
Ceftriaxone sodium is an antibiotic used clinically.We have developed a strategy for evaluation of the consistency of ceftriaxone sodium for injection and used it to replicate the formulation. Comparison of the quality of generic and original raw materials and analysis of the process of drug production revealed that the quality of ceftriaxone sodium raw material is the most important factor in replicating the formulation, and a key factor affecting the quality of raw materials is the ceftriaxone sodium crystallisation process. Secondly, the solution clarity of the formulation, another key aspect, was addressed by controlling the leachable components found in rubber closures used in the packaging. The time to achieve therapeutic efficacy of the preparation could be preliminarily evaluated by determining the rates of salt formation and protein binding. Finally, the key quality control index and drug quality standards were evaluated by systematic comparison of the pharmaceutical index of generic products with that of the original preparation, particularly the impurity profile. On this basis, a strategy for the evaluation of ceftriaxone sodium for injection has been developed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
China is a country where generic drugs are widely used, in particular, most antibiotics used in the country are produced by domestic generic manufacturers. In February 2016, the General Office of the State Council of China issued the “Opinion on the Consistency Evaluation of the Quality and Efficacy of Generic Drug”, proposing that “the generic drugs approved for listing before the implementation of the new classification of chemicals must be evaluated for consistency if they are not approved in accordance with the principle of consistency with the quality and efficacy of the original drug.” The purpose of the generic drug consistency evaluation is to guarantee the quality and therapeutic efficacy of domestically manufactured drugs, to allow the substitution of generic drugs and original drugs in the clinic, to reduce national medical expenses, and to promote globalization of the pharmaceutical industry. In December 2017, the Centre for Drug Evaluation issued “The Technical Requirements for Consistency Evaluation of Listed Chemical Generic Drugs (Injections) (draft for comments)” [1] and officially launched the consistency evaluation of the quality and efficacy of generic drugs for chemical injection. Taking antibiotic injections as an example, we discuss the main concerns with and strategies for the evaluation of consistency evaluation/re-evaluation of injections [2] and propose that “in the generic drug consistency evaluation, we should first have a deep understanding of the critical quality attributes (CQAs) of reference preparations, determine the critical material attributes (CMAs) and critical process parameters (CPPs), reveal the main quality differences between generic and reference preparations, and realise the control of the CQAs of the product and ensure consistency between generic and reference preparations through the control of CMAs and CPPs of generic drugs.” The quality and efficacy information of domestic generic drugs are usually well documented, especially in the national evaluation inspection over the years. In the generic drug consistency evaluation, key quality differences between domestic generic drugs and reference preparations can be identified through systematic literature research, and targeted improvements can be made.
Ceftriaxone sodium is a third-generation cephalosporin developed by the Swiss pharmaceutical company Roche in the 1980s and marketed under the trade name Rocephin. Ceftriaxone sodium is a crystalline powder, and ceftriaxone sodium for injection is manufactured in a sterile packaging process. Therefore, the quality of the preparation depends largely on the quality of the raw materials used. In 1991, ceftriaxone sodium and its preparations were successfully replicated in China. At present, there are 35 domestic manufacturers producing ceftriaxone sodium as a raw material with 35 approval numbers and 104 manufacturers producing ceftriaxone sodium for injection with 336 approval numbers. There are many studies evaluating the differences in quality between generic ceftriaxone sodium for injection preparations and Rocephin in China and abroad [3,4,5].We used this as a demonstration variety to establish an approach for the evaluation of consistency of antibiotic injections as per the recommended strategy and replicated the formulation for the purposes of our study. The feasibility of the strategy and the key factors affecting the evaluation of consistency of the generic drugs are discussed by comparing the generic drugs with the original ones.
Ceftriaxone sodium (C18H16N8Na2O7S3· 3½ H2O) is a crystalline powder containing 3.5 crystalline water molecules and 2 sodium atoms. The national evaluation inspection data showed that incomplete salt-formation was observed in domestically-produced ceftriaxone sodium. This can cause high serum protein binding of the drug, leading to delayed action [6]. Additionally, the clarity of ceftriaxone sodium reconstituted solutions worsens during storage [7], which may be attributed to the specific adsorption of the 2,6-ditert-butylp-methyl phenol (BHT) and total siloxanes in rubber closure by ceftriaxone sodium [8]. This absorption is worsened by the unsuitable crystal structure, resulting in increased turbidity [9].
