
Senna tora (L.) Roxb. syn Cassia tora L., commonly known as sickle pod, is widely distributed in tropical Asian countries. Various parts of the plant are reported for their medicinal values due to the presence of anthraquinones, phenolic compounds, emodin, â-sitosterol and chrysophanol. Asensitive analytical procedure using UHPLC-ESI-MS/MS has been developed and validated for simultaneous quantification of five phenolic compounds in the leaves, stem and root extracts of S. tora, which were also evaluated by the radical scavenging activity for DPPH. The highest scavenging activity (95.3 ± 0.5% at 100 μg/mL) was shown by the methanolic extract of CT-2 leaves collected from Nainital. The developed method was applied to investigate the variation of five phenolic compounds in S. tora samples collected from three geographical locations. The results indicated significant variations in scavenging activity among analyzed samples collected from different locations in India, mainly due to the content of rutin.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1. INTRODUCTION
Senna tora syn. Cassia tora L. (Caesalpiniaceae) is a small annual herb or under shrub growing as common weed in Asian countries. It constitutes preparation “Dadhughnavati” which is among successful antifungal formulations [1, 2] also commonly known as Sicklepod, Coffeepod and Chakwad. Various medicinal properties have been attributed to this plant in the traditional system of Indian medicine. Various parts of the plant are reputed for their medicinal values. The seeds of S. tora have been used in Chinese medicine as an aperient, anti-asthenic and diuretic agent and also to improve visual activity [3, 4]. The leaves of S. tora contain several anthraquinone glycosides which are well known for their therapeutic value and also show purgative action [5]. It has been reported that the extract of this plant has significant antifungal activity [6]. The plant is also reported to have a significant hepatoprotective effect against the toxicity of galactosamine in primary cultured rat hepatocytes [7]. The plant has been used as a laxative and in the treatment of skin disorders [8]. The leaves and seeds of S. tora have been used for the treatment of leprosy, ring worm, flatulence, colic, dyspepsia, constipation, cough, bronchitis and cardiac disorders in the Ayurvedic system of medicine. It was reported that seeds of S. tora have antioxidant activity and contain many active substances including chrysophenol, emodin, rhein, etc. [9]. Many medicinal properties such as antimicrobial, anti-hepatotoxic and anti-mutagenic activities have been attributed to this plant [10,11, – 12].
Literature survey indicated that analytical methods including HPTLC, HPLC and CE have been used for determining the major active components of S. tora [13,14,15, – 16]. However, these methods suffer from low sensitivity, low resolution, long analysis time and large amount of solvent consumption. Ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-ESI-MS/MS) analytical method is a more powerful approach to rapidly quantify multi-component samples due to its rapid separation power, low detection limit, better accuracy, high specificity and resolution [17] as compared to the above-mentioned techniques. Therefore, the present study was aimed to develop and validate a rapid and sensitive UHPLC-ESI-MS/MS method for simultaneous determination of five phenolic compounds in the leaves, stem and root extracts of C. tora, and its application to studying variations in the content of phenolic compounds in samples collected from three different locations in India.
2. EXPERIMENTAL PART
2.1. Plant Material
Senna tora plant was collected from three different locations in India, including Lucknow (U.P.), Nainital (U.K.) and Bhavnagar (Gujarat), coded as CT-1, CT-2 and CT-3, respectively. The plant materials were authenticated at the Herbarium of the CSIR-National Botanical Research Institute, Lucknow, where voucher specimens were deposited. The samples of fresh leaves, stems and roots were collected, washed thoroughly and dried in shade. Each sample (50 g) of leaves, stem, and root was powdered in a mixer-grinder and used for the further study.
2.2. Extraction
Powdered samples of S. tora, were extracted with methanol by cold extraction process (Maceration). Methanol was added to plant material in a conical flask, which was kept at room temperature overnight with occasional shaking. The extract was then filtered using Millipore filter and solvent under reduced pressure at 550C by Buchi evaporator. The extract was stored in dark bottle and kept at 40C until analysis.
