
Methods for determining the quantitative contents of meta-iodobenzylguanidine (MIBG) and iodide ion, the chemical purity of MIBG, and the radiochemical purity of [131I]-MIBG using HPLC with spectrophotometric and radiometric detection were developed. The detection limits were 50 – 250 μg/mL for MIBG, 0.78 – 58.5 μg/mL for NaI, and 300 – 2250 kBq for [131I]-MIBG. Validation of the methods confirmed that their characteristics met the specified requirements.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
meta-Iodobenzylguanidinium sulfate (Fig. 1) labeled with 131I ([131I]-MIBG) is widely used in medical practice for diagnosis and therapy of malignant neoplasms [1, 3].

meta-Iodobenzylguanidinium sulfate.

meta-Iodobenzylammonium chloride.
[131I]-MIBG is a structural analog of noradrenaline and accumulates selectively after i.v. injection in tissues with high adrenergic innervations (stimulated by adrenaline and noradrenaline), especially in neuroectodermal tumors.
The preparation possesses clearly pronounced affinity for the target organ and damaged tissues and accumulates mainly in excretory organs, salivary glands, adrenals, lungs, and spleen [2].
The main processing impurities that should be controlled are the MIBG precursor meta-iodobenzylammonium chloride (MIBA) and the medical radioisotope 131I used in the synthesis. International pharmacopoeias establish the following limits on impurity contents: MIBA, ≤ 1%; 131I, ≤ 5% for diagnosis and ≤ 7% for therapy. The present article focused on method development for controlling them.
Chemical purity and content of MIBG. Nonaqueous titration and HPLC can be used to determine the quantitative content of main ingredient. Each method has its pluses and minuses. HPLC is a relative method that must be calibrated using a reference standard of the drug substance, which is not required for titration because the titration is measured using common compounds. The absoluteness of the method allows the compound concentration to be determined immediately without using calibration curves and mathematical models. However, nonaqueous titration places high demands on the experimental set up and the quality of used reagents. Also, impurities that affect the analytical results must be masked. Mixtures of aromatic amines were titrated before [3,4,5]. However, HPLC can determine the content and chemical purity in one analysis.
HPLC methods for determining the contents of main ingredients and chemical purity are known from European (EuPha) [6], US (USP) [7], and International Pharmacopoeias [8] and the scientific literature [9,10,11]. However, they had different drawbacks in practice so we decided to develop our own.
Radiochemical purity can be determined by planar chromatography and electrophoresis with detection by β- or γ-spectrometry and HPLC with β - and γ -radiation detectors. The accuracy of planar chromatography methods depends strongly on the collimator slit width and geometry for β - and γ -scanners and on the distance from the plate to the collimator or on the size of the plate tracks and paper, the activity of which is measured using a spectrometer. Such problems are not observed for HPLC. However, the Raytest Ramona Star detector used by us gave a flattened and broadened analyte peak for γ -rays because of the measurement cell geometry and hindered rapid analyses. The lower background of β -particles (as compared with γ -photons) was considered in selecting an analytical method and detector. Because of this and an existing developed method for determining the chemical purity and content of MIBG, HPLC with β -spectrometric detection was selected.
Experimental Part
Samples of MIBG drug substance, finished preparation [131I]-MIBG, and processing impurities MIBA, NaI, and Na131I were used for the research.
A pharmacopoeial standard (EuPha: Iobenguane sulfate, cat. Code 10328000) was used to develop the method for determining MIBG content. The standard for developing a method to determine the content of iodide ions was working reference standard (WRS) NaI (analytically pure, Reakhim). The standard for developing a method to determine the radiochemical purity of [131I]-MIBG was a certified Na131I solution (KPRI).
Solutions of actual samples were prepared from MIBG and MIBA (KPRI).
The studies used a Shimadzu LC-20AD HPLC with a Shimadzu SPD-20A UV detector and a Shimadzu LC-20AT HPLC (Shimadzu Corp., Japan) with a Raytest Ramona Star radiometric detector (Elysia-Raytest GmbH, Germany) with a cell for detecting β-radiation.
The ranges of studied concentrations and activities were “cold” MIBG, 50 – 250 μg/mL; NaI, 0.78 – 58.5 μg/mL; and [131I]-MIBG + Na131I, 300 – 2250 kBq.
All validation tests were planned, set up, and executed in compliance with State Pharmacopoeia XIVth Ed. (SP XIV) [12], RMG 61-2010 “Accuracy, trueness, and precision measures of the procedures for quantitative chemical analysis” (RMG 61-2010) [13]. Calculations were carried out according to SP XIV, RMG 61-2010, MI 2083-90 “Indirect measurements. Determination of measurement results and estimation of their errors” (MI 2083-90) [14] and GOST R 8.736-2011 “Multiple direct measurements. Methods for processing measurement results” [15]. Dixon and Grubbs criteria were used to check for outliers; Fisher criterion, the model adequacy; Pearson criterion, correlation functions.
The significance of coefficients in regression equations was checked using the Student and p-criteria. Critical values of Student and Fisher statistical distributions and Grubbs criteria were taken from Tables of Mathematical Statistics [16] and GOST R ISO 5725-2-2002 “Accuracy (trueness and precision) of measurement methods and results” [17].
Results and Discussion
Review of existing analytical methods. The pharmacopoeial monograph in the EuPha 9.0 proposes using wavelength 254 nm for detection. Research showed that the absorption maximum was located near 230 nm (Fig. 3). The mobile phase composition with a pH of 8 raised issues because pH ≤ 7.5 is critical if silica gel without a grafted phase is used. The USP does not justify the choice of rather large-grained sorbent (10 μm) and the semi-preparative column size. The phosphoric acid in the mobile phase limits the ability to use the method with evaporative and aerosol detectors. Poor reproducibility of analytical results was observed during selection of the separation conditions. The EuPha method gave a highly flattened asymmetric analyte peak that eluted much later than the regulated time, i.e., 13 min using a Merck Purospher STAR Si column (5 μ, 125 × 4 mm) vs. 7 min using a silica gel column (5μ, 250 × 4 mm). MIBA and MIBG in the tested mixture could not be separated using the USP method.