Ceftriaxone sodium can form a variety of impurities during production and storage [10,11,12]. Data of the national evaluation inspection data revealed that the impurity profile of ceftriaxone sodium for injection manufactured in different enterprises in China varied greatly, showing 4(14 various impurities (unpublished information). Various impurities in cephalosporins cause different toxic reactions [13] and it is suggested that adverse drug reactions may vary based on the impurity profile of the products. Based on the above analysis and the process of preparation of ceftriaxone sodium for injection, the quality of raw materials is thought to be the most critical factor in the replication of the formulation. The most important factor affecting the quality of the raw material is the process of crystallisation of ceftriaxone sodium.
Basic strategies for the effective production of ceftriaxone sodium for injection are as follows:
(1) Raw materials with similar crystallisation processes and similar quality were selected. X-ray diffraction (XRD) spectra of the powder were used to screen the crystallisation process of the raw materials. According to Xue, et al. [5], the XRD pattern of the raw material powder should be basically consistent with that of the reference preparation. In addition, the wet product and moisture content of ceftriaxone sodium were determined by referring to the literature [14].
(2) The quality of the rubber closures used for the packaging was also evaluated. The quantitative relationship between the adsorption amount of migrant BHT and total siloxane (hexamethylcyclotrisiloxane, octaethylcyclotetrasiloxane and decamethylcyclopentasiloxane), and the solution clarity was used to determine the maximum BHT and total siloxane content permissible in the rubber closure [8]. These data were used to establish the quality control standard for the rubber closure used in ceftriaxone sodium for injection.
(3) The key quality control indexes and drug quality standards were determined by comparison of pharmaceutical indexes, particularly the impurity profiles, with those of reference preparations.
Thus, a strategy for the evaluation of generic ceftriaxone sodium for injection was developed as is depicted by the diagram in Fig 1.

Strategy of evaluation of the generic ceftriaxone sodium for injection.
EXPERIMENTAL
Chemicals and Reagents
The reference ceftriaxone sodium for injection was purchased from F. Hoffmann-La Roche Ltd. (Welwyn Garden City, UK; batch number B0467B03). The domestic reference preparation, ceftriaxone sodium for injection, was purchased from Shanghai Roche Pharmaceutical Ltd. (Shanghai, China; batch number SH6550). Three batches of self-developed generic preparations (batch numbers 22190501, 22190502, and 22190503) were used and ten samples of ceftriaxone sodium for injection from different manufacturers were collected from the market inspection in 2017(2018. Ceftriaxone reference substance (batch number: 130480-201504, content: 83.9%), ceftriaxone E-isomer reference substance (batch number: 130660-201702), and ceftriaxone impurity C reference substance (batch number: 130661-201001, content: 99.1%) were provided by the National Institutes for Food and Drug Control. Impurities B, D, and E were synthesised by Sundia MediTech Company Ltd. (Shanghai, China).
Acetonitrile (chromatographic grade, 99.99%) was purchased from Thermo-Fisher Scientific (Waltham, MA, USA; LOT 190262). Octylamine (ALDRICH, LOT STBF6957V), human serum albumin (HAS; content ≥96%; SIGMA, LOT SLBT3708), and BHT (SIGMA, LOT BCCB4438) were purchased from Sigma Aldrich (St. Louis, MO, USA). Hexamethylcyclotrisiloxane (TCI, LOT RIYJI-SH), octamethylcyclotetrasiloxane (TCI, LOT 073XB-GC), and decamethylcyclopentasiloxane (TCI, LOT KAF2O-CK) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Sodium elemental standard material (1000 μg/mL, batch number 191068-3) was provided by the National Analysis and Testing Centre for Nonferrous Metals and Electronic Materials.
Experimental Techniques
Powder x-ray diffraction. The following settings were used for XRD analysis: anode target, Cu; voltage, 45 kV; current, 200 mA, monochromator, Ni filter; detector, D/teX Ultra 250 one-dimensional array detector, 2θ:3(50°; scanning interval, 0.01°; scanning rate, 8°/min; slit system, Soller Slit 5.0°; IS, 1/2°; ISL, 10.0 mm; RS1, 20.0 mm; and RS2, 20.0 mm.