2.3. Reagents and Chemicals
Acetonitrile, methanol (LC-MS grade) and formic acid (analytical grade) purchased from Fluka, Sigma-Aldrich (St. Louis, MO, USA) were used in mobile phase and sample preparation throughout the LCMS analysis. Ultra pure water, obtained from Direct-Q system (Millipore, Milford, MA, USA) was used throughout the analysis. Gallic acid, syringic acid, p-coumaric acid, ferulic acid and rutin were purchased from Sigma-Aldrich.
2.4. Sample Preparation
Dried residues (1 mg) of each sample were weighed accurately, dissolved in 1 mL of methanol and sonicated using ultrasonicator (Bandelin SONOREX, Berlin). The solutions were filtered through 0.22 μm syringe filter (Millex-GV, PVDF, Merck Millipore, and Darmstadt, Germany). The filtrates were diluted with acetonitrile to final working concentration, vortexed for 30 sec, and 2 μL aliquot was injected into the UPLC-MS/MS system for analysis.
2.5. Preparation of Standard Solution s
A mixed standard stock solution (1 mg/mL) of selected analytes was prepared in methanol. The working standard solutions were prepared by diluting the mixed standard solution with acetonitrile to a series of concentrations within the ranges from 0.5 to 200 ng/mL used for plotting calibration curve. The standard stock and working solutions were stored at –20°C until use and vortexed prior to injection.
2.6. Instrumentation and Analytical Conditions
The ultra-high performance liquid chromatography electrospray ionization tandem mass spectrometry (UHPLC-ESI-MS/MS) analysis was performed on Waters Acquity UPLC system (Waters, Milford, MA, USA) interfaced with hybrid linear ion trap triple-quadrupole mass spectrometer (API 4000 QTRAP MS/MS system from AB Sciex, Concord, ON, Canada) equipped with electrospray (Turbo V) ion source. TheWaters Acquity UPLC system was equipped with a binary solvent manager, sample manager, column oven and photodiode array detector (PDA). AB Sciex Analyst software version 1.5.1 was used to control the LC-MS/MS system and for data acquisition and processing. All the statistical calculations related to quantitative analysis were performed using Graph Pad Prism software version 5.
UPLC conditions. The chromatographic separation of selected analytes was achieved on an Acquity UPLC BEH C18 column (50 mm × 2.1 mm id, 1.7 μm) at a column temperature of 25°C. Analysis was completed with gradient elution of 0.1% formic acid in water (A) and acetonitrile (B) as mobile phase at a flow rate of 0.3 mL/min. The 2.5 min UPLC gradient system was as follows: 0 – 1.5 min, 20 – 58% B; 1.5 – 1.8 min, 58 – 62% B; 1.8 – 2.0 min, 62 – 62% B; 2.0 – 2.5 min, 62 – 20% B; equilibration time 2 min. The sample injection volume was 2 μL.
MS conditions. All analytes were detected in negative electrospray ionization mode using precursor ion scan, and the mass spectra were recorded in the range of m/z 100 – 1000 at a cycle time of 9s and a step size of 0.1 Da. Nitrogen was used as the nebulizer, heater, and curtain gas as well as the collision activation dissociation (CAD) gas. Optimized source parameters were as follows: ion spray voltage set at -4200 V, curtain gas, nebulizer gas (GS1) and heater gas (GS2) set at 20, 20 and 20 psi, respectively, with a source temperature of 450°C. The CAD gas was set at medium, and the interface heater was on. Simultaneous quantification of analytes was carried out using multiple reaction monitoring (MRM) acquisition mode at unit resolution and its conditions were optimized for each compound during infusion. The transitions and optimized compound dependent MRM parameters: declustering potential (DP), entrance potential (EP), collision energy (CE) and cell exit potential (CXP) for each analyte are listed in Table 1.