UV spectrum of MIBG
Separation process. The roles of the mobile-phase modifiers were defined during development of the analytical method. HOAc accelerated desorption of the compound from the column. Et2NH enhanced adsorption and the peak asymmetry diminished (Fig. 4).

Effect of modifiers on separation of MIBG and MIBA.
MIBG could be expected to have strongly basic properties because of the guanidine moiety in the cation. However, studies of MIBG solubility found that the benzene ring contributed much more to the chemical properties than the guanidine group. Small amounts of added acid and amine (≤ 0.5 mL/100 mL of eluent) had positive effects (decreased asymmetry, narrower width, shorter retention time). Addition of 1 mL/100 mL of eluent and greater caused a slight reduction in the separation parameters, possibly because of the increased ionic strength of the solution and increased competition of the analyte and solvent in the eluent layer on the sorbent particle surface [18].
The effect of pH on the distribution of MIBG in the mobile and stationary phases was studied. The retention time of the salt was shorter in acidic solution; longer, in basic. The dependences between pH and modifier concentration and separation parameters were nonlinear (Fig. 5).

Effect of pH on broadening and retention time of the peak.
Selection and optimization of MIBG analytical conditions. Validation and study of method applicability limits. The research and development on the separation conditions for determining the MIBG content in several batches of samples found the optimal conditions to be:
Column | Merck ChromoLith High Resolution RP-18 100 × 4.6 mm |
Mobile phase | 25%v/v CH3CN: 75%v/v H2O : 0.2%v/v CH3COOH : 0.1%v/v (C2H5)2NH |
Flow rate | 2.0 mL/min |
Temperature | 45°C |
Detector | spectrophotometric, 230 nm |
Sample volume | 20 μL |
Analysis time | 2 min |
Validation of the process found that MIBG concentrations in the range 50 – 250 μg/mL were described most accurately by the equation C = 7.17 × 10–5 × x, where C is the MIBG concentration (μg/mL); x, peak area. The direct dependence of the concentration on peak area had a high correlation coefficient (r = 0.9999). The regression model described the obtained results with a determination coefficient of 0.9999 (Figs. 6 and 7). Table 1 presents the precision as the confidence interval limits; the trueness as the standard deviation of unexcluded systematic error in the analytical method, and the accuracy as the standard deviation of the uncertainty in the analytical results (total systematic and random errors).

Chromatogram for separation of a mixture of MIBA (0.939 min) and MIBG (1.179 min).

Plot of MIBG sulfate concentration vs. peak area.
The dependence of NaI concentrations in the range 0.78 – 58.5 μg/mL on peak area obeyed the equation: C = 1.51 × 10–5 × x, where C is the NaI concentration (μg/mL); x, peak area. The direct dependence of concentration on peak area had a high correlation coefficient (r = 0.9997) (Fig. 9). A regression model described the results with a determination coefficient of 0.9994. Table 1 presents the precision, trueness, and accuracy.
The radiation stability of MIBG sulfate was studied to determine if the proposed method could be used with MIBG in the presence of various radioprotectors. The method was highly selective and could be used to separate mixtures of MIBG, iodides, Me2CO, methylethylketone, EtOH, and p-hydroxybenzoic acid (Fig. 8).

Chromatogram of a mixture of NaI, Me2CO, MIBA, MIBG, and unknown impurities after sample irradiation (λ = 230 nm).

Plot of NaI concentration vs. peak area.