Impurity profile analysis. Liquid chromatography (LC) was done using an Agilent1200 Infinity series system (Agilent Technologies Inc., Santa Clara, CA, USA), with an Agilent OpenLab2.3 workstation. Conventional chromatographic analysis was carried out using a Kromasil 100-C18 (250 × 4.6 mm, 5 μm) column. Mobile phases A and B were 0.02 mol/L octylamine solution-acetonitrile 73:27 (pH value adjusted to 6.5 using phosphoric acid), and acetonitrile, respectively. A linear elution gradient was used: 0 – 5 min, 100% A; 5 – 45 min, 100 – 70% A; 45 – 55 min, 70 – 60% A; 55 – 56 min, 60 – 100% A; 56 – 65 min, 100% A. The flow rate was set to 1.0 mL/min, wavelength to 254 nm, and column oven temperature to 30°C.
High-resolution mass-spectrometry (HR-MS).
HR-MS measurements were performed with a dual gradient UltiMate3000 HPLC system (Dionex Inc., Sunnyvale, CA, USA) equipped with a Q Extractive Focus LTQ Orbitrap XL high resolution mass spectrometer (Thermo Fisher Scientific Inc.), as per the method described by Jin, et al. [15]. This analysis was performed on Capcell Pak MGII C-18 (150 mm × 4.6 mm, 5 μm) column. Mobile phases C and D were 0.5% formic acid solution and acetonitrile with 0.5% formic acid, respectively. A linear elution gradient was as follows: 0(5.5 min, 100% C; 5.5 – 25 min, 100 – 0% C; 25 – 30 min, 100 – 0% C. Ion source was operated in the electrospray ionization mode. The switch valve quantitative ring volume was set to 500 L. The LC-MS parameters were as follows: spray voltage, 3.0 kV; capillary temp, 350.0°C; sheath gas flow rate, 35 L/h; aux gas flow rate, 10.0 L/h; aux gas heater temperature, 350°C; S-lens radio frequency level, 50.0; scan range, 100 – 1000 m/z. The primary mass was measured in positive polarity at a resolution of 70,000; for the secondary mass, the resolution was 17,500, the isolation window was 3.0 m/z, (N)CE was 10, and the default charge state was 1. Data processing was conducted using the Perlscript (Quant Merge) software.
Relationship between the Adsorption Capacity of Migrates and Solution Clarity
Analysis of sample solution clarity. Ceftriaxone sodium for injection preparations from 10 manufacturers were selected from market inspection records and made into a 10 mg/mL aqueous solution. Visual inspection and turbidimetry were used to assess the clarity of the solution, as described in the Chinese Pharmacopoeia [16]. In the turbidimetry method, a HACH TL23 instrument (Hach, Ames, IA, USA) was used to measure the turbidity of each sample. Three measurements were taken and the average was calculated. The turbidity value of a blank solution was deducted from the mean value to get the turbidity value of sample solution.
Adsorption capacities of migrants. The antioxidant (BHT) and siloxane (hexamethyl cyclotrisiloxane, octaethyl cyclotetrasiloxane, and decamethyl cyclopentasiloxane) in the rubber closure can form an insoluble complex with ceftriaxone sodium, causing the solution to become turbid [8]. The adsorption capacities of migrants in the products from 10 manufacturers were measured using Agilent 7694 – 7890 gas chromatography system (Agilent Technologies Inc., Santa Clara, CA, USA). The chromatographic column was composed of 5% phenyl–95% methylsiloxane (DB-5MS, 30.0 m × 0.25 mm × 0.25 m), the carrier gas was nitrogen, and the flow rate was set to 1.0 mL/min. The programmed temperature of the column was maintained at 50°C for 10 min, then increased at a rate of 10°C/min to 170°C for 20 min, and the headspace condition was 121°C for 30 min. The death time (t0) of the chromatographic system was determined using methane.
Quantitative relationship between the adsorption capacity of migrates and solution clarity was studied taking the peak area of migrates in the samples as the x-axis coordinate and the turbidity values of samples as the y-axis coordinate. This curve was used to determine the quantity (peak area) of migrates in the sample that would produce a solution complying with the clarity requirements specified in the Chinese Pharmacopoeia (turbidity not more than that of standard solution no. 1).