2.7. DPPH Radical Scavenging Activity
The DPPH radical scavenging activity was determined by the method of Shimada [18]. According to this, 2 mL of DPPH methanol solution was added to sample solution (1 mg/mL) at 20, 40, 60, 80 and 100 μL. The mixture was shaken vigorously and allowed to stand at room temperature in the dark for 30 min. Then the absorbance was measured at 517 nm in a spectrophotometer. The same procedure was repeated to obtain the antioxidant capacity of quercetin which was used as positive control.
3. RESULTS AND DISCUSSION
3.1. Optimization of LC Conditions
In order to achieve rapid and efficient analysis, a short chromatographic column Acquity UPLC BEH C18 column (50 mm × 2.1 mm id, 1.7 μm) was employed in the UPLC system. Different mobile phase systems (water–methanol, water–acetonitrile, 0.1% formic acid in water– methanol, 0.1% formic acid in water– acetonitrile, 0.1% formic acid in water – 0.1% formic acid in methanol and 0.1% formic acid in water – 0.1% formic acid in acetonitrile) using different mobile phase compositions in gradient elution at various flow rates (0.2, 0.3, 0.35, 0.4 and 0.5 mL/min) and column temperatures (25, 30, 35, 40 and 50°C) were examined and compared in order to obtain better chromatographic behavior and appropriate ionization. It was found that 0.1% formic acid in water–acetonitrile was better than other systems. Finally, 0.1% formic acid in water–acetonitrile was chosen as the eluting solvent system at a flow rate of 0.3 mL/min with the column temperature of 25°C to give the acceptable separation and ionization within a run time of 2.5 min.
3.2. Optimization of MS Conditions
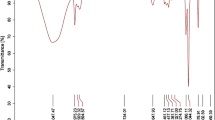
Preliminarily, each targeted analyte was infused into the mass spectrometer and mass spectra were studied in both positive and negative ionization modes. During tuning (Q1 scan) it was observed that all analytes exhibited good signal sensitivity in negative ionization mode. Then, the compound dependent MRM parameters: DP, EP, CE and CXP were optimized for each targeted analyte by injecting the individual standard solution into the mass spectrometer to achieve the most abundant, specific and stable MRM transition shown in Table 1. The source parameters including the curtain gas, GS1, GS2 and ion source temperature were further optimized in order to get the highest abundance of precursor-to-product ions. The optimized compound dependent parameters and source parameters were combined, and finally the optimized UHPLC-ESI-MS/MS method in MRM acquisition mode was applied to quantify five selected bioactive constituents in the leaves, stem and root extract of S. tora. The UHPLC-MRM ion chromatograms of analytes is shown in Fig. 1.

UHPLC-MRM ion chromatograms of analytes extracted from S. tora.
3.3. Identification of Target Analytes
Target analytes in the samples were unambiguously identified by the comparison of their retention times and MS/MS spectra to those of the authentic standard solutions. The mass spectra generated for all the targeted compounds by ESI-MS in the negative ion mode gave the deprotonated molecules [M-H]–. The MS/MS spectra of the five phenolic compounds are shown in Fig. 2. The most abundant fragment ion of each compound was selected for MRM transition. The major fragment ion in the MS/MS spectra of the [M-H]– ions of gallic acid, m/z 169 [M-H]–, and p-coumaric acid m/z 163 [M-H]– was generated due to the loss of CO2 molecule, providing an anion of [M-H-CO2]–. Syringic acid m/z 197 [M-H]– produced major fragment ion at m/z 182 due to loss of methyl radical. Ferulic acid, m/z 193 [M-H]– yielded the predominant product ion at m/z 134 corresponding to [M-H-COO-CH3]. Rutin, m/z 609 [M-H]–, predominantly generated product ion at m/z 301 due to O-glycosidic cleavage.

MS/MS spectra of five selected phenolic compounds extracted from S. tora.
3.4. Validation Procedure for Quantitative Analysis
The proposed UHPLC-ESI-MS/MS method for quantitative analysis was validated according to the guidelines of International Conference on Harmonization (ICH, Q2R1) by determining linearity, limit of detection (LOD), limit of quantification (LOQ), precision, stability and recovery.
