;12:3351–7.
Kaitu'u-Lino TJ, Pattison S, Ye L, Tuohey L, Sluka P, MacDiarmid J, et al. Targeted nanoparticle delivery of doxorubicin into placental tissues to treat ectopic pregnancies. Endocrinology. 2013;154:911–9.
Blundell C, Tess ER, Schanzer ASR, Coutifaris C, Su EJ, Parry S, et al. A microphysiological model of the human placental barrier. Lab Chip. 2016;16:3065–73.
ACKNOWLEDGMENTS AND DISCLOSURES
The authors acknowledge the support of the Israel Science Foundation (ISF) Grant 506/13.
Sara Eyal is affiliated with the David R. Bloom Centre for Pharmacy and Dr. Adolf and Klara Brettler Centre for Research in Molecular Pharmacology and Therapeutics at The Hebrew University of Jerusalem, Israel.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors have no commercial or financial relationships that could be constructed as a potential conflict of interest.
Additional information
Guest Editor: Sara Eyal
Dedicated to (late) Dr. Zvi Ben Zvi, Ben Gurion University of the Negev, Beer Sheva, Israel, for his contribution to the field of maternal-fetal pharmacology.
Electronic supplementary material
ESM 1
(DOCX 45 kb)
Rights and permissions
About this article

Cite this article
Tetro, N., Moushaev, S., Rubinchik-Stern, M. et al. The Placental Barrier: the Gate and the Fate in Drug Distribution. Pharm Res 35, 71 (2018). https://doi.org/10.1007/s11095-017-2286-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11095-017-2286-0