Quality Control of Rubber Closure
To prepare the test sample, a rubber closure was cut into pieces, placed into a 10 mL headspace bottle, sealed with a blank rubber closure, and determined by HS-GC. Reference solutions were prepared by dissolving octamethyl cyclotetrasiloxane in dimethyl sulfoxide (0.04, 0.05, and 0.06 mg/mL). Aliquot (1.0 mL) of each solution was accurately transferred to 10 mL headspace bottle, sealed, and injected by headspace. The peak areas of reference solutions with different concentrations were measured by HS-GC and a linear equation of relationship between the peak area and concentration of reference substance was determined. The quantity of BHT (peak area) in the rubber closure corresponding to the clarity limit stated in the Chinese Pharmacopoeia was determined using the adsorption capacity of migrate–solution clarity curve described in section
This was then used to determine the concentration of reference substance corresponding to this value (0.0518 mg/mL), based on the peak area-reference substance concentration linear equation. One third of the peak area corresponding to this concentration was used as the reference peak area (Aref) for the quality control of the rubber closures. The peak area of BHT should be no more than 3.0 times Aref and the peak area of volatile total siloxane should be no more than 3.5 times Aref.
Analysis of Protein Binding Rate
Human serum albumin (0.66 g) was weighed and transferred to a 10 mL volumetric flask, and water was added to form solution A. Ceftriaxone sodium for injection (16.3 mg) was weighed, transferred to a 25 mL volumetric flask, and water was added to dilute and form solution B. Then, 1.0 mL of each solutions was accurately measured and mixed for 10 min in a water bath at 37°C., Aliquot (250 μL) of this was accurately measured, transferred to a centrifugal filter, centrifuged at 25000 rpm for 90 sec, and the filtrate was taken as the test preparation. Next, 1.0 mL of solution B and 1.0 mL of water were accurately measured, mixed, and prepared in the same way as the standard. Equal volumes (approximately 10 μL) of the test preparation and standard preparation were separately injected into the chromatograph, the chromatograms were recorded, and the protein binding rate was calculated as follows:
where, Asample is the peak area of ceftriaxone in the test preparation, and Astandard is the peak area of ceftriaxone in the standard preparation.
Analysis of the Rate of Salt Formation
Approximately 20 mg of ceftriaxone sodium was transferred to a 50 mL volumetric flask, dissolved in water and diluted to volume. Of this solution, 5.0 mL was transferred to a 50 mL volumetric flask, diluted with water to volume, shaken well, and was used as the test preparation. To prepare the standard solution, sodium single element standard substance (5 mL) was transferred to a 50 mL volumetric flask, diluted with water to volume, and shaken well. Of this solution, 1.0, 2.0, 3.0, 4.0, and 5.0 mL samples were transferred to 100 mL volumetric flasks, diluted with water to volume, shaken well, and used as a series of standard solutions. The content of sodium ions in the samples was determined by atomic absorption spectrophotometry [16] at 589 nm using the standard curve method. The ceftriaxone content in the samples was measured by the method described in the Chinese Pharmacopoeia (2015 edition) [17] and the salt formation rate was calculated.
Comparative Study of Stability
As per the guidelines for stability testing of raw materials and preparations (9001) in the Chinese Pharmacopoeia (2015 edition), accelerated stability testing was carried out at 40 and 75% relative humidity to compare the difference between generic drugs and reference preparations using the samples taken 1, 2, 3 and 6 months from the start of the study. The related substances and polymers were analyzed for impurities as described in section 2.2.2. Changes in the solution color were determined by colorimetry. Other parameters (appearance, identification, acidity or alkalinity, water, particulate matter, weight variation, visible particles, bacterial endotoxin, sterility, and assay) were evaluated as per the methods described in the ceftriaxone sodium for injection monograph in the Chinese Pharmacopoeia (2015 edition) [18].
Rubber closure compatibility test. As per the YBB00142002-2015 guidelines for compatibility testing of drugs and their packaging materials, the product containers were inverted and stored at 40°C and 90% relative humidity. Samples were taken 1, 2, 3, and 6 months after the start of the study. The clarity of solutions was evaluated as per the method described in Section "Relationship between the Adsorption Capacity of Migrates and Solution Clarity".
Adverse drug reaction monitoring. The adverse drug reaction monitoring reports, from 1st January 2010 to 31st December 2018, of ceftriaxone sodium for injection produced by Shanghai Roche Pharmaceutical Co., Ltd., an import enterprise of domestic reference preparation and two domestic enterprises with large production share (enterprise 1 and enterprise 2) were collected from the national database of adverse drug reactions. The organ systems involved the reported serious adverse drug reactions were statistically analyzed, and differences in the types of adverse drug reactions caused by the products from different manufacturers were compared.