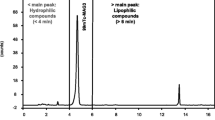
Chromatogram of a mixture of Na 131I and [131I]-MIBG.
Optimization and validation of the method for determining MIBG radiochemical purity. The analytical conditions had to be slightly altered to determine MIBG radiochemical purity because of specifics of the apparatus and the operation of the radiometric detector:
Column | Merck ChromoLith High Resolution RP-18 100 × 4.6 mm |
Mobile phase | 33%v/v CH3CN : 66%v/v H2O : 0.2%v/v CH3COOH |
Flow rate | 1.5 mL/min |
Temperature | 40°C |
Detector | β-spectrometric, channels |
Sample volume | 0 – 1023 |
Analysis time | 20 μL |
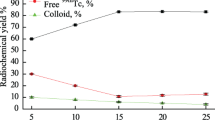
The WRS for [131I]-MIBG was an impurity (Na131I) that was detected by γ-spectrometry because a reference standard could not be obtained. The method of additions using a standard impurity as described in RMG 61-2010 was used to evaluate the quality indicators. A sample with specific activity 768 kBq/mL and an additive with Na131I = 127.6 kBq/mL were used (Fig. 10). Errors were calculated according to MI 2083-90 and GOST R 8.736-2011. The indicator values in percent of the radiochemical purity were precision 2.2%; trueness, 2.93; accuracy, 6.14.
Linearity was experimentally demonstrated in samples with identical radiochemical purity but different specific activities in the range 300 – 2250 kBq/mL. The first stage of statistical processing consisted of reproduction of the regression equation, calculation of statistical criteria for component peak areas as functions of sample activity, and confirmation of the adequacy of the constructed model. The second stage included reproduction of the linear regression equation describing the radiochemical purity as a function of added sample activity. Table 2 presents the values for the statistical criteria and regression models.
References
F. Giammarile, A. Chiti, et al., “EANM procedure guidelines for 131I-meta-iodobenzylguanidine (131I-mIBG) therapy,” Eur. J. Nucl. Med. Mol. Imaging, 35, 1039 – 1047 (2008).
E. Bombardieri, F. Giammarile, et al., “ 131I/127I-Metaiodobenzylguanidine (mIBG) scintigraphy: procedure guidelines for tumor imaging,” Eur. J. Nucl. Med. Mol. Imaging, 37, 2436 – 2446 (2010).
R. Pohloudek-Fabini and K. Konig, Pharmazie, No. 12, 752 – 756 (1958).
I. Denesh, Titration in Nonaqueous Media [Russian translation], Mir, Moscow (1971).
S. Siggia and J. G. Hanna, Quantitative Organic Analysis via Functional Groups, Wiley, New York (1979).
Iobenguane sulfate for radiopharmaceutical preparations (2351), European Pharmacopoeia 9.0, Strasbourg (2018).
Iobenguane I-123 Injection, The United States Pharmacopoeia 42, The National Formulary 37, United Book Press, Inc., Baltimore, MD (2018).
Iobenguane (131I) injection, The International Pharmacopoeia, 8th Ed. (2018).
T. J. Mangner, Potential Artifacts in the Chromatography of Radiopharmaceuticals. Analytical and Chromatographic Techniques in Radiopharmaceutical Chemistry, Springer, New York (1986).
J. J. R. Mertens, W. Vanryckeghem, and L. Carlsen, in: Progress in Radiopharmacy. Proceedings of the Second European Symposium on Radiopharmacy and Radiopharmaceuticals, P. H. Cox, et al. (eds.), (1986), p. 101 – 109.
D. M. Wieland, J. Wu, L. E. Brown, et al., J. Nucl. Med., 21, 349 (1980).
State Pharmacopoeia of the Russian Federation, XIIIth Ed., Vol. 1 – 3, Moscow (2015).
RMG 61-2010. Accuracy, trueness, and precision measures of the procedures for quantitative chemical analysis, Standartinform, Moscow (2013).
MI 2083-90. Indirect measurements. Determination of measurement results and estimation of their errors, USSR Committee for Standardization and Metrology, Moscow (1991).
GOST R 8.736-2011. Multiple direct measurements. Methods for processing measurement results, Standartinform, Moscow (2013).
L. N. Bol’shev and N. V. Smirnov, Tables of Mathematical Statistics [in Russian], Nauka, Moscow (1983).
GOST R ISO 5725-2-2002. Accuracy (trueness and precision) of measurement methods and results, Standartinform, Moscow (2009).
O. B. Rudakov, The Chromatographer’s Companion. Methods of Liquid Chromatography [in Russian], Vodolei, Voronezh (2004).
Acknowledgments
The work used equipment at the Unikum VVR-ts, KPRI.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 54, No. 2, pp. 54 – 59, February, 2020.
Rights and permissions
About this article

Cite this article
Kapustyan, A.V., Buryak, A.A. & Boiko, V.M. Development and Validation of an Analytical Method for Meta-Iodobenzylguanidine Sulfate Radiolabeled with 127I and 131I. Pharm Chem J 54, 201–207 (2020). https://doi.org/10.1007/s11094-020-02178-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-020-02178-y