Linearity, LOD and LOQ. The stock solution was diluted with methanol to provide a series of concentrations in the range of 0.5-200 ng/mL for the construction of calibration curves. The linearity of calibration was performed by the analytes peak area ratios versus the nominal concentration and the calibration curves were constructed with a weight (1/x2) factor by least-squares linear regression. The applied calibration model for all curves was y = a x + b, where y = peak area of analyte, x = concentration of analyte, a = slope of the curve, and b = intercept. The LODs and LOQs were measured with S/N of 3 and 10, respectively, as criteria. The results are summarized in Table 2. All calibration curves indicated good linearity with correlation coefficients (r2) within 0.9985 – 0.9997 in the indicated test ranges. The LOD for each analyte varied from 0.12 to 0.41 ng/mL and the LOQ varied from 0.36 to 1.24 mg/mL.
Precision, stability and recovery. The intra-day and inter-day variations, which were chosen to determine precision of the developed method, were investigated by determining five selected analytes in six replicates during a single day and by duplicating the experiments on three consecutive days. Variations of the peak area were taken as the measures of precision and expressed as percentage relative standard deviations (RSD). The overall intra-day and inter-day precisions were not more than 1.93%. Stability of sample solutions stored at room temperature was investigated by replicate injections of the sample solution at 0, 2, 4, 8, 12 and 24 h. The RSD values of stability of the 5 selected analytes were ≤2.73%. The recovery test was applied to evaluate the accuracy of this method. Three different concentration levels (high, middle and low) of the analytical standards were added into the samples in triplicate and the corresponding average recoveries were determined. The analytical method developed had good accuracy, with overall recovery in the range from 98.50 to 102.50% (RSD ≤ 1.91%) (Table 2).
Quantitative analysis of samples. The developed UHPLC-ESI-MS/MS method in MRM mode was applied to determine the content of five phenolic compounds in leaf, stem and root extracts of Senna tora. Data on the content of five bioactive constituents are summarized in Table 3. Quantitative analysis showed that rutin was the predominant (34.2 ± 0.3)–(6540 ± 1.0) μg/g component in all samples except CT-1R, CT-2R and CT-3R. Ferulic acid was second abundant constituent with the content range of (31.6 ± 0.1 – 95.2 ± 0.3) μg/g. All constituents except gallic acid and syringic acid were detected in all investigated samples. Gallic acid was detected only in CT-1S, CT-3S and CT-3R, whereas syringic acid was detected in CT-1S, CT-1R, CT-2S, CT-2R and CT-3R. The overall quantitative analysis indicated that the highest total content (6704.0 μg/g) of five phenolic compounds was detected in CT-2L.
3.5. Antioxidant Activity
DPPH radical-scavenging activity. DPPH scavenging activity is based on the ability of sample to donate hydrogen which reacts with the DPPH radical. When a solution of DPPH is mixed with a substance that can donate a hydrogen atom or transfer electron to DPPH, thus neutralize the free radical character and then this gives rise to the reduced form DPPH (non-radical) with the loss of the violet color. Radical scavenging activity increases with increasing percentage of the free radical inhibition [19]. The color change from violet to yellow and fall in absorbance of the stable radical DPPH was measured for different concentrations. In the present study, the Methanolic extract of CT-2 Leaves showed higher antioxidant activity. This is due to the presence of the most bioactive compounds such as polyphenols including tannins, flavonoids in higher polar fraction [20].
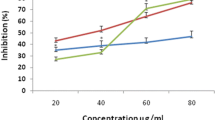
Plants are potential sources of natural antioxidants. It produces various antiradical compounds to counteract reactive oxygen species (ROS) in order to survive [21]. In our study, the maximum DPPH antiradical activity (95.3 ± 0.5)% was shown by CT-2 (Nainital), methanolic extract of S. tora leaves at 100 μg/mL as shown (Fig. 3).

Histogram of the scavenging activity of leaf, stem and root extracts of S. tora samples CT-1, CT-2, and CT-3 at different concentrations (μg/mL).