RESULTS
Powder x-ray Diffraction Analysis
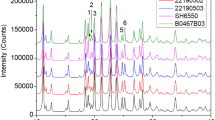
The domestic generic preparation, domestic reference preparation, and reference preparation were analyzed by powder x-ray diffraction (Fig. 2). The number and positions of the diffraction peaks of each sample are seen to overlap, indicating that their crystal forms were essentially the same. Referring to the work by Xue, et al. [5], the differences in the diffraction peaks at 2θ = 11 – 13° and 2θ = 18 – 23° were further compared. The diffraction peaks in the range of 2θ = 18 – 23° showed some variations: the ratio of the rela tive peak strength of I2/I1 was approximately 1.5, and that of I3/I1 was approximately 2 in the reference preparation; meanwhile, I2/I1 of the three batches of generic preparations was about 0.8~1.0, and I3/I1 was approximately 1.0 and 1.2. Variations in the relative peak intensity of specific diffraction peaks in the three batches of generic preparations indicated that the process control level of raw material crystallization was lacking.

Powder x-ray diffraction patterns of domestic generic preparations (a, b, c), domestic reference preparation (d), and reference preparation (e). The ratio of the relative peak strength of I2/I1 was approximately 1.5 and that of I3/I1 was approximately 2 in the reference preparation; I2/I1 of the generic materials was approximately 0.8~1.0 and I3/I1 was approximately 1.0 and 1.2.
Impurity Profile Analysis
Related substances found in the reference and domestic preparations (five manufacturers) of ceftriaxone sodium for injection were compared (Fig. 3). More than 10 kinds of impurities were identified in the product; those in the domestic generic preparations were analyzed using an impurity reference substance combined with column switching HR-MS.

(a) Chromatogram of known impurities in ceftriaxone sodium. (b) Chromatogram of reference preparation. (c) Chromatogram of generic preparation. The remaining are typical chromatograms of domestic preparations.
The main impurities of ceftriaxone sodium are listed in Table 1. Mass spectrometric analysis of impurities for which no reference compounds were available is shown in the SUPPLEMENTARY MATERIALS section. Compared with those in the products from other domestic manufacturers, the types of impurities in the generic formulation were significantly lower and equivalent to those found in the reference preparation. The only impurity that was found in a concentration greater than 0.1% was the ceftriaxone triazine ring (impurity C). Impurity peak 3 (ceftriaxone impurity E, 0.05%) and impurity peak 7 (dimer hydrolysate, 0.03%) were unique impurities that were found only in the domestic products but not in the reference formulation. In the accelerated stability experiment, the changes in the content of related substances in the reference, domestic reference, and the three batches of self-developed generic preparations were essentially the same. The content of impurity C increased over time, while the increases in other single and polymer impurities was not obvious (Table 1). The impurity profiles of generic preparations were found consistent with that of the reference drug.
Quantitative Relationship between the Adsorption of Migrates and Solution Clarity
Gas chromatographic analysis of the sample solutions prepared from the rubber closures used by 10 different manufacturers of ceftriaxone sodium for injection was carried out, and the types and contents of volatile substances in the rubber closures used by the manufacturers were found to be significantly different (Fig. 4). The differences in the total siloxane and BHT content are shown in Table 2.

Gas chromatograms of rubber closures from different manufacturers: (1) hexamethyl cyclotrisiloxane, (2) octamethyl cyclotrasiloxane, (3) decamethyl cyclotrasiloxane, (4) butylated hydroxytoluene (BHT).
The quantitative relationship between the migrant adsorption amount and solution clarity (turbidity value) of ceftriaxone sodium for injection from 10 different manufacturers was analyzed (the turbidity of solution and the peak area of volatile were determined twice for each batch of samples, and the average value was taken), and the results showed a good correlation between the total siloxane chromatographic peak area and solution clarity, BHT chromatographic peak area and solution clarity, and the sum of siloxane and BHT peak area and solution clarity (Fig. 5). According to the principles of the 0902-clarity test method described in the Chinese Pharmacopoeia (2015 edition) [16], the absorbance (A) of the turbidity standard solution (No. 1) at 550 nm was found to be 0.12 – 0.15, and its turbidity value (NTU) was in the range 5.4 – 6.6. According to the quantitative relationship between the adsorption of migrates and solution clarity, the maximum adsorption of migrates allowed in the sample (maximum peak area) that met the Chinese Pharmacopoeia solution clarity standard (turbidity not more than that of the standard solution) could be calculated. The total siloxane peak area, BHT peak area, and the sum of siloxane and BHT peak area were in the ranges of 25(34, 20(28, and 30(40, respectively. The total siloxane peak area should be not more than 3.5 times Aref, the BHT peak area should be not more than 3.0 times Aref, and the peak area of the sum of total siloxane and BHT should be not more than 4.0 times Aref. Therefore, by regulating the amount of volatile substances in the butyl rubber closure, the maximum adsorption of migrates in the product can be limited in order to ensure that turbidity of the sample solution is not more than that of the standard solution (No. 1).

Correlation between the total siloxane peak area and turbidity value, butylated hydroxytoluene (BHT) peak area and turbidity value, and peak area of the sum of total siloxane and BHT and turbidity value.
Comparison of the Generic and Reference Preparations
Comparison of key indicators. Key properties of the reference and generic preparations of ceftriaxone sodium for injection were compared. The key indicators of generic preparations were found to be the same as those of the domestic reference preparation, but slightly different from those of the reference preparation. Results are shown in Table 3 (the protein binding rate was measured five times for each batch of samples and the average value was taken; the content of water, value of pH and salt formation rate were measured twice for each batch of samples and averaged. In the accelerated stability test and compatibility test, 5 bottles of each batch of sample were taken at each sampling time point; after mixing the contents, related substances and polymer were measured twice and averaged. The clarity of sample in 5 bottles was measured and averaged.
Comparison of Stability
Outcomes of the accelerated stability tests of three batches of generic preparation, one batch of domestic reference preparation, and one batch of reference preparation were compared. All the pharmacopeial quality control indexes (appearance, identification, acidity or alkalinity, water, particulate matter, weight variation, visible particles, bacterial endotoxin, and sterility) were found to be within acceptable limits. The drug content in the three batches of generic preparation decreased by 3 – 10% in 6 months of the stability experiment; this was similar to that of the domestic reference preparation, but the content of the reference preparation remained almost unchanged (Table 3). Variations of the impurity content in all the preparations were similar: all impurities increased slightly during the experiment (Table 3). Increase in the total polymer content for three batches of generic preparation (0.09%) was slightly higher than that of the domestic reference and the reference preparation (0.02%).
The change in color of the generic preparation was better than that of the domestic reference and reference prepara tions. On day 0, the color of three batches of generic preparation and domestic reference preparation was lighter than the yellow-green No.7 standard solution, while the reference preparation was darker than the yellow-green No.7 standard solution. After 3 months, the domestic reference preparation was darker than the yellow-green No.7 standard solution. After 6 months, two batches of the generic preparation were found to be darker than the yellow-green No.7 standard solution, while one batch remained lighter than the yellow-green No.7 standard solution. Differences in color values of each sample solution through the course of the stability experiment are presented in Table 4.
In the test for compatibility with the rubber closures, the rate of change of turbidity of the generic preparation was slightly higher than that of the reference domestic reference preparations, which were found to be similar. The turbidity values of all the samples were much lower than that of the turbidity standard solution (No.0.5).
Comparison of Adverse Drug Reactions
Analysis of the serious adverse reactions data of ceftriaxone sodium for injection from 2010 to 2018 in the National Database of Adverse Drug Reactions showed that there were significant differences in the types of adverse reactions caused by products from different manufacturers. There were 20,254, 31,112, 19,498 and 18,138 adverse drug reaction reports for the domestic original, imported, and the two other domestic products, respectively. Among them, 2407, 2908, 2189 and 1153, respectively were serious adverse reactions.
The incidence of cardiovascular events (palpitation, cyanosis, hypertension, hypotension, arrhythmia, tachycardia, myocardial ischemia, cardiac arrest, bradycardia, etc.) caused by the domestic preparations was approximately twice as high as that caused by the imported products; the cases of respiratory damage (dyspnoea, cough, shortness of breath, laryngeal oedema, asthma, bronchial spasms, respiratory depression, etc.) were approximately 2(3 times those caused by the imported products; and the incidence of other systemic side effects (chest pain, chills, fever, periorbital oedema, oedema, high fever, paleness, fatigue, pain, oral oedema, discomfort, fainting, systemic oedema, etc.) was approximately 1.5 times higher than that caused by the imported products. These data are summarized in Table 5.