In concluding, the present study was devoted to quantification of five phenolic compounds in methanolic extracts of leaves, stems and roots of S. tora using UHPLC-ESI-MS/MS method. The developed method was applied to investigate geographical variations among samples in term of content of quantified compounds. Results indicated that the highest total content of five phenolic compounds was detected in the extract of S. tora leaves collected from Nainital, Uttarakhand, India. Rutin was the predominant compound in all samples except root extracts. Therefore this validated method and study may be helpful in quality control of S. tora. It is suggested that the methanolic extract of S. tora leaves collected from Nainital is a potential source of natural antiradicals. This methanolic extract exhibited the highest radical scavenging activity, probably due to the maximum content of flavonoids present in CT-2 leaves.
References
T. K. Acharya and I. B. Chatterjee, Lloydia, 38, 218 – 220 (1975).
T. Hatano, H. Uebayashi, S. Shiota, et al., Chem. Pharm. Bull. (Tokyo), 47, 121 – 127 (1991).
K. R Kirtikar and B. D. Basu’s Illustrated Indian Medicinal Plants, ed. by E. Blatter, J. E. Caius and K. S. Mhaskar, (1975), Vol. 11, pp. 878 – 879.
L. Asolkar, K. K. Kakkar, and O. J. Chakre, Second Supplement to Glossary of Indian Medicinal Plants (1992), pp. 180 – 181.
M. Pal, D. K. Ro and P. R. Pal, Indian J. Pharm., 39, 116 – 117 (1977).
P. K. Mukherjee, K. Saha, B. P. Saha, M. Pal. Das, Phytother. Res., 10, 551 – 552 (1996).
S. M. Wong, M. M. Wong, O. Seligmann, H. Wagner, Phytochemistry, 28, 211 – 214 (1989).
J. D. Hooker, The Flora of British India, Reeve and Co. (1879), Vol. 11, p. 26.
K. C. Huang, The Pharmacology of Chinese Herbs, CRC Press, Boca Raton (1993), p. 103.
S. M. Wong, M. M. Wong, O. Seligmann, and H. Wagner, Planta Medica, 55, 276 – 280. (1989).
J. S. Choi, H. J. Lee, K. Y. Park, et al., Planta Medica, 63, 11 – 14 (1997).
G. C. Yen and D. Y. Chung, J. Agric. Food Chem., 47, 1326 – 1332 (1999).
E. Priya, P. Sanmuga, S. Senthamil, and P. P. Prakash, J. Liq. Chromatogr. Relat. Technol., 36, 2525 – 2533 (2013).
Qiwei Zhang, Y. Jian and Z. Jun, Chin. Trad. Herbal Drugs, 2, 007 (1996).
Z. H. O. U Jia-ru, J. Anhui Agri. Sci., 21, 023 (2008).
J. Koyama, I. Morita, K. Kawanishi, et al., Chem. Pharm. Bull., 51, 418 – 420 (2003).
X. Liang, L. Li, J. Tian, et al., Phytochem. Anal., 25, 537 – 543 (2014).
K. Shimada, K. Fujikawa, K. Yahara, T. Nakamur, J. Agric. Food Chem., 40, 945 – 948 (1992).
M. Schreiner, B. Beyene, A. Krumbein and H. Stutzel, J. Agric. Food Chem., 57, 7259 – 7263. (2009).
H. A. El-Shemy, M. A. Aboul-Soud, A. A. Nassr-Allah, et al., Curr. Med. Chem., 17, 129 – 138 (2010).
F. Lu and L. Y. Foo, Toxicological aspects of Food Antioxidants, Marcel Dekker: New York (1995).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shukla, S., Pandey, R., Kumar, B. et al. Phenolic Compounds and Radical Scavenging Activity of Senna tora (L.) Roxb. Herbs Collected from Various Geographical Locations of India. Pharm Chem J 55, 684–690 (2021). https://doi.org/10.1007/s11094-021-02478-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-021-02478-x