DISCUSSION
The raw material of ceftriaxone sodium is thought to be the key factor affecting the quality of the final product. This is produced in a crystallisation process, which not only determines whether salt formation has gone to completion but also influences compatibility with the rubber closure, and consequently, the type and content of impurities in the product. This study systematically compared the generic and reference preparations for consistency (Table 3). Powder x-ray diffraction was used to compare the crystallization process of ceftriaxone sodium raw material with the reference preparation; the rate of salt formation and protein binding were used to further evaluate the difference between ceftriaxone sodium raw material and the reference preparation; and the content of ceftriaxone (C18H16N10O7S3) and water in ceftriaxone sodium were taken as the routine quality control indicators of the raw material, which could also be used for evaluating the quality of ceftriaxone sodium for injection. Therefore, the quality of the raw materials used in ceftriaxone sodium for injection could be ensured, and consistent CQAs of the generic and reference preparations could be maintained.
Solution clarity is a CQA of ceftriaxone sodium for injection. The clarity of the domestic preparation was significantly different from that of the original preparation [7]. Gas chromatography analysis was used to evaluate the content of transferable siloxane and BHT in the butyl rubber closures, and the permissible limit was established using the linear relationship between solution clarity and the content of volatile components. Making sure that the content of BHT and siloxanes in the closures did not exceed this limit would help ensure that the clarity of the solution remains unchanged during storage.
The national evaluation inspection data showed that the different of impurity profile between domestic ceftriaxone sodium for injection was great. Impurities with different structures result in different toxic reactions [13]. The triazirine ring on the 3rd-position side chain of ceftriaxone sodium is the main toxic functional group [19, 20] and can regulate the expression of various gene pathways in vivo [20]. The E-isomer of ceftriaxone is evidently more toxic than ceftriaxone [19]. 7-ACA and its derivatives are important impurities that can lead to cardiac toxicity [21, 22]. Analysis of the data from the National Database of Adverse Drug Reactions showed that there were significant differences in the types of adverse reactions caused by products from different manufacturers (Table 5). The types of adverse reactions caused by the domestic original products were similar to those caused by the imported products, suggesting that they represent the inherent adverse effects of ceftriaxone sodium. The types of adverse reactions caused by the two domestic preparations were significantly different to those caused by the imported products. The types of adverse reactions also differed between the products from the two domestic companies. This is an interesting observation, since the impurity profiles of the generic and reference preparation were found to be the same, and the raw materials and production processes were controlled. Therefore, the adverse reactions caused by the domestic products would be expected to be similar to those caused by the reference preparation.
In concluding, this systematic study evaluated generic preparations of ceftriaxone sodium for injection and found that the key factor restricting consistency evaluation is the availability of basic technology in domestic pharmaceutical enterprises. For cephalosporins, the process of crystallization is the most critical factor in the production process of raw materials. The crystallinity of the raw material affects the physical and chemical properties, stability, and efficacy of the final preparation. Ensuring consistency of the crystal form of the raw material with that of the reference preparation is the basis of ensuring the consistency in the final product. The impurity profile, rate of salt formation, and protein binding rate are the key parameters to be evaluated in ceftriaxone sodium for injection. By systematically comparing the physical and chemical parameters (such as water, pH, and particulate matter) and stability of the generic preparation with those of the original preparation, differences in the quality, efficacy, and safety of the generic and original preparations can be understood, and a comprehensive quality control standard can be established. According to the generic strategy of ceftriaxone sodium for injection proposed in this study, the product achieved the expected effect. The generic products were found to be similar to the domestic original product, but differed slightly from the original preparation. The continuous improvement of the crystallisation process by domestic raw material enterprises is the key to ensuring consistent product quality and promoting further improvements in quality.
References
Center for drug Evaluation, Notice on soliciting public opinions for “The technical requirements for consistency evaluation of listed chemical generic drugs (injections)”, December 22, 2017; http://www.cde.org.cn/news.do?method=largeInfo&id=314268.
C. Q. Hu, Chin. J. Antibiot., 44(3), 281 – 288 (2019).
P. A. Lambert and B. R. Conway, J. Chemother., 15(4), 357 – 368 (2003).
I. Arnet, M. Altermatt, Y. Roggo, and G. Schnetzler, J. Chemother., 27(6), 337 – 342 (2015).
J. Xue, Y. H. Jia, J. Li, et al., Acta Pharm. Sin., 49(7), 1034 – 1038 (2014).
L. Tu, Quality Evaluation of Domestic Ceftriaxone Sodium for Injection, China Pharmaceutical University (2002).
D. S. Zhang, J. Xue, S. C. Yao, et al., Chin. J. New Drugs, 25(22), 2606 – 2613 (2016).
X. M. Chong, X. Dong, S. C. Yao, et al., Drug Dev. Ind. Pharm., 45(1), 159 – 167 (2019).
X. M. Chong, X. Dong, S. C. Yao, et al., Chin. J. Antibiot., 44(8), 942 – 945(2019).
L. Lu, Dissertation, National Institutes for Food and Drug Control, Beijing (2011).
H. T. Qu, Dissertation, Shenyang Pharmaceutical University, Shenyang (2011).
J. Yu, L. H. Yang, C. Q. Hu, et al., Central South Pharmacy, 12(02), 106 – 109 (2014).
Y. Han, B. Chen, Y. Liu, et al., Chin. J. New Drugs, 28(19), 2353 – 2359 (2019).
Y. Zhao, C. Q. Hu, S. C. Yao, et al., 11(5), 8 – 18 (2021).
J. Li, L. X. Wang, S. C. Yao, et al., Curr. Pharm. Anal., 9(2), 145 – 158 (2013).
Chinese Pharmacopoeia Commission, Chinese Pharmacopoeia (2015 Edition), General Chapter 0902, Clarity of Solution.
Chinese Pharmacopoeia Commission, Chinese Pharmacopoeia (2015 Edition), General Chapter 0406, Atomic Absorption.
Chinese Pharmacopoeia Commission, Chinese Pharmacopoeia (2015 Edition), pp. 252 – 253.
J. P. Zhang, J. Q. Qian, J. W. Tong, et al., Chem. Res. Toxicol., 26(8), 1168 – 1181 (2013).
J. Q. Qian, Y. Han, J. Li, et al., Toxicol. In Vitro, 46,137 – 147 (2018).
B. Chen, Z. Q. Gao, Y. Liu, et al., Front. Pharmacol., 26(8), 403 (2017).
Y. Han, B. Chen, J. P. Zhang, et al., Toxicol. Appl. Pharm., 27(347), 33 – 44 (2018).
Funding
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Xiaomeng Chong and Lixin Wang. The first draft of the manuscript was written by Xiaomeng Chong, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
SUPPLEMENTARY MATERIAL
SUPPLEMENTARY MATERIAL
Impurities 3 and 7 were analyzed by column switching and high-resolution mass spectrometry. The +enhanced full mass scan (EMS) spectrum and the secondary mass spectrum of +enhanced product ions (EPI) m/z = 372.0402 [M+H]+ of impurity 3 are shown in Fig. 1-1. The ions at m/z = 372.01015, 394.02192 and 409.99565 in the +EMS spectrum were predicted to appear as [M+H]+, [M+Na]+ and [M+K]+ ion peaks, respectively. Therefore, the molecular weight of impurity 3 was expected to be 371.01015 and it was predicted to be impurity E (EP). The structure and mass fragmentation pathway of impurity 3 are shown in Fig. 1-2.

Fig. 1-1. The +EMS (1) and EPI (2) spectra of impurity 3.

Fig. 1-2. The structures and mass fragmentation pathways of impurity 3.
The +EMS spectrum and secondary mass spectrum of +EPI m/z = 950.08069 [M+H]+ of impurity 7 are shown in Fig. 2-1. The ions at m/z = 950.08069 and 988.0360 in the +EMS spectrum were predicted to appear as [M+H]+ and [M+K]+ ion peaks, respectively. Therefore, the molecular weight of impurity 7 was expected to be 949.08069. Based on fragment ions in the secondary mass spectrum, it was suggested to be a dimer formed by the hydrolysis of lactone on the 3-side chain of ceftriaxone and the release of the ring with ceftriaxone. The structure and mass fragmentation pathway of impurity 7 are shown in Fig. 2-2.

Fig. 2-1. The +EMS (1) and EPI (2) spectra of impurity 7.

Fig. 2-2. The structures and mass fragmentation pathways of impurity 7.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chong, X., Wang, L., Hu, C. et al. Formulation of Ceftriaxone Sodium Under the Guidance of Drug Quality Analysis. Pharm Chem J 56, 1512–1525 (2023). https://doi.org/10.1007/s11094-023-02823-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-023-02823-2